
Abstract
Severe spinal cord injury causes permanent loss of function and sensation throughout the body. The trauma causes a multifaceted torrent of pathophysiological processes which ultimately act to form a complex structure, permanently remodeling the cellular architecture and extracellular matrix. This structure is traditionally termed the glial/fibrotic scar. Similar cellular formations occur following stroke, infection, and neurodegenerative diseases of the central nervous system (CNS) signifying their fundamental importance to preservation of function. It is increasingly recognized that the scar performs multiple roles affecting recovery following traumatic injury. Innovative research into the properties of this structure is imperative to the development of treatment strategies to recover motor function and sensation following CNS trauma. In this review, we summarize how the regeneration potential of the CNS alters across phyla and age through formation of scar-like structures. We describe how new insights from next-generation sequencing technologies have yielded a more complex portrait of the molecular mechanisms governing the astrocyte, microglial, and neuronal responses to injury and development, especially of the glial component of the scar. Finally, we discuss possible combinatorial therapeutic approaches centering on scar modulation to restore function after severe CNS injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
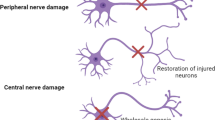
The failure of repair following spinal cord injury (SCI) is due to factors both intrinsic to cellular components and extrinsic, surrounding the site of injury. In the 1980s, the Aguayo lab first showed that long-distance axon regeneration from CNS neurons within a peripheral nerve graft was abruptly halted at the border between the Schwann cell-laden peripheral nervous system (PNS) and astrocyte-rich central nervous system (CNS) (David and Aguayo 1981). This key finding suggested the importance of extrinsic factors in adversely affecting regeneration following CNS injury. Decades of research have shown that the hypertrophic lesion penumbra which forms the following injury to the CNS (encapsulating reactive astrocytes, activated microglia, and oligodendrocyte progenitor cells) comprises one of three inhibitory cellular compartments which impede axon regeneration both at PNS-to-CNS graft interfaces and within the lesion parenchyma. This is traditionally termed the glial scar (Fig. 1). The second barrier to regeneration, which also resides near the lesion penumbra but lies inside the glial scar, is the fibrotic scar — a structure replete with fibroblasts or fibroblastic-like cells including those derived from the meninges, mural/adventitial sources, or pericytes (Guimarães-Camboa et al. 2017; Riew et al. 2021) (Fig. 1). Swirls of rigid basal lamina membrane form between the astrocytic and fibroblastic layers of the scar further preventing axonal growth (Rudge and Silver 1990; Li et al. 2020b). The third barrier is the lesion epicenter itself, which comprises mostly systemically derived inflammatory cells such as activated macrophages (Busch et al. 2009; Kigerl et al. 2009; Popovich 2014). While all these compartments are dynamic in composition, their formation permanently alters the cellular landscape and extracellular matrix of the spinal cord up to centimeters rostral and caudal from the initial impact. With the accessibility of next-generation sequencing technologies, innovations in SCI research have provided new understanding of the cellular and molecular diversity of cells and structures which encompass the remodeled CNS environment after trauma. This appreciation is stimulating the development of clinically relevant treatment strategies to aid recovery of motor and sensory function.

Cellular constituents of the glial/fibrotic scar. Following spinal cord injury, pro-inflammatory cascades activate the multitude of cells found at the spinal cord lesion. The glial scar itself is composed of astrocytes, NG2 + oligodendrocyte progenitor cells, and microglia, among others. Astrocytes and oligodendrocyte progenitor cells of the lesion penumbra are especially important in remodeling the extracellular matrix and upregulating axon-inhibitory chondroitin sulfate proteoglycans (CSPGs). Cells of the lesion penumbra work in concert to cordon off the pro-inflammatory lesion epicenter. The lesion epicenter predominantly includes macrophages and fibroblasts which are sequestered to prevent the spread of inflammation after injury
Here, we review how recent single-cell RNA sequencing and genomic-targeting techniques have expanded our appreciation of the barriers that oppose or promote axon regeneration and functional recovery. This will include a brief review of the differences in regeneration capacity between divergent phyla and neonatal versus adult regeneration after mammalian SCI concentrating on the new insights that emerging techniques reveal about both glia and non-glia in the context of scar formation. We will additionally examine how wound and glial scar-associated products affect neurons. Our review will end with a discussion of the potent growth inhibitory effects of one especially critical family of extracellular matrix molecules, the chondroitin sulfate proteoglycans (CSPGs), and how alleviating these glycoproteins in combination with other strategies can help promote functional recovery.
Changes in the capacity of CNS regeneration through different phyla and across ages
Simple invertebrates are capable of regenerating whole-body structures and organs de novo (Fig. 2). Following repeated amputations Lineus sanguineus (species ribbon worm, phylum Nemertea) are able to completely regenerate the anterior aspect of their bodies meaning 200,000 worms could be generated following a similar number of dissections, each just 1/200,000th the volume of the original animal (Zattara et al. 2019). However, these remarkable regenerative capabilities are not universal across the ~ 1200 species within the phylum and the defining factors causing this regenerative capacity are not well understood. Zebrafish (Danio rerio) (Becker and Becker 2014; Cigliola et al. 2020) efficiently regenerate their spinal cords as adults after injury (Mokalled et al. 2016; Tsata et al. 2020; Klatt Shaw et al. 2021). Among most mature amphibians, complete structural regeneration is lost, but Urodeles (newts and salamanders) regenerate their spinal cords throughout life (Ferretti et al. 2003) (Fig. 2). The ability to regenerate in fish is, in part, due to their capacity for scar-free wound healing (Tsata et al. 2020). The loss of regeneration in amphibians occurs at stages when they begin developing scar-like tissue, likely due to maturation of the immune system (Bertolotti et al. 2013, Edwards-Faret et al. 2021).

Schematic phylogenetic relationship between regenerating animals. Simplified representation of the phylogenetic relationship between selected species capable of spinal cord regeneration (either within development or through their life span) following injury. Zebrafish, salamanders, and nemertea can regenerate their spinal cords throughout life following injury (red), frogs can do so at the tadpole stage (orange). Some higher-order animals are capable of this in the days following birth (green) including the opossum (until P17) and mouse (until P2). A number of other species (black) with common ancestors to these species are not known to regenerate their spinal cords following injury at any stage of development
In mammals, the capacity for CNS regeneration sharply declines with age (Fig. 3). This is strikingly illustrated by the South American opossum (Monodelphis domestica) which is born precociously. Up to 12 days after birth, cervical segments of the opossum cord do not scar (Fig. 2). Regeneration fails rostrally after this time while the lumbar segments, which mature and scar later than the rostral sections, retain the ability to regenerate until 17 days of age (Mladinic and Wintzer 2002). After these developmental stages, scarring occurs globally, and regeneration capacity in the opossum is lost. In mice, the capacity for robust regeneration after birth with scar-free wound healing (following very fine crush injuries to the spinal cord) is limited to the early post-nate (see below sect. “The biology and transcriptomic changes of microglia after injury” for more details). However, following a moderate-to-severe spinal cord transection injury which produces more lesion core-associated inhibitors, even neonatal mice rarely regrow axons, and locomotor and bladder functions never return (Gearhart et al. 1979). Among rats (Sprague-Dawley), robust sprouting (rather than frank regeneration) of the corticospinal tract can occur just after birth, but this capacity for plasticity is lost in the surgically hemisected spinal cord (an extensive lesion) at the end of the first week post-partum (Kunkel-Bagden et al. 1992).

Mammalian neurons lose regeneration potential with age. Embryonic and immature mammalian neurons have a higher potential for axon regeneration after injury due to increased pro-regenerative intrinsic factors and decreased regeneration-inhibitory extrinsic factors compared to the adult mammalian neuron. Immature neurons additionally possess pro-regenerative molecular signals/transcriptome, which is turned off as the neurons age into adulthood
In mammals and birds, the capacity for neurons to regenerate is the greatest during embryonic development (Smith et al. 1987; Steeves and Tetzlaff 1998). For example, neurons derived from embryonic brains transplanted into the more mature CNS are capable of elaborate regrowth and connection-forming abilities (Wictorin and Björklund 1992). Other classic examples of this capacity are shown by Iwashita et al. who implanted embryonic spinal cord grafts into the resected spinal cords of neonatal rats and observed lengthy axonal regeneration with recovery of locomotor function in adulthood (Iwashita et al. 1994). These results were confirmed by Lu et al. who have documented impressive amounts of long-distance regeneration from dissociated neural stem cells harvested from embryonic rats and placed into the spinal cord of adults (Lu et al. 2014). These cells were transplanted in a fibrin matrix impregnated with a host of growth factors to minimize scar and maximize graft integration.
Younger neurons have different responses to extrinsic cues that allow for enhanced axonal regeneration during mammalian development. These include changes in response to inhibitory myelin signaling (Poplawski et al. 2020). Embryonic rat dorsal root ganglion neurons are less sensitive to the inhibitory myelin protein, Nogo-A, whereas by post-natal day 6, the same rat neuron growth cones collapse when exposed to the molecule (Bandtlow 2003). The decreased presence of axon-inhibitory structures such as the scar or perineuronal nets laden with growth inhibitory CSPGs (which do not form until the end of the critical period) plays a vital role (Celio et al. 1998; Takesian and Hensch 2013; Warren et al. 2018a). Specific CSPGs, such as phosphacan, are more highly expressed in the glial scar of the adult compared to the wound response in the neonate (McKeon et al. 1995). It is possible that immature neurons respond differently than adult neurons to CSPGs. Indeed, lack of growth inhibition by CSPGs, as well as their receptors in immature neurons, has been reported (Laabs et al. 2005; Busch et al. 2009; See et al. 2010; Haas and Fischer 2014).
What might be the additional cadre of molecular changes that occur between development and adulthood in the mammal to induce a regeneration-inhibitory “switch?” During development, axons first need to extend toward their intended target. Forward growth then needs to be altered to allow for branching, synaptogenesis, and integration into circuits. This program may, in part, be dictated by interactions between CSPGs and their receptor protein tyrosine phosphatase sigma (PTPRσ) which signal via a newly appreciated relationship with autophagic flux (Sakamoto et al. 2019; Tran et al. 2020). However, the host of transcriptional or molecular switches that allow this change in regeneration capacity with age will surely be multifaceted. Tedeschi et al. hypothesized that genes inhibiting lengthy axon regrowth in the later stages of embryonic development and beyond might be related to mechanisms that control the intrinsic switch from rectilinear growth to that of branching and synapse formation (Tedeschi et al. 2016). Using bulk RNA sequencing, they found that the synaptogenesis-related voltage-gated calcium channel subunit α-2-δ inhibits axon outgrowth at both the end of embryogenesis and throughout adulthood while regulating the differentiation of presynaptic terminals (Kurshan et al. 2009). Blocking the α-2-δ subunit with pregabalin was able to promote CNS (dorsal column) axon regeneration in dorsal root ganglion neurons of adult mice after conditioning sciatic nerve crush lesions. It will be interesting to look for transcription factor(s) and promoter regions that initiate this transcriptomic switch and whether other genes may be involved to cumulatively decrease regeneration capacity with age. These same transcriptomic switches may prove therapeutically beneficial if they can be transiently “turned off” in specific cell populations in adulthood to recapitulate an embryonic state to encourage regeneration.
In addition to the decrease in regeneration capacity from development to adulthood, a growing body of literature suggests that a second, progressive switch occurs between adulthood and old age, further inhibiting regeneration (Geoffroy et al. 2017). One of the cellular mechanisms governing this further regeneration decline may be the scar-enhancing effects of neuroinflammation (Fitch and Silver 2008), which increases in intensity with age (Russo et al. 2011). Chronic neuroinflammation decreases neurogenesis in the hippocampus (Russo et al. 2011) and is responsible for neural stem cell decline in the aging mouse brain (Kalamakis et al. 2019). Stem cell niche-derived cytokines including IFN, CXCL10, and the Wnt agonist, sFRP5, induce a quiescence of stem cells within the subventricular zone. This preserves a small pool of neural stem cells in the aging brain but decreases the amount of neurogenesis compared to younger brains. Work in progress by Paramos-de-Carvalho et al. suggests that, in the aged adult, neurons become senescent following inflammation from SCI and that inhibiting their depletion using a senolytic treatment may improve functional recovery (Paramos-de-Carvalho et al. 2020). Indeed, neuroblast senescence in aged brains increases as it becomes more inflammatory (Jin et al. 2021). Clearly, with age come innate molecular changes based on cellular transcriptional heterogeneity as well as differences in immune responses to injury and exacerbation of inflammation-induced glial scar formation (Fitch and Silver 2008). In the following sections, we will explore how next-generation sequencing technologies with accompanying experimental manipulations are beginning to characterize and define critical molecular differences that lead to regenerative failure in the context of CNS injury or disease with an emphasis on SCI.
Spinal cord lesions and their cellular constituents
Traumatic SCI begins with physical (primary) injury to the spinal cord, which immediately causes axon shearing, bleeding, and cell death. This leads to the release of alarmins, which recruit local microglia and systemically circulating immune cells that pass through the damaged blood-spinal cord barrier to propagate inflammation and secondary damage (Popovich et al. 1997; Gadani et al. 2015). This ensures that formation of the scar is triggered, in part, by inflammatory processes and requires the concerted effort of a myriad of different cell types as well as transcriptomic and molecular changes driven initially by inflammation-induced reactivity. Here, we will focus on astrocytes, microglia, and neurons. For an in-depth discussion of the fibrotic component of scar, see review by Jae K Lee in this issue.
Next-generation transcript sequencing has emerged as a powerful tool to assess system-wide cellular changes following injury for a broad molecular view of SCI. SCI transcriptomics was assessed as bulk RNA using microarrays and other gene-biased methods. Single-cell RNA sequencing now offers a more sensitive (Chen et al. 2013) and unbiased examination of some RNA transcripts in a variety of cell types to identify novel biomarkers for injury progression, characterize cell heterogeneity/changes after injury, and potentially enhance targeted translation of therapies for SCI. For example, single-cell RNA sequencing revealed that the glial scar penumbra is replete with proliferating oligodendrocyte progenitor cells, which greatly contributes to CSPG deposition and remodeling of the lesion site (Milich et al. 2020). In fact, recent findings suggest that astrocytes, while a major contributor of CSPGs early after injury, are not the predominant CSPG producer over time after SCI (Yang et al. 2020).
However, single-cell RNA sequencing is not without its pitfalls. Rare cell types may be difficult to sequence without specific methods of enrichment for the depth of gene coverage, that is the number of affected genes positively identified in a single cell. The sheer complexity of data from any one experiment makes meaningful interpretation of genomic changes a challenge especially in linking specific gene changes back to biological significance. Added to this, the increased use of this technology among disparate labs with no common naming convention or taxonomy to categorize these newly identified subcellular populations makes collaboration and understanding of the system as a whole difficult moving forward. Nonetheless, recent use of next-generation sequencing has revealed novel insights into cell-specific differences in transcriptomic response after SCI in the following cell types we will focus on here: astrocytes, microglia, and neurons.
The biology and transcriptomic changes of astrocytes after injury
The regional heterogeneity of astrocytes and their differences in response to injury or pro-inflammatory molecules such as beta-amyloid (Canning et al. 1993) have been further substantiated at the molecular level by next-generation sequencing technologies (Zeisel et al. 2018).
Having established innate molecular diversity among astrocytes, researchers have recently begun to ask how different astrocyte subpopulations respond to injury or inflammation. Huang et al. for example, found that a subset of reactive astrocytes expresses the transcription factors OCT4 and KLF yielding expression of neural stem cell-related markers nestin and sox2 as well as Hippo/Yap pathway activation after SCI (Huang et al. 2020). Overexpression of these transcription factors further conferred improved motor function after SCI in mice. In a further exploration of astrocyte heterogeneity, Zamanian et al. genetically profiled reactive astrocytes termed “A1” derived from mice treated with a systemic LPS injection (Zamanian et al. 2012). Using single-cell microfluidic qPCR, Liddelow et al. found that it was not LPS itself that activated astrocytes (since they do not express TLR4 and MYD88 surface receptors) but rather injury-activated microglia through 1L-1α, TNF, and C1q signaling. Activated A1 astrocytes were found to do the following: upregulate complement cascade genes, were destructive to synapses, showed reduced phagocytic ability, and induced apoptosis in neurons and oligodendrocytes (Zamanian et al. 2012; Liddelow et al. 2017). This is in contrast to “A2” astrocytes, which were described as potentially neuroprotective by upregulating neurotrophic factors (Liddelow et al. 2017). Of note, like the M1/M2 convention of naming reactive macrophages, which has been repeatedly critiqued for its over simplicity (Martinez and Gordon 2014; Ransohoff 2016; Mesquida-Veny et al. 2020), the “A1” and “A2” paradigm, too, flattens the molecular heterogeneity of astrocytes and their diverse response to injury. Thus, while useful, it is important to recall that astrocyte reactivity, as well as their transcriptomic diversity, likely exists on a spectrum as they respond near to or further from a lesion at different ages (Escartin et al. 2021). For example, reactive astrocytes far from a lesion in regions undergoing Wallerian degeneration upregulate GFAP but do not markedly change their orientation, at least within the first few months after injury, and can support robust regeneration even of adult axons (Davies et al. 1999; Li et al. 2020a, b). This is in contrast to astrocytes in the scar surrounding the lesion which upregulate GFAP, undergo hypertrophy, and dramatically change their orientation and density, which ultimately helps the astrocyte block regeneration (Davies et al. 1997; Grimpe et al. 2005; Zukor et al. 2013).
In addition to considerations of innate astrocyte heterogeneity and spatial relationship to the lesion, age is a factor in understanding how astrocytes react to injury. Mature astrocytes transplanted into brains stimulate fibroblast and macrophage entry and enhanced cavitation within (and basal lamina formation around) the lesion site (Smith et al. 1987; Smith and Silver 1988; Filous et al. 2010). Mature astrocytes activated by flow-sorted amyloid plaques upregulate the GABA transmitter, ATP, and glutamate (among others) to impair memory function in mouse models of Alzheimer’s disease (Jo et al. 2014). On the other hand, neonatal astrocytes implanted into the adult, diminished scar formation, and stimulated the regrowth of injured fibers, promoting locomotor function following SCI (Smith et al. 1987; Smith and Silver 1988; Joosten et al. 2004). While immature astrocytes still undergo reactive gliosis following injury (Smith et al. 1987; Balasingam et al. 1994), they do not pack as tightly nor allow for such exuberant basal lamina formation as mature astrocytes. Transcriptional changes to astrocyte precursors after brain stab injury suggest an increase in astrocytic differentiation rather than traditional hypertrophy-associated changes associated with gliosis (Domowicz et al. 2011).
Although astrogliosis occurs in all age groups, the mechanism and potential for glial repair after injury differ greatly between neonatal and adult astrocytes. Neonatal astrocytes secrete neuroprotective factors after ischemic stroke such as PDGF, IGF, and VEGF, which encourage neuronal survival (Wagenaar et al. 2018; Revuelta et al. 2019). Despite their gliotic changes after injury in embryonic and neonatal injuries, transplantation of embryonic astrocytes into the adult lesion site after SCI is a beneficial treatment (Davies et al. 2006, 2011). Various types of neurons seeded onto substrates containing reactive wound tissue harvested from neonatal rat brains (Rudge and Silver 1990; McKeon et al. 1991) extend far longer neurites than those grown on CSPG containing basal lamina-rich wound substrates derived from adult brains. Immature astrocytes were also observed to form bridges between the lesion and tissue border enabling axon outgrowth (Smith and Silver 1988; Filous et al. 2010; Haas et al. 2012). Thus, in contrast to mature astrocytes, especially in the vicinity of older lesions, immature astrocytes are permissive for axon outgrowth.
The biology and transcriptomic changes of microglia after injury
The roles of microglia and macrophages have often been conflated in SCI research because of the difficulty in distinguishing the two cell populations with immunostaining techniques alone. These two cell populations, however, are distinct. Macrophages, but not microglia, present at the lesion cause dieback of the dystrophic axon tip towards its soma (Horn et al. 2008; Evans et al. 2014). Using a transgenic mouse line that specifically labeled microglia, Bellver-Landete et al. found that microglia proliferated around the lesion site and peaked at 2 weeks post-SCI (Bellver-Landete et al. 2019). This is in contrast to infiltrating monocytes in rats, which peak at 1 week post-injury and again around 60 days post-injury (Popovich et al. 1997; Milich et al. 2020). Activated microglia were also marked by an amoeboid shape and expression of CD68, suggesting increased phagocytic behavior (Janda et al. 2018). By 2 weeks post-injury, microglia serve as a physical barrier between infiltrating leukocytes and astrocytes, which become reactive in response to microglia-derived factors including IGF-1 (Bellver-Landete et al. 2019). These findings suggest that microglia and astrocytes work in concert to seal off infiltrating immune cells and fibroblasts, respectively, to contain inflammation after SCI (Bundesen et al. 2003; Soderblom et al. 2013). Further, microglia-corralling and scar-forming capabilities require an upregulation of the semaphorin receptor Plexin-B2, which peaks by 2 weeks post-injury and wanes by 21 days post-injury to promote motility (Zhou et al. 2020). Depleting microglia with a stimulating growth factor receptor antagonist, PLX5622 (Bellver-Landete et al. 2019; Fu et al. 2020), or conditionally knocking out Plexin-B2 specifically in microglia (Zhou et al. 2020) resulted in an increase in systemic immune cell infiltration, increased neuronal and oligodendrocyte apoptosis, disruption of glial scar formation primarily through disorganized and delayed astrocyte repopulation, and worsened behavioral outcomes after SCI. Therefore, in adulthood, microglia work together with other immune cells such as macrophages and other glia (including astrocytes) to aid in effective scar formation. Recent findings have even revealed that astrocytes and microglia coordinate to maintain brain homeostasis by working in concert to phagocytose specified territories of apoptosed neurons (Damisah et al. 2020). The ability of microglia and astrocytes to efficiently clear apoptosed cells and other debris declines with age (Damisah et al. 2020), which could further inflammatory processes over time. Our current understanding corroborates that glial scar formation is an immediately beneficial process, comprising many cell types (beyond that which is discussed here) to contain and limit the spread of inflammation. However, over time, the adult scar and its extracellular matrix components impede regeneration (see below and (Tran et al. 2018b)).
While microglia were shown to coordinate scar formation during adulthood, a recent seminal study by Li et al. highlights the difference between microglia-driven scar-forming, regeneration blocking, wound healing in the adult and scar-free, and regeneration promoting healing in the neonate (Li et al. 2020b). Li et al. performed narrow spinal crush injuries in neonatal (post-natal day 2) mice, which led to a lesion-associated increase of loosely organized GFAP-positive astrocytes, but with little obvious hypertrophy and with an absence of classic adult scar-associated matrix constituents. This contributed to wound healing without scar formation and allowed for robust growth of serotonergic and cortico-spinal axons directly through the lesion. Notably, axon growth largely failed to extend past the injury site when lesions were made slightly later (by post-natal day 7), or in the adult. While the authors could not determine whether axon growth was due to regeneration from injured cells or the arrival of late projecting fibers, it was clear that the scar-free wound healing response in the neonate allows for axonal growth into and well beyond the lesion. Importantly, there was total absence of an adult-like scar with little deposition of CD68 + immune cells or basal lamina constituents such as fibronectin, collagen type I, laminin, or CSPGs. It was further demonstrated that axon growth-blocking glial scar formation of the post-injury neonatal spinal cord was critically dependent on immature microglia as their depletion through conditional knock down, or treatment with a drug to inhibit colony-stimulating factor 1 receptor allowed for much stronger astrocytic hypertrophy and packing density. These treatments dramatically reduced but did not completely eliminate, the efficacy of axon growth into or through the lesion site. The vast majority of failed regenerating serotonergic axons now halted their growth directly abutting the dense astrocytic wall, revealing clear evidence that astrocytes (likely in and of themselves) can build a regeneration-blocking scar barrier whose inhibitory properties at this stage appear to arise largely from the mere physical arrangement and packing density of the cells (see (Xu et al. 1999) for the role of GFAP in the compaction of scar astrocytes). Importantly, the lack of potently inhibitory basal lamina constituents such as collagens and CSPGs likely allowed for a small number of neonatal axons (with their enhanced growth machinery compared to adult neurons) to pass through the barrier.
Using single-cell RNA sequencing, Li et al. found five transcriptionally distinct microglia populations after injury, confirming a heterogeneous response. Further analysis suggested that neonatal microglia become transiently reactive after injury, then revert to a homeostatic state by 3 days post-injury (Li et al. 2020b). As part of this transiently reactive response, neonatal injury-activated microglia first secreted fibronectin and remodeled the extracellular matrix to close the wound and enable an axon growth-permissive bridge. This vital response to enable regeneration is curtailed in the adult where fluid-filled cysts and the glial scar form (Rooney et al. 2009). The initiation of inflammation and engagement of the CNS immune system to propagate regenerative processes also bring to mind Ernst Haeckel’s adage that “ontogeny recapitulates phylogeny” as the zebrafish regeneration response system also relies on an initial inflammatory signal to start neural stem cell proliferation and regeneration after a brain injury or SCI (Kyritsis et al. 2012). Increasingly, recent findings are highlighting a major difference in injury responses between aged and immature immune cells. Young neutrophils (Ly6Glo), characterized after being activated by zymosan, were found to drive retinal ganglion cell axon regeneration by secreting growth factors such as NGF and IGF-1 (Sas et al. 2020). Transplantation of this cell type into the sciatic nerve enabled dorsal root ganglion axon regeneration into the lacerated dorsal column of the thoracic spinal cord (Sas et al. 2020). Immature microglial cell transplantation into the injured adult spinal cord also improved wound healing without scar and permitted axonal regeneration beyond the lesion (Li et al. 2020b).
Li et al. (2020a, b) emphasize the importance of proteases in scar formation. In the neonate, microglia produce endogenous proteinase inhibitors to promote healing. They found that serine protease inhibitors and cystein peptidases such as Cstb encoding cystatin b, which inhibits lysosomally derived cathepsin proteases, were upregulated (Li et al. 2020a, b). Blocking proteinases with inhibitors prior to transplanting mature microglia into the lesioned adult cord led to reductions in collagen I and CSPG deposition which, in turn, resulted in some regenerating axons, although fewer than that which occurred after transplanting purified immature microglia. This suggests the likely existence of other scar-free wound healing factors. Rampant protease secretion from immune cells propagates their entry through the blood-brain barrier after traumatic injury (Noble et al. 2002) or neurodegenerative disorders (Crapser et al. 2020) contributing to secondary damage and loss of synapses. However, the fine control of local protease release from growth cones or the leading processes of migrating cells is critically important during developmental pathfinding (Brooks et al. 2013) as well as during regeneration or sprouting in the adult (Krystosek and Seeds 1984; Luo et al. 2018; Tran et al. 2018a; Carulli and Verhaagen 2021; Tran and Silver 2021).
The response of neurons after SCI
At the acute stage following injury, sheared axons undergo a process called dieback that drives their projections back towards the soma. Axons at the lesion epicenter can be cut by the shear force of traumatic SCI as well as massively damaged by invading macrophages (Fitch et al. 1999). In response to injury, severed axons die back from the lesion epicenter upon physical contact with infiltrating, reactive macrophages (Horn et al. 2008; Busch et al. 2009). This process is dependent on macrophages directly contacting axons and the subsequent secretion of proteases such as MMP-9 (Busch et al. 2009, 2011). Treatment of macrophages with MMP-9 specific inhibitors, anti-inflammatory drugs, or other inflammatory-modulating substances was able to greatly reduce the macrophage-induced dieback phenomenon (Busch et al. 2011; DePaul et al. 2015). Thus, dieback and ensuing axon damage are a consequence of the pro-inflammatory environment shortly after SCI. Interestingly, embryonic neurons are resistant to attack by activated macrophages (Busch et al. 2009).
In response to the formation of the glial scar, the growth cone tips under attack by macrophages eventually cease retracting and become “entrapped” in the reactive astrocyte/OPC/pericyte penumbra of the lesion (Davies et al. 1997; Filous et al. 2014; Son 2015) where they can reside in a stable, dystrophic state for decades (Ruschel et al. 2015). These growth cones, trapped indefinitely in the glial scar penumbra, may be able to do so because they develop synaptic-like contacts on CSPG producing (NG2) glia that colocalize with synaptic markers such as PSD95 and SNAP25 (see below and (Filous et al. 2014)).
Transcriptomic changes of neurons after SCI
Next-generation sequencing has revealed a greater diversity of spinal cord neurons than previously considered. Sathyamurthy et al. created an atlas of the adult mouse lumbar spinal cord and identified more than 40 distinct neuronal subpopulations (Sathyamurthy et al. 2018). Of note, five neuronal clusters of the ventral horn and one along the mid-cord expressed the perineuronal net CSPGs aggrecan and brevican (Sathyamurthy et al. 2018), showing that perineuronal nets surround previously unidentified ventral neurons (Galtrey et al. 2008). The molecular diversity of lumbar spinal cord neurons is surprisingly heterogeneous — this may also be the case with neurons of other regions of the spinal cord, but how and which important molecular differences underpin their unique responses to SCI or scarring are yet to be determined. For example, SCI researchers have long observed that, within the CNS, serotonergic neurons respond most robustly to various forms of therapeutic treatments, are less likely to die back due to macrophage attack, and are most likely to sprout following CNS lesions (Li et al. 2004; Pearse et al. 2004; Alilain 2009; Alilain et al. 2011; Hawthorne et al. 2011; Lang et al. 2015; Jin et al. 2016; Warren et al. 2018b). Propriospinal interneurons have also been reported to regenerate their axons directly through the early lesion environment after thin midline cuts to the spinal cord (Fenrich and Rose 2009). In contrast, a classic example of a regeneration-refractory and poorly sprouting pathway is the corticospinal tract, an important system involved in voluntary motor control (Welniarz et al. 2017).
Recent studies revealed insights into the growth characteristics of the corticospinal tract, which matures relatively late in development with elongation and synaptogenesis continuing post-natally (Bates and Stelzner 1993). In rodents, late-arriving corticospinal axons can circumnavigate lesions during the first post-natal week (Schreyer and Jones 1983). However, regeneration of this tract in the adult does not occur. To better understand this tract’s unique response following SCI, Tsujioka et al. performed a unilateral pyramidotomy to track changes and compared gene expression of the cervical cord between 7-day-old and 8-week-old mice at 3 days post-pyramidotomy (Tsujioka and Yamashita 2019). Tsujioka et al. found that the inflammatory response was different between neonatal and adult mice: adults showed an increase of Iba1 + microglia/macrophages near the denervated tract after pyramidotomy whereas neonatal mice did not show any such long-term accumulation (see also Li et al. 2020b). The neonatal group also increased gene networks responsible for axon regeneration, myelination, and cell proliferation. In the adult, Tsujioka et al. found enrichment of genes involved in lysosomal activity and phagocytosis, toll-like receptor signaling, and coagulation and complement pathways by microglia/macrophages. Adults showed high inflammatory response activation marked by Ccl5 and Cd52 and an increase in cell proliferation and cell cycle-regulating genes. While this study identified which genes were up- or downregulated following injury, the dosage or extent of gene expression especially between the two different age groups when genes themselves may be innately subject to changes in activation or downregulation could not be assessed. This caveat, however, could be addressed by comparing the transcriptome of the same age group under different conditions such as with a control, non-regenerating group, and a treatment group where axon regeneration is encouraged.
In this regard, Poplawski et al. (2020) performed a dorsal column lesion at the cervical level to transect the corticospinal tract in adult mice. In one group, neural progenitor cells derived from embryonic day 12 mice (which had been shown to allow for corticospinal tract regeneration and synapse reformation Lu et al. 2012, 2014)) were grafted into the lesion, and mRNAs enriched from corticospinal neurons were subjected to single-cell RNA sequencing at 10, 14, and 21 days after injury. A remarkably pro-regenerative gene network similar to an embryonic transcriptional growth state (embryonic day 18 corticospinal neurons) was initiated by injury alone, although it disappeared as soon as 2 weeks following injury. However, animals with neural progenitor cell grafts were able to sustain this pro-regenerative response. In cases of successful corticospinal axon regeneration, it was found that there were increases in both the neuroprotective genes early after injury and the expression of genes responsible for inducing synapse formation and axon guidance. Notably, genes such as Htt encoding the Huntington protein (Htt), which aids embryo survival and selective autophagy (Rui et al. 2015), were identified as essential to this pro-regenerative response. The discovery of the Htt protein as a component of axon regeneration (Poplawski et al. 2020) further pinpoints the importance of autophagy as one of the many processes essential to axon regeneration (Tran et al. 2020). Interestingly, upregulation of this embryonic response is sustained by neural progenitor cell grafts instead of recruiting other genes not formally part of the embryonic response. Thus, recapitulation of an embryonic response and maintenance of this response may be a meaningful therapeutic approach moving forward.
SCI and plasticity of the glial scar
The glial/fibroblastic scar forms from the synergistic interactions of astrocytes and microglia in cooperation with several additional cell types including, but not limited to macrophages, oligodendrocyte progenitor cells, pericytes/fibroblasts, ependymal (Meletis et al. 2008), and endothelial cells. These cells work in tandem under a highly inflammatory environment following traumatic SCI to proliferate, activate, and cordon off further inflammatory propagation from the lesion epicenter (Tran et al. 2018b). The remarkable increase in density of the scar-forming reactive astroglia that (after many months) eventually fill the territory left after removal of cell debris (Silver and Miller 2004) as well as the remodeled extracellular matrix (in particular the basal lamina constituents, CSPGs, and collagens) strongly inhibit the regeneration of axons (Davies et al. 1997; Bradbury et al. 2002; Hara et al. 2017; Dias et al. 2018). However, in very special circumstances (after thin lesions that limit basal lamina production or lesion core inflammation or during the early formative stages of scar development), the astrocyte component of scar is not an absolute barrier and, if they bridge the lesion, can even allow for some regeneration when combinations of strongly intrinsic growth-promoting and/or extrinsic inhibitory factors are modulated (Rudge and Silver 1990; McKeon et al. 1995; Liu et al. 2010; Zukor et al. 2013; Anderson et al. 2016; Silver 2016).
Recent work further contextualizes this view characterizing key differences between the subacute (2 weeks) and chronic (8 weeks) post-SCI glial scar (Li et al. 2020a). Tandem mass tag-based quantitative proteomic analysis of the glial scar secretome was performed over time in a model of complete transected thoracic SCI in Sprague-Dawley rats. The subacute glial scar secreted up to three times the amount of growth factors such as bFGF, VEGF, and PDGF compared to the chronic scar. As previously shown (Andrews et al. 2012; Yi et al. 2012), the expression of CSPGs in the lesion penumbra changes with time, with at least four times the amount of detected CSPG-glycosaminoglycans (GAGs) forming in the chronic than subacute glial scar. Among other axon-inhibitory extracellular matrix proteins, Hara and colleagues found that collagens (Hara et al. 2017) were also upregulated in the chronic scar. Pharmacological blockade of the interaction between reactive astrocytes and type I collagen prevented glial scar formation, allowing for a more loosely arranged glial architecture which, in turn, aided axonal regrowth and functional recovery following SCI in the mouse (Hara et al. 2017). Importantly, surgical removal of the chronic scar (8 weeks post-injury) produced significant ascending sensory tract axons regenerating into the lesion site with the aid of BDNF as assessed by cholera toxin B tracing. These were the same neuronal subtypes identified, and techniques used in research as failing to regenerate following acute astrocyte ablation which releases the highly inhibitory early lesion core inflammatory population (Anderson et al. 2016; Silver 2016). Thus, while the subacute astroglial scar is important in wound healing and corralling inflammatory processes, it contributes to (but is not the only constituent of) the chronic impediment to axon regeneration. The glial scar should be viewed as a dynamic, time-dependent, and constantly remodeling structure. Reactive and scar astrocytes can, for a time, be highly plastic cells whose growth supportive or inhibitory functions are determined by local signaling events (DePaul et al. 2017). For example, grafting skin precursor-derived Schwann cells into a chronically contused spinal cord can alter the formation of the extremely dense and molecularly inhibitory, barrier-forming scar at the astroglial/Schwann cell interfaces through stimulating the astrocytes to decrease inhibitory ECM secretion, re-orient, and migrate well into the graft (Assinck et al. 2020). This dynamic action allowed for the regeneration of catecholaminergic axons into and beyond the lesion epicenter.
CSPGs impede regeneration/plasticity
A key inhibitory component of the glial scar are the CSPGs, secreted early on by reactive astrocytes and in the chronic phase by various cell types (Tran et al. 2018b). Other extracellular matrix proteins of the chronic glial scar (collagens, tenascins, semaphorins, and ephrins) additionally provide inhibition to regeneration following SCI (Tran et al. 2018b). CSPGs are extracellular matrix proteins composed of a protein core and a varying number of sugar moieties called glycosaminoglycan (GAG) chains (Fig. 4). They include three major subtypes: secreted lecticans, membrane-bound phosphacan (PTPRZ1), and NG2 (CSPG4).

Mechanisms of axon regeneration failure. Acutely after spinal cord injury, infiltrating macrophages come into physical contact with the tips of sheared axons causing the injured axon to dieback to the neuron soma. Chronically, chondroitin sulfate proteoglycans (CSPGs) of the glial scar cause growth cone dystrophy of approaching axons. CSPGs consist of a lectican group (brevican, neurocan, versican, and aggrecan) and phophocan and NG2 (not pictured here). The glycosaminoglycan (GAG) chains, notably sulfation patterns CS-A and CS-E, are especially inhibitory to axon regeneration. GAG chains of CSPGs bind to protein tyrosine phosphatase receptor sigma (PTPRS) promoting monomerization of the receptor at the axon growth cone to cause dystrophy and chronic regeneration failure
The lecticans are also found endogenously in specialized extracellular matrix structures called perineuronal nets (PNNs), which surround the soma and proximal dendrites of certain subpopulations of neurons including fast-spiking parvalbumin and GABAergic neurons (Fawcett et al. 2019). During development, PNN formation closes the critical period to ensure neural circuit stability (e.g., in the visual pathway) (Liu et al. 2013). In the spinal cord, the majority of PNNs are found in the ventral motor horn (Irvine and Kwok 2018). Circuit stability is conferred by CSPGs (Warren and Alilian 2018; Warren et al. 2018a; Carulli and Verhaagen 2021). Perhaps through dampening autophagic flux (Tran et al. 2020), CSPGs through protein tyrosine phosphatase receptor sigma (PTPRσ) binding promote initial adhesion and receptor recruitment necessary for synaptogenesis (Han et al. 2016; Bomkamp et al. 2019). It has recently been found that the PNN-PTPRσ complex exerts its inhibitory action on neuronal plasticity, at least in part, by restricting signaling of the neurotrophic receptor tyrosine kinase 2 (TRKB) (Lesnikova et al. 2021).
The LAR family receptors mediate the inhibitory actions of CSPGs
Leukocyte common antigen-related tyrosine phosphatase receptor (LAR) (Fisher et al. 2011) and PTPRσ (Shen et al. 2009) both confer inhibitory CSPG signaling (Fig. 4). The CS-E motif of CSPGs through PTPRσ binding dampens autophagic flux (Martin et al. 2011) through dephosphorylation of the actin cytoskeleton-binding protein cortactin, perturbing autophagosome and lysosomal fusion (Sakamoto et al. 2019). By imposing autophagic dysfunction, especially in the context of the post-SCI environment, CSPGs induce axon growth cone dystrophy and axon regeneration failure (Tom et al. 2004; Sakamoto et al. 2019; Tran et al. 2020). Notably, PTPRσ is a bifunctional receptor in that both axon-inhibitory CSPGs and axon-promoting heparan sulfate proteoglycans (HSPGs) bind (Coles et al. 2011, 2014). CSPG-PTPRσ binding promotes monomerization of the receptor to increase its dephosphorylation activity, while HSPG-PTPRσ binding promotes clustering of the receptor and a decrease in dephosphorylating activity (Wu et al. 2017). In the extracellular matrix milieu, the attention of the growth cone to the ratio of HSPGs to CSPGs as well as other extracellular proteins such as reelin (Zluhan et al. 2020) or laminin (McKeon et al. 1995) is, therefore, important in determining whether axon outgrowth proceeds. Following SCI, however, approaching axons are strongly inhibited by the preponderance of upregulated CSPGs found radiating from the glial scar (Davies et al. 1997). Importantly, the interaction of CSPGs with PTPRσ causes axonal growth cones to enter an entrapped dystrophic state, preventing regeneration after SCI not through repulsion but, rather, through an overly adhesive mechanism (Lang et al. 2015).
Perturbing CSPG inhibition promotes regeneration/sprouting and functional recovery
Directly cleaving GAG chains from CSPGs using chondroitinase ABC has proven effective in restoring plasticity, axon regeneration/sprouting, and functional recovery following SCI (Bradbury et al. 2002; Alilain et al. 2011; Warren et al. 2018b, Warren et al. 2021). Perturbing CSPG signaling through PTPRσ is an another effective strategy to enhance regeneration of axons and myelin. Pleiotrophin binding to glypican-2 forms a complex with CSPGs to abrogate CSPG binding to PTPRσ (Paveliev et al. 2016). Enoxaparin has recently been shown to promote functional recovery after SCI by antagonizing PTPRσ (Ito et al. 2021). Our own work in synthesizing a peptide, ISP (intracellular sigma peptide), modeled against the regulatory wedge domain of PTPRσ to “turn off” CSPG-PTPRσ signaling resulted in the rescue of the axon growth cone from becoming entrapped and dystrophic and remarkably enhanced bladder and coordinated locomotor recovery following systemic application in rat models of contusive SCI (Lang et al. 2015; Rink et al. 2018). Treatment of dorsal root ganglion neurons and oligodendrocyte progenitor cells (Luo et al. 2018; Tran et al. 2018a) with ISP-induced autologous localized protease release, which was capable of immediate degradation of CSPGs to enhance process outgrowth or maturation and cell survival, respectively. In dorsal root ganglion neurons, ISP-induced focal release of the lysosomal protease, cathepsin B, was effective in digesting CSPGs located around the advancing growth cone to encourage outgrowth past a CSPG gradient (Tran et al. 2018a). In oligodendrocyte progenitor cells, the ISP-induced protease was MMP-2, which also relieved CSPG-inhibition through degradation, encouraged digestion of CSPGs in demyelinated lesions, and promoted remyelination through restoration of oligodendrocyte migration, maturation, and homeostasis (Luo et al. 2018; Tran et al. 2018a). In both cell types, cell migration was increased. This may in part be through due to disruption of CSPG-PTPRσ signaling which dampens autophagy (Sakamoto et al. 2019; Tran et al. 2020).
During development, increases in cytoskeletal stability are typically associated with differentiation of an axonal rather than dendritic phenotype (Witte et al. 2008). It has been shown that further increasing microtubule stability in the adult can promote axonal regeneration after traumatic SCI (Hellal et al. 2011; Ruschel et al. 2015; Ruschel and Bradke 2018). Interestingly, administration of tubulin stabilizing pharmaceutical therapies (taxol and epothilone B and D) also acts to rearrange the cytoskeleton of surrounding astrocytes at the site of injury, reducing CSPG secretion and thus facilitating axonal growth (Hellal et al. 2011). It is now important to determine how and where in the dynamic microtubules of the cytoskeleton this stabilizing effect occurs to promote this duel neuronal and glial effect facilitating long-range transport (Ertürk et al. 2007) and alteration of the extracellular matrix.
CSPGs impact on the immune system
CSPGs contribute to proinflammatory signaling in immune cells through the CD44 receptor, which results in enhanced TNFα secretion by microglia/macrophages (Rolls et al. 2008). This further prolongs the post-SCI inflammatory environment and secondary damage to tissue. Depletion of GAGs with chondroitinase ABC is able to encourage microglia/macrophages toward an alternative inflammatory phenotype, which conferred neuroprotection and improved recovery after acute SCI (Bartus et al. 2014; Didangelos et al. 2016). So, too, in a mouse model of inflammatory multiple sclerosis, experimental autoimmune encephalomyelitis (EAE), CSPGs exacerbate cell-damaging inflammatory processes by stimulating macrophages to produce proinflammatory cytokines (Stephenson et al. 2018). Analysis of a multiple sclerosis genome-wide associated screen led to the discovery of the glycosyltransferase exotosin-like 2 gene, which normally limits the cellular production of CSPGs. Knockout of this gene resulted in an increase of CSPG deposition within lysolecithin-induced focal demyelinated lesions (Pu et al. 2020). This resulted in the increased recruitment and activation of microglia/macrophages to demyelinated lesions and further axon loss. Importantly, reduction of CSPGs in mouse models of demyelination through inhibition of its synthesis (Keough et al. 2016) or by enhancing oligodendrocyte maturation and autologous production of CSPG-degrading proteases via the use of ISP (Luo et al. 2018) encouraged regeneration of myelin and functional recovery.
Combinatorial strategies to promote functional recovery after SCI
Emphasis on the multitude of cells involved in the formation of the glial/fibrotic scar highlights the many hurdles required for post-SCI regeneration and ultimately functional recovery. These obstacles, for the most part, can be encompassed by two problems: an intrinsic one whereby the adult neuron lacks a robust “motor” for axon process outgrowth compared to their immature counterparts (Zukor et al. 2013; Andrews et al. 2016; Cheah et al. 2016; Fawcett 2017), and an extrinsic one where the adult spinal cord suffers from a dearth of substrate adhesion molecules or their receptors while the remodeled chronic SCI environment is replete with inhibitory extracellular matrix proteins. Moving forward, combinatorial strategies that address these major obstacles will be necessary to best encourage functional recovery. In a striking example of the challenges present in restoring functional recovery after injury, Bei et al. illustrated how promoting axon regeneration through boosting retinal ganglion neurons’ intrinsic growth motor using a PTEN and SOCS3 co-deletion is itself insufficient after optic tract transection as conductance of de novo; unmyelinated axons were lacking (Bei et al. 2016). However, PTEN and SOCS3 co-deletions in combination with myelin-promoting growth factors OPN, IGF1, and CNTF improved optomotor function. Still, other studies have sought to recapitulate developmental programs enabling axon growth (Filbin 2006; Lee et al. 2013; Hilton and Bradke 2017; Courtine and Sofroniew 2019).
Conclusions
In the past decades, our understanding of the glial scar has grown to encompass much more than reactive astrocytes around the lesion penumbra. We currently appreciate that the glial/fibrotic scar comprises a myriad of cells including (but not limited to) macrophages, microglia, oligodendrocyte progenitor cells, pericytes/fibroblasts, ependymal cells, and endothelial cells, with neuron growth cones entrapped in the lesion penumbra. Here, we have discussed how next-generation sequencing technologies have revealed how complicated this post-injury structure is at the cellular, molecular, and transcriptomic levels with an emphasis on the pre- and post-astrocytic, microglial, and neuronal cellular heterogeneity. Acutely, this structure is indispensable to the wound healing process to limit further secondary tissue damage and to resolve rampant inflammatory processes. Chronically, however, the typical glial/fibrotic scar that forms after contusive or extensive surgical lesions with a balance of extracellular matrix proteins tipped in the direction of inhibition, including CSPGs, poses a hugely inhibitory biochemical and physical barrier to successful axon regeneration and functional recovery. Our understanding of the uniquely heterogeneous composition of the scar and its evolution over time advocates for a combinatorial strategy to encourage axon regeneration and functional recovery. Treatment strategies that simultaneously target both the intrinsic problem of decreased axon outgrowth machinery as well as the extrinsic hurdles of increased axon-inhibitory proteins coupled with targeted rehabilitation are our best options going forward.
References
Alilain W (2009) Shedding light on restoring respiratory function after spinal cord injury. Front Mol Neurosci 2:1–6
Alilain WJ, Horn KP, Hu H, Dick TE, Silver J (2011) Functional regeneration of respiratory pathways after spinal cord injury. Nat 475:196–200
Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532:195–200
Andrews EM, Richards RJ, Yin FQ, Viapiano MS, Jakeman LB (2012) Alterations in chondroitin sulfate proteoglycan expression occur both at and far from the site of spinal contusion injury. Exp Neurol 235:174–187
Andrews MR, Soleman S, Cheah M, Tumbarello DA, Mason MR, Moloney E, Verhaagen J, Bensadoun JC, Schneider B, Aebischer P, Fawcett JW (2016) Axonal localization of integrins in the CNS is neuronal type and age dependent. eNeuro 3
Assinck P, Sparling JS, Dworski S, Duncan GJ, Wu DL, Liu J, Kwon BK, Biernaskie J, Miller FD, Tetzlaff W (2020) Transplantation of skin precursor-derived Schwann cells yields better locomotor outcomes and reduces bladder pathology in rats with chronic spinal cord injury. Stem Cell Rep 15:140–155
Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW (1994) Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci 14:846–856
Bandtlow CE (2003) Regeneration in the central nervous system. Exp Gerontol 38:79–86
Bartus K, James ND, Didangelos A, Bosch KD, Verhaagen J, Yáñez-Muñoz RJ, Rogers JH, Schneider BL, Muir EM, Bradbury EJ (2014) Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J Neurosci 34:4822–4836
Bates CA, Stelzner DJ (1993) Extension and regeneration of corticospinal axons after early spinal injury and the maintenance of corticospinal topography. Exp Neurol 123:106–117
Becker T, Becker CG (2014) Axonal regeneration in zebrafish. Curr Opin Neurobiol 27:186–191
Bei F, Lee HHC, Liu X, Gunner G, Jin H, Ma L, Wang C, Hou L, Hensch TK, Frank E, Sanes JR, Chen C, Fagiolini M, He Z (2016) Restoration of visual function by enhancing conduction in regenerated axons. Cell 164:219–232
Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle ME, Vernoux N, Tremblay M, Fuehrmann T, Shoichet MS, Lacroix S (2019) Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun 10:518
Bertolotti E, Malagoli D, Franchini A (2013) Skin wound healing in different aged Xenopus laevis. J Morphol 274:956–964
Bomkamp C, Padmanabhan N, Karimi B, Ge Y, Chao JT, Loewen CJR, Siddiqui TJ, Craig AM (2019) Mechanisms of PTPσ-mediated presynaptic differentiation. Front Synaptic Neurosci 11:17
Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, Mcmahon SB (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nat 416:636–640
Brooks JM, Su J, Levy C, Wang JS, Seabrook TA, Guido W, Fox MA (2013) A molecular mechanism regulating the timing of corticogeniculate innervation. Cell Rep 5:573–581
Bundesen LQ, Scheel TA, Bregman BS, Kromer LF (2003) Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci 23:7789–7800
Busch SA, Hamilton JA, Horn KP, Cuascut FX, Cutrone R, Lehman N, Deans RJ, Ting AE, Mays RW, Silver J (2011) Multipotent adult progenitor cells prevent macrophage-mediated axonal dieback and promote regrowth after spinal cord injury. J Neurosci 31:944–953
Busch SA, Horn KP, Silver DJ, Silver J (2009) Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci 29:9967–9976
Canning DR, Mckeon RJ, Dewitt DA, Perry G, Wujek JR, Frederickson RC, Silver J (1993) beta-Amyloid of Alzheimer’s disease induces reactive gliosis that inhibits axonal outgrowth. Exp Neurol 124:289–298
Carulli D, Verhaagen J (2021) An extracellular perspective on CNS maturation: perineuronal nets and the control of plasticity. Int J Mol Sci 22(5):2434
Celio MR, Spreafico R, De Biasi S, Vitellaro-Zuccarello L (1998) Perineuronal nets: past and present. Trends Neurosci 21(12):510–515
Cheah M, Andrews MR, Chew DJ, Moloney EB, Verhaagen J, Fässler R, Fawcett JW (2016) Expression of an activated integrin promotes long-distance sensory axon regeneration in the spinal cord. J Neurosci 36:7283–7297
Chen K, Deng S, Lu H, Zheng Y, Yang G, Kim D, Cao Q, Wu JQ (2013) RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS ONE 8(8):e72567
Cigliola V, Becker CJ, Poss KD (2020) Building bridges, not walls: spinal cord regeneration in zebrafish. Dis Model Mech 13(5)
Coles CH, Mitakidis N, Zhang P, Elegheert J, Lu W, Stoker AW, Nakagawa T, Craig AM, Jones EY, Aricescu AR (2014) Structural basis for extracellular cis and trans RPTPsigma signal competition in synaptogenesis. Nat Commun 5:5209
Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR (2011) Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Sci 332(6028):484–488
Courtine G, Sofroniew MV (2019) Spinal cord repair: advances in biology and technology. Nat Med 25(6):898–908
Crapser JD, Spangenberg EE, Barahona RA, Arreola MA, Hohsfield LA, Green KN (2020) Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58:102919
Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, Grutzendler J (2020) Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv 6(26):eaba3239
David S, Aguayo AJ (1981) Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Sci 214:931–933
Davies JE, Huang C, Proschel C, Noble M, Mayer-Proschel M, Davies SJ (2006) Astrocytes derived from glial-restricted precursors promote spinal cord repair. J Biol 5:7
Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J (1997) Regeneration of adult axons in white matter tracts of the central nervous system. Nat 390:680–683
Davies SJ, Goucher DR, Doller C, Silver J (1999) Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 19:5810–5822
Davies SJ, Shih CH, Noble M, Mayer-Proschel M, Davies JE, Proschel C (2011) Transplantation of specific human astrocytes promotes functional recovery after spinal cord injury. PLoS ONE 6(3):e17328
DePaul MA, Lin CY, Silver J, Lee YS (2015) Peripheral nerve transplantation combined with acidic fibroblast growth factor and chondroitinase induces regeneration and improves urinary function in complete spinal cord transected adult mice. PLoS ONE 10(10):e0139335
DePaul MA, Lin CY, Silver J, Lee YS (2017) Combinatory repair strategy to promote axon regeneration and functional recovery after chronic spinal cord injury. Sci Rep 7:9018
Dias DO, Kim H, Holl D, Solnestam BW, Lundeberg J, Carlen M, Goritz C, Frisen J (2018) Reducing pericyte-derived scarring promotes recovery after spinal cord injury. Cell 173:153–165
Didangelos A, Puglia M, Iberl M, Sanchez-Bellot C, Roschitzki B, Bradbury EJ (2016) High-throughput proteomics reveal alarmins as amplifiers of tissue pathology and inflammation after spinal cord injury. Sci Rep 6:21607
Domowicz MS, Henry JG, Wadlington N, Navarro A, Kraig RP, Schwartz NB (2011) Astrocyte precursor response to embryonic brain injury. Brain Res 1389:35–49
Edwards-Faret G, González-Pinto K, Cebrián-Silla A, Peñailillo J, García-Verdugo JM, Larraín J (2021) Cellular response to spinal cord injury in regenerative and non-regenerative stages in Xenopus laevis. Neural Dev 16:2. https://doi.org/10.1186/s13064-021-00152-2
Ertürk A, Hellal F, Enes J, Bradke F (2007) Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci 27(34):9169–9180
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, Macvicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A (2021) Reactive astrocyte nomenclature definitions and future directions. Nat Neurosci 24(3):312–325
Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, Huang AY, Silver J (2014) High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol 254:109–120
Fawcett JW (2017) An integrin approach to axon regeneration. Eye(Lond) 31(2):206–208
Fawcett JW, Oohashi T, Pizzorusso T (2019) The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat Rev Neurosci 20(8):451–465
Fenrich KK, Rose PK (2009) Spinal interneuron axons spontaneously regenerate after spinal cord injury in the adult feline. J Neurosci 29(39):12145–12158
Ferretti P, Zhang F, O’Neill P (2003) Changes in spinal cord regenerative ability through phylogenesis and development: lessons to be learnt. Dev Dyn 226(2):245–256
Filbin MT (2006) Recapitulate development to promote axonal regeneration: good or bad approach? Philos Trans R Soc Lond B Biol Sci 361(1473):1565–1574
Filous AR, Miller JH, Coulson-Thomas YM, Horn KP, Alilain WJ, Silver J (2010) Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev Neurobiol 70(12):826–841
Filous AR, Tran A, Howell CJ, Busch SA, Evans TA, Stallcup WB, Kang SH, Bergles DE, Lee SI, Levine JM, Silver J (2014) Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J Neurosci 34(49):16369–16384
Fisher D, Xing B, Dill J, Li H, Hoang HH, Zhao Z, Yang XL, Bachoo R, Cannon S, Longo FM, Sheng M, Silver J, Li S (2011) Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J Neurosci 31(40):14051–14066
Fitch MT, Doller C, Combs CK, Landreth GE, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19(19):8182–8198
Fitch MT, Silver J (2008) CNS injury glial scars and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol 209(2):294–301
Fu H, Zhao Y, Hu D, Wang S, Yu T, Zhang L (2020) Depletion of microglia exacerbates injury and impairs function recovery after spinal cord injury in mice. Cell Death Dis 11(7):528
Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J (2015) The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85(4):703–709
Galtrey CM, Kwok JCF, Carulli D, Rhodes KE, Fawcett JW (2008) Distribution and synthesis of extracellular matrix proteoglycans, hyaluronan, link proteins and tenascin-R in the rat spinal cord. Eur J Neurosci 27(6):1373–1390
Gearhart J, Oster-Granite ML, Guth L (1979) Histological changes after transection of the spinal cord of fetal and neonatal mice. Exp Neurol 66(1):1–15
Geoffroy CG, Meves JM, Zheng B (2017) The age factor in axonal repair after spinal cord injury: a focus on neuron-intrinsic mechanisms. Neurosci Lett 652:41–49
Grimpe B, Pressman Y, Bunge MB, Silver J (2005) The role of proteoglycans in Schwann cell/astrocyte interactions and in regeneration failure at PNS/CNS interfaces. Mol Cell Neurosci 28(1):18–29
Guimarães-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, Dalton ND, Rockenstein E, Masliah E, Peterson KL, Stallcup WB, Chen J, Evans SM (2017) Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 20(2):345–359
Haas C, Fischer I (2014) Transplanting neural progenitors to build a neuronal relay across the injured spinal cord. Neural Regen Res 9(12):1173–1176
Haas C, Neuhuber B, Yamagami T, Rao M, Fischer I (2012) Phenotypic analysis of astrocytes derived from glial restricted precursors and their impact on axon regeneration. Exp Neurol 233(2):717–732
Han KA, Woo D, Kim S, Choii G, Jeon S, Won SY, Kim HM, Heo WD, Um JW, Ko J (2016) Neurotrophin-3 regulates synapse development by modulating TrkC-PTPσ synaptic adhesion and intracellular signaling pathways. J Neurosci 36(17):4816–4831
Hara M, Kobayakawa K, Ohkawa Y, Kumamaru H, Yokota K, Saito T, Kijima K, Yoshizaki S, Harimaya K, Nakashima Y, Okada S (2017) Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spinal cord injury. Nat Med 23(7):818–828
Hawthorne AL, Hu H, Kundu B, Steinmetz MP, Wylie CJ, Deneris ES, Silver J (2011) The unusual response of serotonergic neurons after CNS injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. J Neurosci 31(15):5605–5616
Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Sci 331(6019):928–931
Hilton BJ, Bradke F (2017) Can injured adult CNS axons regenerate by recapitulating development? Dev 144(19):3417–3429
Horn KP, Busch SA, Hawthorne AL, Van Rooijen N, Silver J (2008) Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci 28(38):9330–9341
Huang X, Wang C, Zhou X, Wang J, Xia K, Yang B, Gong Z, Ying L, Yu C, Shi K, Shu J, Cheng F, Han B, Liang C, Li F, Chen Q (2020) Overexpression of the transcription factors OCT4 and KLF4 improves motor function after spinal cord injury. CNS Neurosci Ther 26(9):940–951
Irvine SF, Kwok JCF (2018) perineuronal nets in spinal motoneurones: chondroitin sulphate proteoglycan around alpha motoneurones. Int J Mol Sci 19(4):1172
Ito S, Ozaki T, Morozumi M, Imagama S, Kadomatsu K, Sakamoto K (2021) Enoxaparin promotes functional recovery after spinal cord injury by antagonizing PTPRσ. Exp Neurol 340:113679
Iwashita Y, Kawaguchi S, Murata M (1994) Restoration of function by replacement of spinal cord segments in the rat. Nat 367(6459):167–170
Janda E, Boi L, Carta AR (2018) Microglial phagocytosis and its regulation: a therapeutic target in Parkinson’s disease? Front Mol Neurosci 11:144
Jin WN, Shi K, He W, Sun JH, Van Kaer L, Shi FD, Liu Q (2021) Neuroblast senescence in the aged brain augments natural killer cell cytotoxicity leading to impaired neurogenesis and cognition. Nat Neurosci 24(1):61–73
Jin Y, Dougherty SE, Wood K, Sun L, Cudmore RH, Abdalla A, Kannan G, Pletnikov M, Hashemi P, Linden DJ (2016) Regrowth of serotonin axons in the adult mouse brain following injury. Neuron 91(4):748–762
Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896
Joosten EA, Veldhuis WB, Hamers FP (2004) Collagen containing neonatal astrocytes stimulates regrowth of injured fibers and promotes modest locomotor recovery after spinal cord injury. J Neurosci Res 77(1):127–142
Kalamakis G, Brüne D, Ravichandran S, Bolz J, Fan W, Ziebell F, Stiehl T, Catalá-Martinez F, Kupke J, Zhao S, Llorens-Bobadilla E, Bauer K, Limpert S, Berger B, Christen U, Schmezer P, Mallm JP, Berninger B, Anders S, Del Sol A, Marciniak-Czochra A, Martin-Villalba A (2019) Quiescence modulates stem cell maintenance and regenerative capacity in the aging brain. Cell 176(6):1407–1419
Keough MB, Rogers JA, Zhang P, Jensen SK, Stephenson EL, Chen T, Hurlbert MG, Lau LW, Rawji KS, Plemel JR, Koch M, Ling CC, Yong VW (2016) An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat Commun 7:11312
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29(43):13435–13444
Klatt Shaw D, Saraswathy VM, Zhou L, Mcadow AR, Burris B, Butka E, Morris SA, Dietmann S, Mokalled MH (2021) Localized EMT reprograms glial progenitors to promote spinal cord repair. Dev Cell 56(4):613–626
Krystosek A, Seeds NW (1984) Peripheral neurons and Schwann cells secrete plasminogen activator. J Cell Biol 98(1):773–776
Kunkel-Bagden E, Dai HN, Bregman BS (1992) Recovery of function after spinal cord hemisection in newborn and adult rats: differential effects on reflex and locomotor function. Exp Neurol 116(1):40–51
Kurshan PT, Oztan A, Schwarz TL (2009) Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat Neurosci 12(11):1415–1423
Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D, Iltzsche A, Brand M (2012) Acute inflammation initiates the regenerative response in the adult zebrafish brain. Sci 338(6112):1353–1356
Laabs T, Carulli D, Geller HM, Fawcett JW (2005) Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol 15(1):116–120
Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J (2015) Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nat 518(7539):404–408
Lee YS, Lin CY, Jiang HH, DePaul M, Lin VW, Silver J (2013) Nerve regeneration restores supraspinal control of bladder function after complete spinal cord injury. J Neurosci 33(26):10591–10606
Lesnikova A, Casarotto P, Biojone C, Castrén E (2021) Perineuronal net receptor PTPσ regulates retention of memories. bioRxiv 2021.02.26.433000
Li X, Li M, Tian L, Chen J, Liu R, Ning B (2020a) Reactive astrogliosis: implications in spinal cord injury progression and therapy. Oxid Med Cell Longev 2020:9494352
Li Y, Carlstedt T, Berthold CH, Raisman G (2004) Interaction of transplanted olfactory-ensheathing cells and host astrocytic processes provides a bridge for axons to regenerate across the dorsal root entry zone. Exp Neurol 188(2):300–308
Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, Yang Z, Chen B, Shi Z, Meng H, Zhou S, Zhu J, Jacobi A, Swarup V, Popovich PG, Geschwind DH, He Z (2020b) Microglia-organized scar-free spinal cord repair in neonatal mice. Nature 587:613–618
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nat 541(7638):481–487
Liu H, Xu H, Yu T, Yao J, Zhao C, Yin ZQ (2013) Expression of perineuronal nets, parvalbumin and protein tyrosine phosphatase σ in the rat visual cortex during development and after BFD. Curr Eye Res 38(10):1083–1094
Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z (2010) PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13:1075–1081
Lu P, Wang Y, Graham L, Mchale K, Gao M, Wu D, Brock J, Blesch A, Rosenzweig ES, Havton LA, Zheng B, Conner JM, Marsala M, Tuszynski MH (2012) Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell 150(6):1264–1273
Lu P, Woodruff G, Wang Y, Graham L, Hunt M, Wu D, Boehle E, Ahmad R, Poplawski G, Brock J, Goldstein LS, Tuszynski MH (2014) Long-distance axonal growth from human induced pluripotent stem cells after spinal cord injury. Neuron 83(4):789–796
Luo F, Tran AP, Xin L, Sanapala C, Lang BT, Silver J, Yang Y (2018) Modulation of proteoglycan receptor PTPσ enhances MMP-2 activity to promote recovery from multiple sclerosis. Nat Commun 9:4126
Martin KR, Xu Y, Looyenga BD, Davis RJ, Wu CL, Tremblay ML, Xu HE, Mackeigan JP (2011) Identification of PTPsigma as an autophagic phosphatase. J Cell Sci 124(5):812–819
Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13
McKeon RJ, Höke A, Silver J (1995) Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol 136(1):32–43
McKeon RJ, Schreiber RC, Rudge JS, Silver J (1991) Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci 11(11):3398–3411
Meletis K, Barnabé-Heider F, Carlén M, Evergren E, Tomilin N, Shupliakov O, Frisén J (2008) Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol 6(7):e182
Mesquida-Veny F, Del Río JA, Hervera A (2020) Macrophagic and microglial complexity after neuronal injury. Prog Neurobiol 101970
Milich LM, Choi J, Ryan C, Yahn SL, Tsoulfas P, Lee JK (2020) Single cell analysis of the cellular heterogeneity and interactions in the injured mouse spinal cord. bioRxiv
Mladinic M, Wintzer M (2002) Changes in mRNA content of developing opossum spinal cord at stages when regeneration can and cannot occur after injury. Brain Res Rev 40(1–3):317–324
Mokalled MH, Patra C, Dickson AL, Endo T, Stainier DY, Poss KD (2016) Injury-induced ctgfa directs glial bridging and spinal cord regeneration in zebrafish. Sci 354(6312):630–634
Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z (2002) Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 22(17):7526–7535
Paramos-de-Carvalho D, Martins I, Cristóvão AM, Dias AF, Neves-Silva D, Pereira T, Chapela D, Farinho A, Jacinto A, Saúde L (2020) Depletion of senescent cells improves functional recovery after spinal cord injury. bioRxiv
Paveliev M, Fenrich KK, Kislin M, Kuja-Panula J, Kulesskiy E, Varjosalo M, Kajander T, Mugantseva E, Ahonen-Bishopp A, Khiroug L, Kulesskaya N, Rougon G, Rauvala H (2016) HB-GAM (pleiotrophin) reverses inhibition of neural regeneration by the CNS extracellular matrix. Sci Rep 6:33916
Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB (2004) cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 10(6):610–616
Poplawski GHD, Kawaguchi R, Van Niekerk E, Lu P, Mehta N, Canete P, Lie R, Dragatsis I, Meves JM, Zheng B, Coppola G, Tuszynski MH (2020) Injured adult neurons regress to an embryonic transcriptional growth state. Nat 581(7806):77–82
Popovich PG (2014) Neuroimmunology of traumatic spinal cord injury: a brief history and overview. Exp Neurol 258:1–4
Popovich PG, Wei P, Stokes BT (1997) Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 377(3):443–464
Pu A, Mishra MK, Dong Y, Ghorbanigazar S, Stephenson EL, Rawji KS, Silva C, Kitagawa H, Sawcer S, Yong VW (2020) The glycosyltransferase EXTL2 promotes proteoglycan deposition and injurious neuroinflammation following demyelination. J Neuroinflammation 17:220
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19(8):987–991
Revuelta M, Elicegui A, Moreno-Cugnon L, Bührer C, Matheu A, Schmitz T (2019) Ischemic stroke in neonatal and adult astrocytes. Mech Ageing Dev 183:111147
Riew TR, Jin X, Kim S, Kim HL, Lee MY (2021) Temporal dynamics of cells expressing NG2 and platelet-derived growth factor receptor-β in the fibrotic scar formation after 3-nitropropionic acid-induced acute brain injury. Cell Tissue Res 1–17
Rink S, Arnold D, Wöhler A, Bendella H, Meyer C, Manthou M, Papamitsou T, Sarikcioglu L, Angelov DN (2018) Recovery after spinal cord injury by modulation of the proteoglycan receptor PTPσ. Exp Neurol 309:148–159
Rolls A, Shechter R, London A, Segev Y, Jacob-Hirsch J, Amariglio N, Rechavi G, Schwartz M (2008) Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med 5(8):e171
Rooney GE, Endo T, Ameenuddin S, Chen B, Vaishya S, Gross L, Schiefer TK, Currier BL, Spinner RJ, Yaszemski MJ, Windebank AJ (2009) Importance of the vasculature in cyst formation after spinal cord injury. J Neurosurg Spine 11(4):432–437
Rudge JS, Silver J (1990) Inhibition of neurite outgrowth on astroglial scars in vitro. J Neurosci 10(11):3594–3603
Rui YN, Xu Z, Patel B, Cuervo AM, Zhang S (2015) HTT/Huntingtin in selective autophagy and Huntington disease: a foe or a friend within? Autophagy 11(5):858–860
Ruschel J, Bradke F (2018) Systemic administration of epothilone D improves functional recovery of walking after rat spinal cord contusion injury. Exp Neurol 306:243–249
Ruschel J, Hellal F, Flynn KC, Dupraz S, Elliott DA, Tedeschi A, Bates M, Sliwinski C, Brook G, Dobrindt K, Peitz M, Brustle O, Norenberg MD, Blesch A, Weidner N, Bunge MB, Bixby JL, Bradke F (2015) Axonal regeneration: systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science 348:347–352
Russo I, Barlati S, Bosetti F (2011) Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem 116(6):947–956
Sakamoto K, Ozaki T, Ko YC, Tsai CF, Gong Y, Morozumi M, Ishikawa Y, Uchimura K, Nadanaka S, Kitagawa H, Zulueta MML, Bandaru A, Tamura JI, Hung SC, Kadomatsu K (2019) Glycan sulfation patterns define autophagy flux at axon tip via PTPRσ-cortactin axis. Nat Chem Biol 15(7):699–709
Sas AR, Carbajal KS, Jerome AD, Menon R, Yoon C, Kalinski AL, Giger RJ, Segal BM (2020) A new neutrophil subset promotes CNS neuron survival and axon regeneration. Nat Immunol 21(12):1496–1505
Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ (2018) Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 22(8):2216–2225
Schreyer DJ, Jones EG (1983) Growing corticospinal axons by-pass lesions of neonatal rat spinal cord. Neurosci 9(1):31–40
See J, Bonner J, Neuhuber B, Fischer I (2010) Neurite outgrowth of neural progenitors in presence of inhibitory proteoglycans. J Neurotrauma 27(5):951–957
Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG (2009) PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Sci 326(5952):592–596
Silver J (2016) The glial scar is more than just astrocytes. Exp Neurol 286(2016):147–149
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5(2):146–156
Smith GM, Miller RH, Silver J (1987) Astrocyte transplantation induces callosal regeneration in postnatal acallosal mice. Ann N Y Acad Sci 495(1):185–205
Smith GM, Silver J (1988) Transplantation of immature and mature astrocytes and their effect on scar formation in the lesioned central nervous system. Prog Brain Res 78:353–361
Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, Lee JK (2013) Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci 33(34):13882–13887
Son YJ (2015) Synapsing with NG2 cells (polydendrocytes) unappreciated barrier to axon regeneration? Neural Regen Res 10(3):346–348
Steeves JD, Tetzlaff W (1998) Engines accelerators and brakes on functional spinal cord repair. Ann N Y Acad Sci 860(1):412–424
Stephenson EL, Mishra MK, Moussienko D, Laflamme N, Rivest S, Ling CC, Yong VW (2018) Chondroitin sulfate proteoglycans as novel drivers of leucocyte infiltration in multiple sclerosis. Brain 141(4):1094–1110
Takesian AE, Hensch TK (2013) Balancing plasticity/stability across brain development. Prog Brain Res 207:3–34
Tedeschi A, Dupraz S, Laskowski CJ, Xue J, Ulas T, Beyer M, Schultze JL, Bradke F (2016) The calcium channel subunit alpha2delta2 suppresses axon regeneration in the adult CNS. Neuron 92(2):419–434
Tom VJ, Doller CM, Malouf AT, Silver J (2004) Astrocyte-associated fibronectin is critical for axonal regeneration in adult white matter. J Neurosci 24(42):9282–9290
Tran AP, Silver J (2021) Cathepsins in neuronal plasticity. Neural Regen Res 16(1):26–35
Tran AP, Sundar S, Yu M, Lang BT, Silver J (2018a) Modulation of receptor protein tyrosine phosphatase sigma increases chondroitin sulfate proteoglycan degradation through cathepsin B secretion to enhance axon outgrowth. J Neurosci 38(23):5399–5414
Tran AP, Warren PM, Silver J (2018b) The biology of regeneration failure and success after spinal cord injury. Physiol Rev 98(2):881–917
Tran AP, Warren PM, Silver J (2020) Regulation of autophagy by inhibitory CSPG interactions with receptor PTPsigma and its impact on plasticity and regeneration after spinal cord injury. Exp Neurol 328:113276
Tsata V, Kroehne V, Wehner D, Rost F, Lange C, Hoppe C, Kurth T, Reinhardt S, Petzold A, Dahl A, Loeffler M, Reimer MM, Brand M (2020) Reactive oligodendrocyte progenitor cells (re-)myelinate the regenerating zebrafish spinal cord. Dev 147(24)
Tsujioka H, Yamashita T (2019) Comparison of gene expression profile of the spinal cord of sprouting-capable neonatal and sprouting-incapable adult mice. BMC Genomics 20:619
Wagenaar N, Martinez-Biarge M, Van Der Aa NE, Van Haastert IC, Groenendaal F, Benders M, Cowan FM, De Vries LS (2018) Neurodevelopment After Perinatal Arterial Ischemic Stroke. Pediatrics 142(3)
Warren PM, Alilian W (2018) Plasticity induced recovery of breathing occurs at chronic stages after cervical contusion. J Neurotrauma 36(12):1985–1999
Warren PM, Dickens SM, Gigout S, Fawcett JW, Kwok JCF (2018a) Regulation of CNS plasticity through the extracellular matrix. Oxford University Press
Warren PM, Kissane RW, Egginton S, Kwok JCF, Askew GN (2021) Oxygen transport kinetics underpin rapid and robust diaphragm recovery following chronic spinal cord injury. J Physiol 599(4):1199–1224
Warren PM, Steiger SC, Dick TE, Macfarlane PM, Alilain WJ, Silver J (2018b) Rapid and robust restoration of breathing long after spinal cord injury. Nat Commun 9:4843
Welniarz Q, Dusart I, Roze E (2017) The corticospinal tract: evolution development and human disorders. Dev Neurobiol 77(7):810–829
Wictorin K, Björklund A (1992) Axon outgrowth from grafts of human embryonic spinal cord in the lesioned adult rat spinal cord. NeuroReport 3(12):1045–1048
Witte H, Neukirchen D, Bradke F (2008) Microtubule stabilization specifies initial neuronal polarization. J Cell Biol 180(3):619–632
Wu CL, Hardy S, Aubry I, Landry M, Haggarty A, Saragovi HU, Tremblay ML (2017) Identification of function-regulating antibodies targeting the receptor protein tyrosine phosphatase sigma ectodomain. PLoS ONE 12(5):e0178489
Xu K, Malouf AT, Messing A, Silver J (1999) Glial fibrillary acidic protein is necessary for mature astrocytes to react to beta-amyloid. Glia 25(4):390–403
Yang T, Dai Y, Chen G, Cui S (2020) Dissecting the dual role of the glial scar and scar-forming astrocytes in spinal cord injury. Front Cell Neurosci 14:78
Yi JH, Katagiri Y, Susarla B, Figge D, Symes AJ, Geller HM (2012) Alterations in sulfated chondroitin glycosaminoglycans following controlled cortical impact injury in mice. J Comp Neurol 520(15):3295–3313
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32(18):6391–6410
Zattara EE, Fernández-Álvarez FA, Hiebert TC, Bely AE, Norenburg JL (2019) A phylum-wide survey reveals multiple independent gains of head regeneration in Nemertea. Proc Biol Sci 286(1898):20182524
Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, Van Der Zwan J, Häring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S (2018) Molecular architecture of the mouse nervous system. Cell 174(4):999–1014
Zhou X, Wahane S, Friedl MS, Kluge M, Friedel CC, Avrampou K, Zachariou V, Guo L, Zhang B, He X, Friedel RH, Zou H (2020) Microglia and macrophages promote corralling, wound compaction and recovery after spinal cord injury via Plexin-B2. Nat Neurosci 23(3):337–350
Zluhan E, Enck J, Matthews RT, Olson EC (2020) Reelin counteracts chondroitin sulfate proteoglycan-mediated cortical dendrite growth inhibition. eNeuro 7(4)
Zukor K, Belin S, Wang C, Keelan N, Wang X, He Z (2013) Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci 33(39):15350–15361
Funding
JS was funded by NINDS (NS25713), the Ohio Department of Higher Education-Third Frontier Program, the Brumagin-Nelson Fund, Kaneko Family Fund, and the Hong Kong Spinal Cord Injury Fund. APT is funded by the Brotman Baty Institute. PMW is funded by a King’s Prize Fellowship and a research grant from the Medical Research Council (UKRI MR/S011110/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
A patent (9,937,242) filed by CWRU has been granted for ISP and licensed to NervGen Pharma Corp. The remaining authors declare no competing conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tran, A.P., Warren, P.M. & Silver, J. New insights into glial scar formation after spinal cord injury. Cell Tissue Res 387, 319–336 (2022). https://doi.org/10.1007/s00441-021-03477-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-021-03477-w