
Abstract
Prions are proteinaceous pathogens responsible for a wide range of neurodegenerative diseases in animal and human. Prions are formed from misfolded, ß-sheet rich, and aggregated conformers (PrPSc) of the host-encoded prion protein (PrPC). Prion replication stems from the capacity of PrPSc to self-replicate by templating PrPC conversion and polymerization. The question then arises about the molecular mechanisms of prion replication, host invasion, and capacity to contaminate other species. Studying these mechanisms has gained in recent years further complexity with evidence that PrPSc is a pleiomorphic protein. There is indeed compelling evidence for PrPSc structural heterogeneity at different scales: (i) within prion susceptible host populations with the existence of different strains with specific biological features due to different PrPSc conformers, (ii) within a single infected host with the co-propagation of different strains, and (iii) within a single strain with evidence for co-propagation of PrPSc assemblies differing in their secondary to quaternary structure. This review summarizes current knowledge of prion assembly heterogeneity, potential mechanisms of formation during the replication process, and importance when crossing the species barrier.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prion diseases are a group of expanding fatal, infectious, neurodegenerative disorders affecting humans and wild-life or farmed animals. These diseases include Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in a wide range of cervids (Collinge 2001; Prusiner 1998). Prion diseases have a worldwide distribution. In humans, they are considered as rare diseases; sporadic CJD has a worldwide incidence of 1.5 cases per year per million. In animals, the situation is more contrasted. Up to 12,000 cases of classical BSE (C-BSE) were reported each month in the UK at the peak of the “mad cow” epidemic in the early 1990s. In certain regions of North America, CWD is endemic, reaching a > 70% prevalence in captive herds (Moazami-Goudarzi et al. 2021). Classical scrapie, which is endemic in Europe, had prevalence estimates in sheep that varied from 0 (Portugal) to 245 ‰ (Cyprus) before implementation of control measures at the beginning of the twenty-first century (Fediaevsky et al. 2008). Prion diseases are constantly (re-)emerging. While circulating only in North America and for a limited period in Korea, CWD has suddenly emerged in Scandinavia in 2016 (Benestad et al. 2016) and is now threatening Europe (Hazards et al. 2019). A camelid prion disease has been discovered in Maghreb in 2018 (Babelhadj et al. 2018). In humans, the last identified prion disease is called variably protease-sensitive prionopathy (VPSPr). This very rare disease was discovered in 2008 (Notari et al. 2018).
Prions, the causative pathogens of prion diseases (Prusiner 1982), can propagate between different species. Prions have a zoonotic potential or are truly zoonotic agents, as exemplified by the emergence of variant CJD (vCJD) in humans due to the consumption of C-BSE contaminated food. There are currently great uncertainties about the exact number of vCJD asymptomatic individuals more than 20 years after emergence (Gill et al. 2020), because the molecular determinants of the species (or transmission) barrier that limits inter-species prion propagation remain mostly unknown.
Prions are formed from abnormally folded conformers (PrPSc) of the cellular form of the prion protein (PrPC). In the mature form, PrPC is a ~ 210-amino-acid-long, monomeric, glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein with high primary and tertiary structure identities across mammals. While PrPC N-terminus is unstructured, PrPC C-terminus is folded into a globular domain composed of three α-helices and a small two-stranded β-sheet (Eghiaian et al. 2004; Riek et al. 1996). PrPC is ubiquitously expressed in the organism; the highest levels are found in the brain. PrPC exerts a growing number of signaling functions in healthy individuals, from neuroprotection to stem cell biology (Halliez et al. 2015). PrPC is also involved in other pathologies including cancer (Mouillet-Richard et al. 2021) and Alzheimer’s disease (Lauren et al. 2009). Abnormally folded PrPSc is enriched in β-sheet content and assembles into polydisperse amyloidogenic assemblies. Cryo-electron microscopy near-atomic-resolution structures of purified PrPSc are currently emerging; they suggest a reorganization of PrPC in parallel in-register assembly within a fibrillar supra-organization (Kraus et al. 2021; Manka et al. 2022). PrPSc assemblies deposit mostly in the CNS. They also accumulate at variable levels, and in a strain-dependent manner, in many peripheral tissues, including notably the lymphoid tissue (spleen, lymph nodes, etc.).
As for conventional pathogens, PrPSc replicates and its biological activity or infectivity can be titrated, by bioassays in bioindicator animals or permissive cells or by cell-free amplification assays (Moudjou et al. 2020). PrPSc self-replicates by templating PrPC conversion and polymerization. In sporadic cases, host PrPC would, for unknown reason, misfold spontaneously into a replicative conformer initiating the self-replication process. In inherited cases, host PrPC would misfold due to mutations in the PrP encoding gene (PRNP). In acquired cases, the initial PrPSc conformer would be acquired accidentally.
The quantitative aspects of prion accumulation (PrPSc levels or infectivity) during neuroinvasion have served to elaborate prion replication models. In essence, these models state that monomeric PrPC is constantly recruited by PrPSc assemblies, allowing a cooperative production and accumulation of further PrPSc assemblies. The most popular models remain so far the autocatalytic conversion model by Griffith (1967) and the nucleated-polymerization model by Lansbury and Caughey (1995). While these two models describe qualitatively the cooperativity of PrPSc accumulation, they consider PrPSc assemblies as a static object. Moreover, they fail to describe the process of prion structural diversification and evolution. In this review, we show evidence that PrPSc assemblies are structurally heterogeneous. PrPSc heterogeneity will be considered at different scales, at the host population level with different circulating PrPSc conformations or strains but also within a single host and within a single strain. In the second part of the review, we will elaborate on the mechanisms of PrPSc diversification. In the third part of the review, we will discuss the importance of PrPSc heterogeneity in prion cross-species transmission.
Structural diversity of PrPSc assemblies
PrPSc assemblies conformational heterogeneity must be considered at three different scales. At the population level, this diversity corresponds to the existence of different strains circulating in different hosts from the same species. Strains are conformational variants of PrPSc with specific biochemical and biological phenotypes. At the individual host level (i.e., field isolate), this corresponds to the coexistence of strains. One may be dominant and impose its phenotype but subdominant strains may co-propagate. At the strain level, this corresponds to different PrPSc subpopulations varying not only in their quaternary structures, but also at lower levels of structuration (tertiary and secondary), and impacting in turn their replicative and biochemical properties.
Structural diversity of PrPSc assemblies at the host population level
In prion-susceptible host population, multiple prion strains are recognized due to structurally distinct PrPSc assemblies. Biochemically, these PrPSc species can differ in their posttranslational modifications (e.g., their relative ratio of glycoforms), their relative resistance to protease digestion or relative stability towards chaotropic treatment (e.g., urea or guanidine hydrochloride) or heat treatment (for review, see Beringue et al. 2008b). This points to profound differences at different structural levels. Phenotypically, in both infected hosts and in laboratory animals (notably transgenic mice expressing the mammalian PrPC of interest), prion strains exhibit specific and synchronous incubation times, stereotyped clinical signs, and neuropathology, including the deposition of PrPSc and of vacuolar lesions in specified regions of the CNS, and specific tropism for peripheral tissues including the lymphoid tissue (Beringue et al. 2008b).
Based on biochemical and neuropathological analyses in affected individuals and/or on strain typing studies in laboratory animals, at least eight different strains have been isolated from scrapie-infected sheep and goats (including atypical/Nor98 strain), three strains from bovines with BSE (the classical one responsible for the “mad cow” crisis (C-BSE) and two atypical, putatively sporadic strains), five strains from North American cervids infected with CWD, three strains from Scandinavian CWD, and ten strains from humans with prion diseases (Fig. 1a). This points to the fact that a given PrPC primary structure—from a given host—can stably adopt different strain structural determinants (SSD) in the PrPSc state. Where the SSD are located in PrPSc assemblies and how the SSD specify different biological phenotypes remain poorly understood.

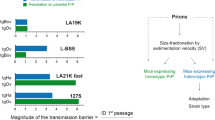
The many scales of PrPSc assemblies structural diversity. a Inventory of the major prion strains identified so far in permissive host species such as sheep, cattle, cervids, and humans. Prion strains are different PrPSc conformations with specific biological and biochemical attributes in infected host. In cervids, the strains in italic are specifically found in Scandinavia. In humans, the major CJD strains are grouped according to Bishop et al. categorization (Bishop et al. 2010) depending on PrP codon 129 genotype (M or V) and PrPSc electrophoretic signature (types 1 or 2). M1 strain regroups MM1 and MV1 cases, V2 strain regroups VV2 and MV2 cases, M2c and M2t strains correspond to MM2 cortical and thalamic forms respectively, and V1 strain corresponds to VV1 cases. Note that VV2 and MV2 may correspond to different strains (Jaumain et al. 2016). FFI, fatal familial insomnia; GSS, Gerstmann-Sträussler-Scheinker; VPSPr, variable protease-sensitive prionopathy. b At the single host level, different prion strains can co-propagate in the SNC or can co-propagate in distinct tissues, due to differential tropism for the lymphoid tissue. c Within a single strain, PrPSc heteroassemblies can be identified by sedimentation velocity (SV)–based size fractionation of PrPSc assemblies coupled with measure of specific infectivity. In this example, PrPSc assemblies originate from brains of terminally-sick ovine PrP transgenic mice infected with two biologically cloned scrapie strains replicating at different pace, LA21K fast and LA19K. The estimated size of the fractionated PrP.Sc assemblies is indicated (data from Tixador et al. 2010)
Structural diversity of PrPSc assemblies at the host level
Within the same infected host, different strains (usually two) can co-propagate, at different rate. Many examples can be found in animal and human prion diseases. Transmission of classical scrapie field isolates from sheep and goats to transgenic mice expressing ovine PrPC showed that a large proportion of the circulating isolates in Europe were composed of two co-propagating strains, which were termed LA19K and LA21K (Fig. 1b (Le Dur et al. 2017)). LA21K was dominant in most scrapie isolates imposing its biochemical phenotype (unglycosylated protease-resistant PrPSc fragment migrating at ~ 21 kDa by SDS-PAGE electrophoresis). To provide insights on the molecular determinants of strain predominance, sheep scrapie isolates containing variable proportions of LA21K and LA19K prions were transmitted experimentally to transgenic mice expressing ovine PrPC at different levels in the brain. Remarkably, LA21K replicated dominantly in transgenic mice expressing near-physiological PrPC levels, whereas LA19K phenotypically outcompeted LA21K in transgenic mice expressing high PrPC levels (Le Dur et al. 2017). These experiments demonstrated that PrPC levels can drive prion strain phenotypic dominance. They also suggested that local variations in native PrPC levels—as observed in response to prion replication (Mays et al. 2014)—may favor the phenotypic selection of a given strain.
In the same infected host, co-propagating strains could preferentially replicate in distinct tissues. In the aforementioned example, LA19K replication was mostly limited to the CNS while LA21K colonized efficiently both the CNS and the lymphoid tissue (Fig. 1b (Beringue et al. 2020)). Such differential tropism may also be linked to different PrPC levels, the spleen expressing ~ 20-fold reduced levels of the normal protein compared to the brain (Beringue et al. 2012).
In another example of co-propagating strains within a single host, the second co-propagating strain was barely detectable. Sheep infected with atypical scrapie (Nor98 strain (Benestad et al. 2003)) were found to co-propagate as minor strain C-BSE (Fig. 1b). C-BSE was identified following experimental transmission of atypical scrapie cases to transgenic mice expressing bovine PrP (Huor et al. 2019) or to pigs (Marin et al. 2021) and by in vitro prion amplification using a C-BSE optimized substrate. Remarkably, C-BSE prions were still detected in Nor98 prions that were biologically cloned by limiting dilution in ovine PrP transgenic mice, suggesting that Nor98 replication intrinsically co-generated C-BSE (Huor et al. 2019).
Transmission of North American CWD prions from diverse cervid sub-species to transgenic mice expressing deer PrP allowed isolation of two strain types, named CWD1 and CWD2. Both strains were found to co-propagate in deer (Fig. 1b (Angers et al. 2010)). Transmission of CWD from infected white-tailed deer to diverse bioindicator mice also lend support to the coexistence of CWD substrains; single polymorphisms at specific codons in the white-tailed deer PrP sequence could impact their relative fitness (Velasquez et al. 2020; Hannaoui et al. 2021).
In human, the co-detection in the brain of two PrPSc signatures termed type 1 and type 2 by immunoblot has been observed in many patients with sporadic CJD (Parchi et al. 1999; Polymenidou et al. 2005; Uro-Coste et al. 2008). Whether this co-occurrence is associated with the co-propagation of two strains or reflects the co-existence of PrPSc molecular subtypes with distinctive physicochemical properties is a debated issue. Recently, transmission of sporadic CJD cases to human PrP transgenic mice showed that each PrPSc signature was associated with a specific strain. In 80% of the investigated cases, the two strain types (M1 and V2) co-propagated in variable amounts (Fig. 1b (Cassard et al. 2020)).
To conclude, strain co-propagation in single infected host may be the rule rather than the exception. As co-propagation identification mostly relies on experimental transmission with more astringent replicative conditions for one of the strains, it cannot be excluded that strain co-propagation results from a unique progenitor strain that spontaneously evolves in these new conditions (see below) or because of intrinsic instability (Bruce and Dickinson 1987; Le Dur et al. 2017). Yet, in the aforementioned examples, there were stable manifestations of “pure” strains on serial transmission to the ad hoc bioindicator mice.
Co-propagation of strains in a single host raises the question of their genesis. Co-propagation is observed in acquired prion diseases, suggesting that multiple infection events could occur. It is also observed in sporadic cases, suggesting that multiple initiator events could occur or that the replication process allows generation of PrPSc assemblies with distinct SSD.
Structural diversity of PrPSc assemblies at the strain level
Evidence for variably protease-resistant PrPSc assemblies
One of the most straightforward demonstrations of within-strain PrPSc heterogeneity is the existence of PrPSc conformers with markedly distinct susceptibility to proteinase K (PK) digestion. By developing a conformation-dependent immunoassay, which detects specifically PrPSc immunoreactivity versus PrPC following chaotropic agent resolved denaturation, Safar and colleagues revealed the existence of protease-sensitive PrPSc alongside the canonical PK-resistant PrPSc conformer (termed PrPres). Protease-sensitive PrPSc was detected in eight different prion strains, including biologically cloned strains (Safar et al. 1998). Co-propagation of PK-sensitive and PK-resistant PrPSc subpopulations within a given strain was also shown with technics substituting PK by other proteases such as thermolysin (Cronier et al. 2008; Owen et al. 2007) or pronase (D'Castro et al. 2010). The exact contribution of PK-sensitive PrPSc conformers to prion physiopathology remains uncertain even if these species could represent up to 80% of total PrPSc content, specially at the early stage of prion replication. Gradual acquisition of PK-resistance by PrPSc, as identified in kinetics studies, is suggestive of a dynamic involvement of PK-sensitive PrPSc in the replication process (Eskandari-Sedighi et al. 2021). At the terminal stage of the disease, depending on the experimental context, PK-sensitive PrPSc was found to be lowly infectious (Cronier et al. 2008) or highly infectious (Berardi et al. 2006) or be able to convert PrP in vitro (Pastrana et al. 2006). These seemingly contradictory observations highlight the fact that the replication process is intrinsically associated to structural diversification.
PrPC post-translational modifications could affect PrPSc structuration PrPSc assemblies
Structural heterogeneity within a given strain may result from PrP backbone modification by prosthetic groups such as the GPI anchor or glycans. Single amino acid modification could conduce to strain mutation; therefore, it would not be surprising that variations in the number and nature of the prosthetic groups affect the conversion pathway and conduce to structural heterogeneity. For example, propagation of biologically cloned mouse prions in transgenic mice devoid of PrP GPI anchor affected their strain properties (Aguilar-Calvo et al. 2017; Mahal et al. 2012) or broadened their host spectrum (Race et al. 2015), suggesting profound structural differences between GPI-free and GPI-anchored PrPSc conformers. Remarkably, a minor proportion (~ 15%) of PrPSc is anchorless in wild-type animals (Stahl et al. 1990). Such presence may contribute to PrPSc conformational landscape during the replication process.
Glycans diversity may also contribute to further broadening of PrPSc structural landscape. PrPC has two asparagine side chains linked to large oligosaccharides with multiple, diverse structures (Rudd et al. 2002). PrPSc, as PrPC, is variably glycosylated at these two sites, which are located at amino acid positions 181 and 197 in the human PrP sequence (Endo et al. 1989). The stoichiometric ratio of PrPSc glycoforms is strain-specific and faithfully maintained during prion passaging in the same host species (Collinge et al. 1996; Somerville and Ritchie 1990), which means that a given strain has a specific preference for certain PrPC glycotypes. Yet, PrPSc from different strains does not appear to differ in glycan composition (Nakic et al. 2021), suggesting that prion SSD are not encoded in glycans. However, PrPSc occupancy by glycans, given their extended size, variable proportion, and composition at each site (Nakic et al. 2021; Rudd et al. 2002), is likely to affect the stability, clearance and the dynamic of the forming assemblies by steric hindrance. Accordingly, transgenic modeling suggests that PrPC glycosylation status can influence the efficacy of intra- and inter-species transmission of prions (DeArmond et al. 1997; Tuzi et al. 2008; Wiseman et al. 2015) and prion strain properties (Cancellotti et al. 2013). However, in cell-free prion amplification assays using unglycosylated PrPC substrate, it was found that glycans were dispensable in specifying prion strain properties (Moudjou et al. 2016; Piro et al. 2009). These opposite results may be due to the different strains studied or to the point mutations inserted to prevent asparagine-linked glycosylation, because trafficking of the PrP mutant can be altered (Salamat et al. 2011) or because PrP post-translational state markedly influences the fate of the aggregates in the brain tissue (Sevillano et al. 2020).
Quaternary structure diversity of PrPSc assemblies, and beyond
Low-resolution structural studies such as sedimentation velocity (SV), size exclusion chromatography, and asymmetric fast-flow-field fractionation have been extensively used to explore the quaternary structure of PrPSc assemblies. In the brain of terminally sick animals solubilized in specific conditions, the size distribution pattern of PrPSc assemblies revealed the existence of subpopulations with distinct quaternary structure (Eskandari-Sedighi et al. 2021; Foliaki et al. 2019; Laferriere et al. 2013; Riesner et al. 1996; Silveira et al. 2005; Tixador et al. 2010). The analysis of different prion strains indicated that PrPSc quaternary structure pattern was strain-specific (Tixador et al. 2010).
When the specific infectivity or seeding activity values (i.e., the amount of infectivity or seeding activity per number of PrP) of size-fractionated assemblies were compared, the variations in the prion titer of the assemblies were decorrelated from size variations (extensively reviewed in Igel-Egalon et al. (2019a)). For example, in rapidly pathogenic scrapie prion strains, PrPSc assemblies with a size equal or below a PrP pentamer exhibited the highest specific infectivity values. These values were 1000 to 10,000-fold higher than those from the bulk of PrPSc assemblies with an estimated size of ~ 30-mers (Fig. 1c). Decorrelation was even more patent when other scrapie strains propagating at slower pace, such as LA19K or Nor98, were considered. Assemblies with the highest specific infectivity values were equivalent to ~ 30- to 60-mers of PrP. They were 1000-fold more infectious than PrP pentamers, which were the richest in terms of objects (Fig. 1c; Igel-Egalon et al. 2019a; Laferriere et al. 2013; Tixador et al. 2010)). Collectively, these data lend strong support to the view that the structural differences between size-fractionated PrPSc assemblies were not only due to quaternary structure variation but also involved variation in lower order structuration, at the tertiary and secondary level. These observations bring compelling evidence that a single prion strain is formed from a spectrum of structurally heterogeneous PrPSc subpopulations.
To conclude this section, there is compelling evidence that prions are formed from a spectrum of heterogeneous PrPSc assemblies. This pleiomorphism can be seen at different scales, in infected host populations, in infected hosts and within a single strain. Whether the intra-strain diversity is in fine similar to the inter-strain diversity remains to be seen. In the future, phenotyping tools with more discriminative power than bioassays may help resolving this question and identifying in PrPSc assemblies the domains responsible for intra- and inter-strain variability.
Mechanisms of PrPSc structural diversification
Theoretical considerations on the mechanisms of prion replication and structural diversification
The existence, in a single host, of different prion strains and, in a single strain, of PrPSc heteroassemblies, raises the question of such structural diversification. As a precise molecular mechanism of prion replication is lacking, only hypotheses can be formulated. The prion replication dogma, which remains mostly theoretical, can be split into three steps (Fig. 2a). The first step is the templating, during which PrPSc assemblies induce host PrPC conformational change through the templating interface. The asymmetric evolution of the templating process (PrPSc is converting PrPC and not the opposite) is only due to the higher stability of PrPSc assemblies compared to the stability of the PrPC fold. The amyloid end-elongation by monomer addition remains until now the most widely accepted mechanism of templating (Collins et al. 2004). In this process, amyloid fibril ends would serve as templating interface for monomeric PrPC and induce its structural rearrangement by a mechanism resembling the induced fit adjustment or conformational selection (Csermely et al. 2010; Koshland 1963). In the end-elongation hypothesis, the number of templating interface remains constant. Thus, an amplification step is required to accommodate the exponential aspect of prion replication (Langevin et al. 2011; Nakaoke et al. 2000). The second step is the amplification of the templating interface, putatively by an assisted fragmentation (Shorter and Lindquist 2006). The third step, responsible for prion dissemination through the infected tissue is the spatial spreading of the templating center. One can easily conceive a stochastic formation of structurally distinct sets of assemblies from different states of prionogenic monomeric PrPC. It is harder to physico-chemically conceive how, during the templating step, structural diversification could take place without any external perturbation (thermal and environmental fluctuations of the PrPC protein) or change in PrPC backbone and/or post-translational modifications. Yet, such external perturbations have to accommodate the reaction mechanism at work in the end-elongation templating. The induced fit adjustment of PrPC on PrPSc can be decomposed in three elementary steps, each one being governed by an equilibrium between backward and forward steps (Fig. 2b). In the first step, PrPSc assemblies will interact with PrPC through single or multiple specific interaction interfaces. This interface could be strain-specific. The N-terminal, polybasic region of PrPC (residues 23–31 (Turnbaugh et al. 2012)) and regions containing residues 89–112 and 136–158 (Moroncini et al. 2004; Solforosi et al. 2007) have been reported to be involved in the interaction between PrPSc and PrPC. The second step, which is concerted with the formation of the PrPSc-PrPC complex, consists in an (at least partial) unfolding of PrPC into PrPU*. Indeed, among all amyloidogenic proteins, PrPC stands apart amid its folded native state (Eghiaian et al. 2004; Riek et al. 1996). Therefore, the templating process inducing PrPC structural transition to PrPSc should first disrupt at least some interactions existing in the native fold (i.e., partial unfolding) prior to the acquisition of any new fold. The importance of this step is experimentally well illustrated in the cell-free formation of synthetic prions where a partial unfolding of recombinant PrP is required to induce PrP fibrils formation (Makarava et al. 2011). It is not straightforward to figure out the impact of external perturbations on these elementary steps. Indeed, as any reaction process—be it irreversible or not—can be decomposed into multiple micro-equilibria, a misfit between PrPSc:PrPC will destabilize the complex and displace the equilibrium toward the dissociation (Fig. 2c).

Prion replication mechanism. Schemes summarizing the current view of a the prion replication process and b the reaction mechanism of the end-elongation templating and induced fit adjustment of PrPC on PrPSc assemblies. The prion replication process is a three-step process with templating, amplification, and spreading of the templating interface. The templating reaction mechanism can be divided in three elementary steps, each one being governed by an equilibrium between backward and forward steps. c An external perturbation can induce a misfit between PrPSc and PrPC, destabilize the complex, and displace the equilibrium toward the dissociation
To conclude, it is difficult to consider a physico-chemically relevant PrPSc diversification process in the frame of the current, theoretical prion polymerization mechanism.
PrPSc assemblies diversification due to prion replication
A change of paradigm is necessary to accommodate PrPSc diversification with the prion replication mechanism. To achieve progress on this issue, we studied kinetically prion assemblies structural diversification during prion replication (Igel-Egalon et al. 2019b). The size distribution analysis (by SV) of PrPSc assemblies from three distinct cloned prion strains (human vCJD, mouse 139A, and scrapie 127S prions) at different time points of the disease showed that the early steps of prion replication generated small oligomeric PrPSc objects (Fig. 3a). The formation of larger-size PrPSc assemblies appeared to be a secondary step in the evolution of the disease and was concerted with the disappearance of the small oligomeric PrPSc objects. The imprecision and the variability of the in vivo experiments required the exploration of the early stage of prion replication through an in vitro bona fide prion replication system such as protein misfolding cyclic amplification (PMCA (Saborio et al. 2001)).

PrPSc diversification during prion replication process by PMCA. a SV-based size distribution of PrPSc assemblies at early and late stages of vCJD prion pathogenesis in the brain of human PrP mice. b SV-based PrPSc size distribution in PMCA products (127S scrapie strain) analyzed immediately at the end of the PMCA reaction or after further quiescent incubation at 37 °C for the indicated period of time (t). This allows identifying two populations of PrPSc assemblies termed Ai and Bj (with i < j). Ai is mostly generated during the active phase of the PMCA reaction. During the quiescent phase, Ai decreases over time in favor of Bj according to a bimodal process (without appearance of assemblies of intermediate size). The inset graph shows the variations of the percentage of Ai and Bj as a function of the quiescent phase (t). The sigmoidal form of the curves is indicative of an autocatalytic reaction process. c Kinetic scheme describing the transformation process of Ai into Bj. Ai and Bj are in equilibrium with their respective suPrP (Igel-Egalon et al. 2017; steps I and II). The best model to account for the cooperative, PrPC-dependent transformation of Ai into Bj implicates the formation of a complex between suPrPA and suPrPB (step III). This complex is at the origin of the secondary templating pathway whereby the transformation of suPrPA to suPrPB is assisted by suPrPB, making the process autocatalytic (data from Igel-Egalon et al. 2019b)
In the PMCA assay, minute amounts of PrPSc are mixed with a substrate containing PrPC. Usually, this substrate is a transgenic mouse brain homogenate or a cell lysate expressing the mammalian PrPC of interest (Moudjou et al. 2016, 2014). The mixture is then submitted to alternative cycles of sonication/quiescent incubation at 37 °C for 1–2 days. Many rounds can be done by refreshing the PrPC substrate. PMCA mimics the prion replication process as, most often, it generates high levels of prion infectivity and maintains prion strain biological properties (Castilla et al. 2008a, b; Moudjou et al. 2016, 2014). As mentioned above, SV coupled with measure of specific biological activity of the fractionated assemblies allows isolating different structural states of PrPSc in the brain of terminally-sick mice. Applying this strategy to PMCA generated prions showed that the prion replication process generated two subsets of structurally different PrPSc assemblies. Their process of formation was intricated and sequential, regardless of the strain considered (Igel-Egalon et al. 2019b). For the three different cloned prion strains studied in vivo (human vCJD, mouse 139A, scrapie 127S), the first replication step generated mostly small PrPSc oligomers (termed Ai). Ai size was below a PrP pentamer. The second step, which required the presence of PrPC, transformed the Ai oligomers into larger ones (termed Bj) (Fig. 3b). This was accompanied by further structural rearrangement at the level of the secondary/tertiary structure, as identified by (1) differences in Ai and Bj specific infectivity values, (2) the irreversibility of the transformation of Ai into Bj (i.e., Bj is not an Ai condensate), and (3) the structural differences in the elementary bricks (Igel-Egalon et al. 2017) constitutive of Ai and Bj assemblies. Kinetic studies and mathematical modeling showed that the transformation process of Ai into Bj assemblies through this secondary templating pathway was cooperative and under the control of PrPC substrate consumption (Fig. 3c). Collectively, these data lend support to the view that the prion replication process is intrinsically a source of PrPSc assemblies diversification within a single strain. This occurs in a deterministic manner, thus contradicting common belief supporting stochastic events or environmental fluctuations at the origin thereof (Weissmann et al. 2011).
To conclude this section, there are at least two possible main ways for prion structural diversification, one due to the replication process itself and the other one due to PrPC itself, under the influence of environmental fluctuations where replication occurs.
PrPSc diversification and prion adaptation
The species barrier phenomenon
Prions can transmit between different species. Yet, this capacity can be restricted by a so-called species or transmission barrier. Prion cross-species transmission issues are highly variable (Fig. 4a). Two extreme issues are well documented; transmission can occur with little or no species barrier (“faithful” or “identical” transmission) or be negative (e.g., Nonno et al. 2006)). In the last case however, prions may persist in the brain for the entire life of the inoculated host because of their pronounced resistance to clearance (Martin et al. 2021). Most often, prion cross-species transmission is difficult, necessitating serial passaging in the new host to adapt and reach 100% attack rate with stabilized disease tempo and consistent neuropathological or biochemical phenotypes. In certain cases, a new prion strain type may suddenly emerge (usually from the second passage onwards), as shown by a drastic reduction of the incubation time to disease and stabilized phenotype (e.g., Chapuis et al. 2016). Usually, this new strain type has lost the capacity to reinfect the parental host. This phenomenon is termed prion “mutation” by analogy with conventional pathogens. There are also reports of prions able to replicate on primary passage in animals expressing PrPC from a foreign species but unable to adapt on further passage, a process referred to as nonadaptive prion amplification (Bian et al. 2017; Duque Velasquez et al. 2020). In short, when prions are confronted to a new host or a new PrPC sequence, all outcomes are seemingly possible.

Molecular determinants of prion species barrier. a Main outcomes observed during experimental prion cross-species transmission (or transmission to heterologous PrPC as done by transgenic modeling). To be pointed out, cross-species transmission of a different prion from species/PrPC 1 may lead to drastically different outcomes. b Conformational selection model to explain prion cross-species transmission at the molecular level. In this model, prions would be composed of a cloud of subcomponents or substrains in varying proportions. The major component would be responsible for the strain phenotype in the parental host species. Other substrains would be co-propagated as minor components. On cross-species transmission, the optimized subcomponent, i.e., the component that lies within the portfolio of acceptable conformations in the new PrPC species, would be preferentially selected. The issue of the transmission will thus mostly depend on the presence of a compatible component and relative concentration. c In the deformed templating model, prion primary passage to species expressing heterologous PrPC would be inefficient, because of structural incompatibility between infecting PrPSc and PrPC. This would lead to generation of PrPSc with atypical conformation (green square) in a reduced number of asymptomatic animals. On subpassage, this conformation would slowly evolve toward an optimized conformation (red triangle), allowing adaptation. d–e In the conformational complementation model (d), prions would be composed of heterogeneous PrPSc assemblies with respect to secondary, tertiary, and quaternary structure. Interaction between these assemblies (red arrows) would allow crossing the species barrier. Mechanistically (e), the complex formed by the suPrPs from the different PrPSc assemblies (here Ai and Bj) would provide an interaction interface with heterologous PrPC that is absent in each assembly or each individual suPrP
Host determinants of the species barrier
Prion cross-species transmission outcome is critically dependent upon host–pathogen interactions. From the host side, the route of infection and the gene encoding PrP are two critical determinants.
During intraspecies transmission events, the efficacy of infection can vary by a million-fold depending on the route of inoculation (Haybaeck et al. 2011; Herzog et al. 2004; Kimberlin and Walker 1978, 1988; Lasmezas et al. 2005, 2001; Taylor et al. 1996). This route-dependent efficacy has been extrapolated to interspecies transmission events, but to our knowledge, no systematic assessment of the minimal infectious dose relative to the route of inoculation has been done.
Transgenic modeling by Prusiner’s group demonstrated that PrP primary structure homology between the host and the infecting prion was sufficient to abrogate prion species barrier. Hamster Sc237 prions which do not induce a clinical disease in wild-type mice propagated readily in transgenic mice expressing hamster PrP (Scott et al. 1989). This seminal experiment paved the way for transgenic mouse models that abrogate prion species barrier in laboratory animals. As another example, sporadic CJD prions, which do not replicate in wild-type mice, faithfully propagate in transgenic mice expressing human PrP (Asante et al. 2002; Beringue et al. 2008a; Collinge et al. 1995). It was later shown that PrP sequence homology is not a prerequisite as sporadic CJD prions replicate in bank vole (Myodes glareolus) with little or no species barrier (Nonno et al. 2006). Yet, bank vole and human PrPs share a 90% amino acid sequence identity. Bank voles or transgenic mice expressing bank vole PrPC may be universal prion acceptors because of their capacity to propagate many strains from many different species (Watts et al. 2014), including prions reputedly difficult to transmit such as those responsible for VPSPr or Gerstmann-Sträussler-Scheinker syndrome (Nonno et al. 2019; Pirisinu et al. 2016).
The fact that expressing PrP with a sequence identical to that of the infecting prion most often if not always abrogates the species barrier in mice or in other, potentially less permissive species like rabbit (Sarradin et al. 2015) or drosophila (Thackray et al. 2018), lends support to the view that they are no non-PrP genes essential to prion cross-species transmission.
It must be noted that within the same species, PRNP polymorphisms can modulate the disease susceptibility to a degree of magnitude like that observed in interspecies transmission event. For example, sheep expressing the ARR allele at PRNP codons 136, 154, and 171 instead of the ancestral ARQ allele (where A, R, and Q stand for alanine, arginine, and glutamine, respectively) are highly resistant to classical scrapie prions (Elsen et al. 1999). In human, a naturally occurring variant at PRNP position 127 (valine instead of glycine) protects against prions responsible for CJD or kuru, an acquired form of CJD due to cannibalistic rituals (Asante et al. 2015). In human, the common polymorphism at PRNP codon 129, where either methionine or valine is present, provides a relative protection, notably against clinical forms of vCJD (Fernandez-Borges et al. 2017; Wadsworth et al. 2004). In cervids infected with CWD, PrPC polymorphisms emerge as important driver of prion selection and evolution, particularly in heterozygous animals (Velasquez et al. 2020; Hannaoui et al. 2021).
Pathogen determinants of the species barrier
From the pathogen side, the prion strain type and obviously the dose inoculated are two critical determinants, the dose being itself critically related to the route of infection.
As mentioned above, strains responsible for the different forms of sporadic CJD do not replicate in wild type mice, despite inoculation at high dose and intracerebral inoculation. Oppositely, vCJD prions can propagate in these animals, the force of the species barrier depending on the genetic background and the associated polymorphisms in the PrP-encoding gene (Bruce et al. 1994).
Strain-dependent susceptibility is similarly observed for the PRNP polymorphisms within the same species. Thus, sheep expressing the ARR allele become fully susceptible to the Nor98 strain responsible for atypical scrapie (Le Dur et al. 2005).
The species barrier is tissue-specific
The capacity of invading prions to replicate extraneurally in the newly infected host is critical as foreign prions can establish easier in the spleen tissue than in the CNS (Beringue et al. 2012; Bian et al. 2021). Transgenic modeling showed that the spleen was 7–tenfold leakier than the brain to prions that adapted with difficulty to foreign PrP species. This included notably C-BSE prions in human PrP transgenic mice expressing methionine at codon 129. Such leakiness of the spleen versus the brain may explain why the number of clinical cases of vCJD is limited, while the number of asymptomatic individuals with PrPvCJD-positive lymphoid tissue is high (Collinge 2012; Gill et al. 2020).
Molecular aspects of the species barrier and prion assemblies diversification
The aforementioned examples indicate that the force of prion species barrier depends on the possibility of interactions between tissue-specific PrPC and the infecting prion strain type. At the molecular level, it is believed that constrained steric interactions between PrPC and PrPSc are the limiting factor. How could this view accommodate PrPSc assemblies diversity? It may be anticipated that the larger the diversity of PrPSc assemblies structures, the greater the probability of interactions with foreign PrPC.
The conformational selection model
Before PrPSc heterogeneity was even unraveled at the strain level, but rather by analogy with the (viral) quasispecies concept, Collinge and Clarke (2007) adapted the conformation selection model (Csermely et al. 2010; Tsai et al. 1999) to explain prion cross-species transmission outcome at the molecular level (Fig. 4b). In essence, this model posits that (i) prions are not clonal but constitute an ensemble with dominant and subdominant PrPSc components or substrains and (ii) host PrPC can accommodate a certain portfolio of PrPSc conformations in the pathological state. On cross-species transmission, if one (major or minor) subcomponent lies within the portfolio of conformations host PrPC can accommodate in the PrPSc state, there will be preferential selection of this compatible conformation and crossing of the species barrier. It is thus expected that the time to disease onset and the attack rate in the new host will mostly reflect the initial concentration of this optimized subcomponent. Above a certain concentration threshold, the attack rate should be relatively high.
Regarding PrPSc assemblies molecular diversity, this model would readily accommodate prion strains co-propagation in a single host, one being preferentially selected on cross-species transmission amid a compatible conformation. It might potentially accommodate PrPSc assemblies heterogeneity at the intra-strain level, as long as one consider that these assemblies can become bona fide strains in the new host. The conformational selection model does not address the molecular mechanism for mutant emergence as such mutant cannot pre-exist in the quasispecies of molecular substrains per se (otherwise, it would be readily selected). The model states in essence that mutation can occur because of the intrinsic instability of certain strains (Collinge 2016).
The Deformed templating model
The deformed templating model is the second model to explain prion species barrier at the molecular level (Fig. 4c). It stems from the difficulty of certain minimalistic preparations of recombinant PrP fibrils to transmit disease to bioindicator animals (Makarava and Baskakov 2013; Makarava et al. 2011, 2016). In these experiments, following recombinant PrP fibrils inoculation, “atypical” forms of PrPSc accumulated in few asymptomatic animals on primary passage. On further passage, virulence gradually increased and “classical” PrPSc species emerged. The deformed templating hypothesis (Makarava et al. 2011) that stems from these results is based on an end-elongation replication process. It considers recombinant PrP assemblies as highly homogenous. It also considers that because bacterial PrP does not express glycans and the GPI anchor, it creates a structural barrier on contact with mammalian PrPC which expresses these post-translational modifications. With these hypotheses, the first step of the cross-species transmission will consist to create a certain degree of heterogeneity on primary passage. After this heterogenization step, the authors made the hypothesis that with several cycles of templating, the templating interface will progressively shift to a more efficient templating interface. This two-step process tentatively explains how a strong species barrier could be crossed and may explain the emergence of prion mutant. Yes, it suffers several limitations. First, other preparations of recombinant PrP fibrils are directly highly infectious, without occurrence of PrPSc structural shift (Choi et al. 2016; Legname et al. 2004). Second, it does not consider PrPSc assemblies diversity. Third, the slow, progressive evolution of the templating interface over passaging should be put in perspective with the number of PrPSc templating events at each passage (approximately 1014 replicating events for hamster 263 K (Igel-Egalon et al. 2017)).
The conformational complementation model
The conformational complementation model is the only model that considers the structural diversity of infecting PrPSc assemblies. In addition, it implicates the existence of synergetic interactions between these differing subpopulations (or a component thereof) (Fig. 4d). This model stems from experiments where the importance of PrPSc assemblies structural diversity in cross-species transmission events was specifically addressed (Igel-Egalon et al. 2020). PrPSc assemblies were separated from each other either by SV-based size-fractionation or by serial dilution before transmission to transgenic mice expressing a foreign PrPC sequence. In the absence of a transmission barrier, separating or diluting PrPSc assemblies was without influence on the disease tempo and prion strain properties (Fig. 5a, b, top graph). In the presence of a species barrier, fractionating PrPSc assemblies by SV overtly delayed and even abrogated priogenesis (Fig. 5a, bottom graph), despite sufficient infectivity load of the isolated assemblies to adapt per se. Dilution had also a severe impact, the efficacy of infection being 10,000-fold decreased compared to the homotypic PrP context or 1000-fold decreased compared to the expected value in the heterotypic PrP context (Fig. 5b, bottom graph).

Effects of fractionating PrPSc assemblies by sedimentation velocity or dilution on prion capacity to cross the species barrier. The cloned scrapie prion strain LA19K propagates without any apparent species barrier from ovine PrP transgenic mice to bovine PrP transgenic mice. The cloned scrapie prion strain LA21K fast can adapt to hamster PrP transgenic mice; achieving a stable biological phenotype in these mice necessitates several passages, indicative of a substantial species barrier. a Effect of SV fractionation on the capacity of LA19K and LA21K fast prions to propagate in heterologous PrPC mice. This was not detrimental to LA19K prions with respect to the disease incidence and size distribution of infectivity (inversely correlated to the mean incubation time shown here) as compared to the original host PrPC. On the opposite, LA21K fast priogenesis was almost completely abrogated with only 3 mice found asymptomatic out of the 300 analyzed (Igel-Egalon et al. 2020). b Effect of dilution on the capacity of LA19K and LA21K fast prions to propagate in heterologous PrPC mice. The limiting dilution values achieved with LA19K prions were similar in the homologous and heterologous PrPC contexts (top panel, compare green and blue lanes). Note that for LA19K in bovine PrP mice, the 10−5 dilution was not tested. At the 10−4 dilution, two-thirds of the mice were affected, as for the 10−5 dilution in ovine PrP mice. On the opposite, the limiting dilution values achieved with LA21K fast were reduced by 10,000-fold in the heterotypic PrPC context (bottom panel, compare blue and green plain lanes). In theory (blue dotted lane), the limiting dilution value in the heterotypic context should have been 1000-fold higher for LA21K fast, as calculated from the attack rate and disease tempo obtained at low dilution and extrapolation from other bioassays (Igel-Egalon et al. 2020). There was thus a strong impact of the dilution on LA21K fast capacity to cross the ovine to hamster transmission barrier
It could be argued that these experiments are congruent with the conformational selection model, the loss of subcomponents by fractionation or dilution resulting in the loss of optimized conformations in the heterotypic PrP context, and thus in delaying/abrogating priogenesis. A first counterargument is that the isolated PrPSc assemblies that finally adapted on serial passage in the new PrP transgenic host did not differ in terms of strain properties from unfractionated prions, meaning that if an optimized conformation pre-existed, it was not lost during fractionation. A second counterargument necessitates to detail one of the experimental paradigms used: The rapidly pathogenic LA21K fast scrapie prion strain (Fig. 1c) was fractionated before transmission to transgenic mice expressing hamster PrP. The pentameric oligomers with the highest specific infectivity values (Figs. 1c and 5a) in the homotypic context had sufficient infectivity levels in the heterotypic context to adapt. They did not elicit a clinical or subclinical disease. The larger assemblies with the lowest specific infectivity values in the homotypic PrP context elicited a subclinical disease in a very low proportion of mice. It could thus be said that these assemblies had the optimized conformation for the hamster PrP sequence. Yet, because there are by far the most populous conformation in the unfractionated prion strains (Tixador et al. 2010), they should have elicited the disease at higher attack rate because of their presence in substantial amounts in the SV fractions.
As a simple selection of optimized PrPSc conformations is unlikely to explain these observations, we thus posited that somehow the assemblies should complement each other to cross the species barrier, i.e., to interact with foreign PrPC. How is complementation mechanistically possible? In its simplest acceptation, it implicates interactions between heteroassemblies to create a new structural information, absent in each single assembly, that allows interaction with heterologous PrPC (Fig. 4e). As mentioned above, structurally distinct PrPSc assemblies are generated by the prion replication process, i.e., Ai and Bj with their specific subunits suPrPA and suPrPB. Mechanistically, we found that the secondary templating pathway that generates, in a PrPC-dependent manner, B from A, involves the formation of a suPrPA/suPrPB heterocomplex (Fig. 3c (Igel-Egalon et al. 2019b)). This is possible because Ai and Bj are constitutively in dynamic equilibrium with their own suPrP (Igel-Egalon et al. 2017, 2019a). The suPrPA/suPrPB heterocomplex may interact with heterologous PrPC because it may have a templating interface that is not present in suPrPA and suPrPB due to the structural constraints imposed by the interactions. On interaction with heterologous PrPC, such a new templating interface would lead to the formation of a de novo suPrPB* with an optimized templating interface for further conversion. The force of the species barrier would thus depend on the stability of the heterocomplex, the possibility of a new interface with heterologous PrPC and on PrPC concentration for the cooperativity of the reaction. This complementation model would best accommodate the within-strain PrPSc assembly diversity and the underlying dynamic mechanism of genesis.
To conclude this section, PrPSc assemblies diversity at the level of the host or the strain must be taken into account when addressing at the molecular level the issue of prion evolution during cross-species transmission. The conformation selection models and the complementation models consider PrPSc diversity with respect to prion substructure or substrains. The second model would go a step further by taking account the dynamics of the assemblies.
Conclusions
This review tentatively unravels the many shades of prion assemblies diversification and the need to bypass the “one assembly fits all” approach to understand prion replication and adaptation at the molecular level. PrPSc assemblies diversity, dynamic of formation, and exchange provide new mechanistic insights into prion replication and adaptation. This provides prions with an evolutionary advantage due to selection of best replicator or mutational events in different environments to finally ensure persistence and diffusion within the host or at the population level.
High-resolution structures of prions purified to homogeneity are beginning to emerge. In essence, they suggest that PrPSc assemblies are organized in a specific manner within a fibrillar supra-organization which is extremely stable. It remains so far difficult to conciliate or to find commonalities between the “deadpan” aspect of these emerging structures and PrPSc diversity and dynamicity. The next challenge will be to provide structures at high-resolution of the assemblies that preserve their diversity.
References
Aguilar-Calvo P, Xiao X, Bett C, Erana H, Soldau K, Castilla J, Nilsson KP, Surewicz WK, Sigurdson CJ (2017) Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 7:43295
Angers RC, Kang HE, Napier D, Browning S, Seward T, Mathiason C, Balachandran A, McKenzie D, Castilla J, Soto C, Jewell J, Graham C, Hoover EA, Telling GC (2010) Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 328:1154–1158
Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, Collinge J (2002) BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 21:6358–6366
Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S, Richard-Londt A, Linehan JM, Brandner S, Alpers M, Whitfield J, Mead S, Wadsworth JD, Collinge J (2015) A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522:478–481
Babelhadj B, Di Bari MA, Pirisinu L, Chiappini B, Gaouar SBS, Riccardi G, Marcon S, Agrimi U, Nonno R, Vaccari G (2018) Prion disease in dromedary camels, Algeria. Emerg Infect Dis 24:1029–1036
Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikoren T (2016) First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res 47:88
Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B (2003) Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec 153:202–208
Berardi VA, Cardone F, Valanzano A, Lu M, Pocchiari M (2006) Preparation of soluble infectious samples from scrapie-infected brain: a new tool to study the clearance of transmissible spongiform encephalopathy agents during plasma fractionation. Transfusion 46:652–658
Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H (2012) Facilitated cross-species transmission of prions in extraneural tissue. Science 335:472–475
Beringue V, Le Dur A, Tixador P, Reine F, Lepourry L, Perret-Liaudet A, Haik S, Vilotte JL, Fontes M, Laude H (2008a) Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE 3:e1419
Beringue V, Tixador P, Andreoletti O, Reine F, Castille J, Lai TL, Le Dur A, Laisne A, Herzog L, Passet B, Rezaei H, Vilotte JL, Laude H (2020) Host prion protein expression levels impact prion tropism for the spleen. PLoS Pathog 16:e1008283
Beringue V, Vilotte JL, Laude H (2008b) Prion agent diversity and species barrier. Vet Res 39:47
Bian J, Khaychuk V, Angers RC, Fernandez-Borges N, Vidal E, Meyerett-Reid C, Kim S, Calvi CL, Bartz JC, Hoover EA, Agrimi U, Richt JA, Castilla J, Telling GC (2017) Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci U S A 114:1141–1146
Bian J, Kim S, Kane SJ, Crowell J, Sun JL, Christiansen J, Saijo E, Moreno JA, DiLisio J, Burnett E, Pritzkow S, Gorski D, Soto C, Kreeger TJ, Balachandran A, Mitchell G, Miller MW, Nonno R, Vikoren T, Vage J, Madslien K, Tran L, Vuong TT, Benestad SL, Telling GC (2021) Adaptive selection of a prion strain conformer corresponding to established North American CWD during propagation of novel emergent Norwegian strains in mice expressing elk or deer prion protein. PLoS Pathog 17:e1009748
Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010
Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H (1994) Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 343:405–411
Bruce ME, Dickinson AG (1987) Biological evidence that scrapie agent has an independent genome. J Gen Virol 68(Pt 1):79–89
Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P, Weissmann C, Manson JC (2013) Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 32:756–769
Cassard H, Huor A, Espinosa JC, Douet JY, Lugan S, Aron N, Vilette D, Delisle MB, Marin-Moreno A, Peran P, Beringue V, Torres JM, Ironside JW, Andreoletti O (2020) Prions from sporadic Creutzfeldt-Jakob disease patients propagate as strain mixtures. mBio 11
Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C (2008a) Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768
Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C (2008b) Cell-free propagation of prion strains. EMBO J 27:2557–2566
Chapuis J, Moudjou M, Reine F, Herzog L, Jaumain E, Chapuis C, Quadrio I, Boulliat J, Perret-Liaudet A, Dron M, Laude H, Rezaei H, Béringue V (2016) Emergence of two prion subtypes in ovine PrP transgenic mice infected with human MM2-cortical Creutzfeldt-Jakob disease prions. Acta Neuropathol Commun 4:10
Choi JK, Cali I, Surewicz K, Kong Q, Gambetti P, Surewicz WK (2016) Amyloid fibrils from the N-terminal prion protein fragment are infectious. Proc Natl Acad Sci U S A 113:13851–13856
Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 24:519–550
Collinge J (2012) Cell biology. The Risk of Prion Zoonoses Science 335:411–413
Collinge J (2016) Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539:217–226
Collinge J, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318:930–936
Collinge J, Palmer MS, Sidle KC, Hill AF, Gowland I, Meads J, Asante E, Bradley R, Doey LJ, Lantos PL (1995) Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 378:779–783
Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (1996) Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690
Collins SR, Douglass A, Vale RD, Weissman JS (2004) Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol 2:e321
Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, Wadsworth JD (2008) Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416:297–305
Csermely P, Palotai R, Nussinov R (2010) Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci 35:539–546
D’Castro L, Wenborn A, Gros N, Joiner S, Cronier S, Collinge J, Wadsworth JD (2010) Isolation of proteinase K-sensitive prions using pronase E and phosphotungstic acid. PLoS ONE 5:e15679
DeArmond SJ, Sanchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB (1997) Selective neuronal targeting in prion disease. Neuron 19:1337–1348
Eghiaian F, Grosclaude J, Lesceu S, Debey P, Doublet B, Treguer E, Rezaei H, Knossow M (2004) Insight into the PrPC– > PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc Natl Acad Sci U S A 101:10254–10259
Elsen JM, Amigues Y, Schelcher F, Ducrocq V, Andreoletti O, Eychenne F, Khang JV, Poivey JP, Lantier F, Laplanche JL (1999) Genetic susceptibility and transmission factors in scrapie: detailed analysis of an epidemic in a closed flock of Romanov. Arch Virol 144:431–445
Endo T, Groth D, Prusiner SB, Kobata A (1989) Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 28:8380–8388
Eskandari-Sedighi G, Cortez LM, Yang J, Daude N, Shmeit K, Sim V, Westaway D (2021) Quaternary Structure Changes for PrP(Sc) Predate PrP(C) Downregulation and neuronal death during progression of experimental scrapie disease. Mol Neurobiol 58:375–390
Fediaevsky A, Tongue SC, Noremark M, Calavas D, Ru G, Hopp P (2008) A descriptive study of the prevalence of atypical and classical scrapie in sheep in 20 European countries. BMC Vet Res 4:19
Fernandez-Borges N, Espinosa JC, Marin-Moreno A, Aguilar-Calvo P, Asante EA, Kitamoto T, Mohri S, Andreoletti O, Torres JM (2017) Protective effect of Val129-PrP against bovine spongiform encephalopathy but not variant Creutzfeldt-Jakob disease. Emerg Infect Dis 23:1522–1530
Foliaki ST, Lewis V, Islam AMT, Ellett LJ, Senesi M, Finkelstein DI, Roberts B, Lawson VA, Adlard PA, Collins SJ (2019) Early existence and biochemical evolution characterise acutely synaptotoxic PrPSc. PLoS Pathog 15:e1007712
Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Brown D, Sinka K, Andrews N, Dabaghian R, Simmons M, Edwards P, Bellerby P, Everest DJ, McCall M, McCardle LM, Linehan J, Mead S, Hilton DA, Ironside JW, Brandner S (2020) Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol 139:965–976
Griffith JS (1967) Self-replication and scrapie. Nature 215:1043–1044
Halliez S, Martin-Lanneree S, Passet B, Hernandez-Rapp J, Castille J, Urien C, Chat S, Laude H, Vilotte JL, Mouillet-Richard S, Beringue V (2015) Prion protein localizes at the ciliary base during neural and cardiovascular development, and its depletion affects alpha-tubulin post-translational modifications. Sci Rep 5:17146
Hannaoui S, Triscott E, Duque Velasquez C, Chang SC, Arifin MI, Zemlyankina I, Tang X, Bollinger T, Wille H, McKenzie D, Gilch S (2021) New and distinct chronic wasting disease strains associated with cervid polymorphism at codon 116 of the Prnp gene. PLoS Pathog 17:e1009795
Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, Mertz K, Zirdum E, Petsch B, Fuchs TJ, Stitz L, Aguzzi A (2011) Aerosols transmit prions to immunocompetent and immunodeficient mice. PLoS Pathog 7:e1001257
Hazards EPoB, Koutsoumanis K, Allende A, Alvarez-Ordonez A, Bolton D, Bover-Cid S, Chemaly M, Davies R, De Cesare A, Herman L, Hilbert F, Lindqvist R, Nauta M, Peixe L, Ru G, Skandamis P, Suffredini E, Andreoletti O, Benestad SL, Comoy E, Nonno R, da Silva Felicio T, Ortiz-Pelaez A, Simmons MM (2019) Update on chronic wasting disease (CWD) III. EFSA J 17
Herzog C, Sales N, Etchegaray N, Charbonnier A, Freire S, Dormont D, Deslys JP, Lasmezas CI (2004) Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet 363:422–428
Huor A, Espinosa JC, Vidal E, Cassard H, Douet JY, Lugan S, Aron N, Marin-Moreno A, Lorenzo P, Aguilar-Calvo P, Badiola J, Bolea R, Pumarola M, Benestad SL, Orge L, Thackray AM, Bujdoso R, Torres JM, Andreoletti O (2019) The emergence of classical BSE from atypical/Nor98 scrapie. Proc Natl Acad Sci U S A
Igel-Egalon A, Bohl J, Moudjou M, Herzog L, Reine F, Rezaei H, Beringue V (2019a) Heterogeneity and architecture of pathological prion protein assemblies: time to revisit the molecular basis of the prion replication process? Viruses 11
Igel-Egalon A, Laferriere F, Moudjou M, Bohl J, Mezache M, Knapple T, Herzog L, Reine F, Jas-Duval C, Doumic M, Rezaei H, Beringue V (2019b) Early stage prion assembly involves two subpopulations with different quaternary structures and a secondary templating pathway. Commun Biol 2:363
Igel-Egalon A, Laferriere F, Tixador P, Moudjou M, Herzog L, Reine F, Torres JM, Laude H, Rezaei H, Beringue V (2020) Crossing species barriers relies on structurally distinct prion assemblies and their complementation. Mol Neurobiol
Igel-Egalon A, Moudjou M, Martin D, Busley A, Knapple T, Herzog L, Reine F, Lepejova N, Richard CA, Beringue V, Rezaei H (2017) Reversible unfolding of infectious prion assemblies reveals the existence of an oligomeric elementary brick. PLoS Pathog 13:e1006557
Jaumain E, Quadrio I, Herzog L, Reine F, Rezaei H, Andréoletti O, Laude H, Perret-Liaudet A, Haïk S, Béringue V (2016) Absence of evidence for a causal link between bovine spongiform encephalopathy strain variant L-BSE and known forms of sporadic Creutzfeldt-Jakob disease in human PrP transgenic mice. J Virol 90:10867–10874
Kimberlin RH, Walker CA (1978) Pathogenesis of mouse scrapie: effect of route of inoculation on infectivity titres and dose-response curves. J Comp Pathol 88:39–47
Kimberlin RH, Walker CA (1988) Pathogenesis of experimental scrapie. Ciba Found Symp 135:37–62
Koshland DE Jr (1963) Correlation of structure and function in enzyme action. Science 142:1533–1541
Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, Raymond GJ, Race B, Baron GS, Caughey B (2021) High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell
Laferriere F, Tixador P, Moudjou M, Chapuis J, Sibille P, Herzog L, Reine F, Jaumain E, Laude H, Rezaei H, Beringue V (2013) Quaternary structure of pathological prion protein as a determining factor of strain-specific prion replication dynamics. PLoS Pathog 9:e1003702
Langevin C, Andreoletti O, Le Dur A, Laude H, Beringue V (2011) Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol Dis 41:219–225
Lansbury PT Jr, Caughey B (1995) The chemistry of scrapie infection: implications of the “ice 9” metaphor. Chem Biol 2:1–5
Lasmezas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, Auvre F, Correia E, Lescoutra-Etchegaray N, Sales N, Wells G, Brown P, Deslys JP (2005) Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783
Lasmezas CI, Fournier JG, Nouvel V, Boe H, Marce D, Lamoury F, Kopp N, Hauw JJ, Ironside J, Bruce M, Dormont D, Deslys JP (2001) Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt– Jakob disease: implications for human health. Proc Natl Acad Sci U S A 98:4142–4147
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132
Le Dur A, Beringue V, Andreoletti O, Reine F, Lai TL, Baron T, Bratberg B, Vilotte JL, Sarradin P, Benestad SL, Laude H (2005) A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc Natl Acad Sci U S A 102:16031–16036
Le Dur A, Lai TL, Stinnakre MG, Laisne A, Chenais N, Rakotobe S, Passet B, Reine F, Soulier S, Herzog L, Tilly G, Rezaei H, Beringue V, Vilotte JL, Laude H (2017) Divergent prion strain evolution driven by PrPC expression level in transgenic mice. Nat Commun 8:14170
Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB (2004) Synthetic mammalian prions. Science 305:673–676
Mahal SP, Jablonski J, Suponitsky-Kroyter I, Oelschlegel AM, Herva ME, Oldstone M, Weissmann C (2012) Propagation of RML prions in mice expressing PrP devoid of GPI anchor leads to formation of a novel, stable prion strain. PLoS Pathog 8:e1002746
Makarava N, Baskakov IV (2013) The evolution of transmissible prions: the role of deformed templating. PLoS Pathog 9:e1003759
Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (2011) Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog 7:e1002419
Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2016) New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol 186:1006–1014
Manka SW, Zhang W, Wenborn A, Betts J, Joiner S, Saibil HR, Collinge J, Wadsworth JDF (2022) 2.7 A cryo-EM structure of ex vivo RML prion fibrils. Nat Commun 13:4004
Marin B, Otero A, Lugan S, Espinosa JC, Marin-Moreno A, Vidal E, Hedman C, Romero A, Pumarola M, Badiola JJ, Torres JM, Andreoletti O, Bolea R (2021) Classical BSE prions emerge from asymptomatic pigs challenged with atypical/Nor98 scrapie. Sci Rep 11:17428
Martin D, Reine F, Herzog L, Igel-Egalon A, Aron N, Michel C, Moudjou M, Fichet G, Quadrio I, Perret-Liaudet A, Andreoletti O, Rezaei H, Beringue V (2021) Prion potentiation after life-long dormancy in mice devoid of PrP. Brain Commun 3:fcab092
Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, Yang J, Grams J, Di Bari MA, Nonno R, Telling GC, Kong Q, Langeveld J, McKenzie D, Westaway D, Safar JG (2014) Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest 124:847–858
Moazami-Goudarzi K, Andreoletti O, Vilotte JL, Beringue V (2021) Review on PRNP genetics and susceptibility to chronic wasting disease of Cervidae. Vet Res 52:128
Moroncini G, Kanu N, Solforosi L, Abalos G, Telling GC, Head M, Ironside J, Brockes JP, Burton DR, Williamson RA (2004) Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci U S A 101:10404–10409
Moudjou M, Castille J, Passet B, Herzog L, Reine F, Vilotte JL, Rezaei H, Beringue V, Igel-Egalon A (2020) Improving the predictive value of prion inactivation validation methods to minimize the risks of iatrogenic transmission with medical instruments. Front Bioeng Biotechnol 8:591024
Moudjou M, Chapuis J, Mekrouti M, Reine F, Herzog L, Sibille P, Laude H, Vilette D, Andreoletti O, Rezaei H, Dron M, Beringue V (2016) Glycoform-independent prion conversion by highly efficient, cell-based, protein misfolding cyclic amplification. Sci Rep 6:29116
Moudjou M, Sibille P, Fichet G, Reine F, Chapuis J, Herzog L, Jaumain E, Laferriere F, Richard CA, Laude H, Andreoletti O, Rezaei H, Beringue V (2014) Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. Mbio 5:e00829-e1813
Mouillet-Richard S, Ghazi A, Laurent-Puig P (2021) The cellular prion protein and the hallmarks of cancer. Cancers (Basel) 13
Nakaoke R, Sakaguchi S, Atarashi R, Nishida N, Arima K, Shigematsu K, Katamine S (2000) Early appearance but lagged accumulation of detergent-insoluble prion protein in the brains of mice inoculated with a mouse-adapted Creutzfeldt-Jakob disease agent. Cell Mol Neurobiol 20:717–730
Nakic N, Tran TH, Novokmet M, Andreoletti O, Lauc G, Legname G (2021) Site-specific analysis of N-glycans from different sheep prion strains. PLoS Pathog 17:e1009232
Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, Sbriccoli M, Lipp HP, Bruce M, Pocchiari M, Agrimi U (2006) Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2:e12
Nonno R, Notari S, Di Bari MA, Cali I, Pirisinu L, d’Agostino C, Cracco L, Kofskey D, Vanni I, Lavrich J, Parchi P, Agrimi U, Gambetti P (2019) Variable protease-sensitive prionopathy transmission to bank voles. Emerg Infect Dis 25:73–81
Notari S, Appleby BS, Gambetti P (2018) Variably protease-sensitive prionopathy. Handb Clin Neurol 153:175–190
Owen JP, Maddison BC, Whitelam GC, Gough KC (2007) Use of thermolysin in the diagnosis of prion diseases. Mol Biotechnol 35:161–170
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR (2006) Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 45:15710–15717
Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS, Gambetti P, Ghetti B, Agrimi U, Nonno R (2016) Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443
Piro JR, Harris BT, Nishina K, Soto C, Morales R, Rees JR, Supattapone S (2009) Prion protein glycosylation is not required for strain-specific neurotropism. J Virol 83:5321–5328
Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 4:805–814
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95:13363–13383
Race B, Phillips K, Meade-White K, Striebel J, Chesebro B (2015) Increased infectivity of anchorless mouse scrapie prions in transgenic mice overexpressing human prion protein. J Virol 89:6022–6032
Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K (1996) NMR structure of the mouse prion protein domain PrP(121–231). Nature 382:180–182
Riesner D, Kellings K, Post K, Wille H, Serban H, Groth D, Baldwin MA, Prusiner SB (1996) Disruption of prion rods generates 10-nm spherical particles having high alpha-helical content and lacking scrapie infectivity. J Virol 70:1714–1722
Rudd PM, Merry AH, Wormald MR, Dwek RA (2002) Glycosylation and prion protein. Curr Opin Struct Biol 12:578–586
Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813
Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4:1157–1165
Salamat MK, Dron M, Chapuis J, Langevin C, Laude H (2011) Prion propagation in cells expressing PrP glycosylation mutants. J Virol 85:3077–3085
Sarradin P, Viglietta C, Limouzin C, Andreoletti O, Daniel-Carlier N, Barc C, Leroux-Coyau M, Berthon P, Chapuis J, Rossignol C, Gatti JL, Belghazi M, Labas V, Vilotte JL, Beringue V, Lantier F, Laude H, Houdebine LM (2015) Transgenic rabbits expressing ovine PrP are susceptible to scrapie. PLoS Pathog 11:e1005077
Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB (1989) Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857
Sevillano AM, Aguilar-Calvo P, Kurt TD, Lawrence JA, Soldau K, Nam TH, Schumann T, Pizzo DP, Nystrom S, Choudhury B, Altmeppen H, Esko JD, Glatzel M, Nilsson KPR, Sigurdson CJ (2020) Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J Clin Invest 130:1350–1362
Shorter J, Lindquist S (2006) Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell 23:425–438
Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B (2005) The most infectious prion protein particles. Nature 437:257–261
Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA (2007) Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 282:7465–7471
Somerville RA, Ritchie LA (1990) Differential glycosylation of the protein (PrP) forming scrapie-associated fibrils. J Gen Virol 71(Pt 4):833–839
Stahl N, Baldwin MA, Burlingame AL, Prusiner SB (1990) Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 29:8879–8884
Taylor DM, McConnell I, Fraser H (1996) Scrapie infection can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J Gen Virol 77:1595–1599
Thackray AM, Andreoletti O, Bujdoso R (2018) Mammalian prion propagation in PrP transgenic Drosophila. Brain 141:2700–2710
Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Beringue V (2010) The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog 6:e1000859
Tsai CJ, Kumar S, Ma B, Nussinov R (1999) Folding funnels, binding funnels, and protein function. Protein Sci 8:1181–1190
Turnbaugh JA, Unterberger U, Saa P, Massignan T, Fluharty BR, Bowman FP, Miller MB, Supattapone S, Biasini E, Harris DA (2012) The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J Neurosci 32:8817–8830
Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, Coghill A, Hart P, Piccardo P, Barron RM, Manson JC (2008) Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol 6:e100
Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, Ironside JW, Haik S, Basset-Leobon C, Lacroux C, Peoch K, Streichenberger N, Langeveld J, Head MW, Grassi J, Hauw JJ, Schelcher F, Delisle MB, Andreoletti O (2008) Beyond PrP9res) type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog 4:e1000029
Velasquez CD, Kim C, Haldiman T, Kim C, Herbst A, Aiken J, Safar JG, McKenzie D (2020) Chronic wasting disease (CWD) prion strains evolve via adaptive diversification of conformers in hosts expressing prion protein polymorphisms. J Biol Chem 295:4985–5001
Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, Welch J, Stone L, Lloyd SE, Hill AF, Brandner S, Collinge J (2004) Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306:1793–1796
Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990
Weissmann C, Li J, Mahal SP, Browning S (2011) Prions on the move. EMBO Rep 12:1109–1117
Wiseman FK, Cancellotti E, Piccardo P, Iremonger K, Boyle A, Brown D, Ironside JW, Manson JC, Diack AB (2015) The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J Virol 89:4738–4747
Funding
BF PhD fellowship is funded by Agence Nationale de la Recherche (ANR-21-CE15-0011–01). Part of the work is funded by the One-Health program from the Ile-de-France Region (PrionDif award) and by a grant from INRAE metaprogram DIGITBIO (PrionDif award).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Informed consent
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Igel, A., Fornara, B., Rezaei, H. et al. Prion assemblies: structural heterogeneity, mechanisms of formation, and role in species barrier. Cell Tissue Res 392, 149–166 (2023). https://doi.org/10.1007/s00441-022-03700-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-022-03700-2