
Abstract
The role of brain-derived neurotrophic factor (BDNF) and its receptor TrkB has been studied in the context of mood disorders and their treatments for a couple of decades. Pharmacologically diverse antidepressant drugs increase the synthesis of BDNF in the cortex (and some subcortical structures) and this effect accounts for their ability to facilitate neurotrophic processes eventually leading into heightened plasticity within the cortex. Induction of BDNF-TrkB signaling has also been associated with the mechanism of action of ketamine and more recently with some other anesthetics, even with ones not thought to possess antidepressant potential. Notably, both ketamine and conventional antidepressants activate TrkB receptor and its downstream signaling rapidly within the same time scale in the brain while electroconvulsive therapy (ECT), among the most potent inducers of BDNF, has not been unequivocally shown to produce such acute effects on TrkB. The ability of antidepressants to regulate TrkB signaling is developmentally regulated and requires an intact central nervous system. The purpose of this review is to highlight and discuss some of these peculiarities associated with the effects of ketamine and classical antidepressants and BDNF on TrkB signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The therapeutic effects of chemically induced seizures against neuropsychiatric disorders, most notably schizophrenia and severe depression, provided a rational basis for the development of electroconvulsive therapy (ECT) (Payne and Prudic 2009). Since its initial clinical implementation in 1938, the antidepressant effects of ECT have been well documented. The first antidepressant drugs, a chlorpromazine derivative imipramine and an isoniazid derivative iproniazid, were discovered in the 1950s. Their pharmacological mode of action was soon traced down to facilitation of monoaminergic tone, which rationalized the subsequent development of pharmacologically similar compounds. Of these drugs, the serotonin/norepinephrine selective reuptake inhibitors are the first-line medications in the treatment of depression today. However, overall remission rates to these medications are low and there is a delayed onset before the effects on the core symptoms of depression become evident (Fava 2003). The relatively recent discovery of the rapid antidepressant effects of ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist (Berman et al. 2000; Aan Het Rot et al. 2012), has generated huge enthusiasm to develop novel antidepressants with better efficacy and a faster onset of therapeutic action. While there is little doubt on the antidepressant effects of ketamine, the precise neurobiological basis of its therapeutic actions—as with classical antidepressants and ECT—remains to be unknown. Emerging experimental data however suggest that induction of intrinsic regenerative and neurotrophic mechanisms and subsequent facilitation of synaptic plasticity significantly contribute to antidepressant effects (Castrén and Rantamäki 2010; Duman and Aghajanian 2012; Castrén and Hen 2013). It is increasingly recognized that upregulation of brain-derived neurotrophic factor (BDNF) synthesis and release and activation of its neurotrophin receptor tropomyosin-related kinase B (TrkB) are at the core of these alterations, as reviewed elsewhere (Duman and Monteggia 2006; Rantamäki and Castrén 2008; Castrén and Rantamäki 2010; Duman and Aghajanian 2012; Castrén and Hen 2013). Less attention has been put on some of the discrepancies and obscurities related to the effects of antidepressants on TrkB. Before highlighting and discussing these, it is important to introduce some of the basic principles of BDNF-TrkB signaling machinery.
An overview of BDNF-TrkB signaling
BDNF belongs to the neurotrophin family of neurotrophic factors (Barde et al. 1982). Neurotrophins are ~ 14-kDa glycoproteins that support the survival and differentiation of specific neuronal populations in the peripheral nervous system during development (Huang and Reichardt 2001). In the developing brain, BDNF regulates the differentiation, maturation and function of many neuronal subtypes, especially the GABAergic (γ-aminobutyric acid) interneurons (Huang et al. 1999). BDNF is the most widely expressed neurotrophin in the adult brain wherein the most prominent expression is observed at the level of the hippocampus and cerebral cortex. BDNF is, however, also found, e.g., in the blood that is thought to arise mainly from circulating platelets (Rosenfeld et al. 1995; Chacón-Fernández et al. 2016). The levels of BDNF in the human serum are in fact very high (~ 5–25 ng/ml; see below). The levels of BDNF in the mouse serum are instead low or undetectable (Chacón-Fernández et al. 2016).
Among neurotrophins, the role of BDNF in regulating synaptogenesis and synaptic plasticity (e.g., long-term potentiation (LTP)) in the developing and adult central nervous system has been best characterized (Thoenen 1995; Hensch 2005; Park and Poo 2013). These effects of BDNF have been associated with the ability of neuronal activity to regulate the expression, process and release of BDNF. While robust neuronal excitation, such as generalized seizures (Isackson et al. 1991), produces prominent increase in BDNF synthesis, physiological stimuli such as light (at the level of visual cortex) (Castrén et al. 1992) and physical exercise (Neeper et al. 1995) also increase bdnf mRNA levels in the adult hippocampus and cortex. Like the other so-called immediate-early genes (e.g., c-fos, arc, zif-268), the bdnf gene is rapidly but transiently activated in response to various stimuli.
The bdnf gene structure is complex and due to alternative splicing, numerous mRNA transcripts can be made (Aid et al. 2007; Pruunsild et al. 2007). These transcripts are, however, translated and processed into identical pro-BDNF protein that is further enzymatically processed into mature protein within or outside the cell (note: the protein sequence of the mature BDNF is essentially identical among humans and mice). LTP-inducing paradigms have been shown to increase bdnf mRNA (Castrén et al. 1993) and concomitant release of pro-BDNF and tissue plasminogen activator (tPA) from neurons resulting in plasmin-mediated cleavage of pro-BDNF within activated synapses in vitro (Pang et al. 2004). Without such conversion, the levels of pro-BDNF may increase extracellularly allowing it to bind to the p75NTR, a member of the TNF family of receptors, whose activation has been associated with opposing effects on neuronal structure and synaptic plasticity as seen with BDNF (Teng et al. 2005; Woo et al. 2005). To which extend this pro-BDNF p75NTR signaling axis influences brain function under physiological context in vivo is unclear, especially in the adult brain where the levels of pro-BDNF are low (Matsumoto et al. 2008) and p75NTR expression is largely confined in the cholinergic neurons of the basal forebrain.
The neurotrophic and neuroplastic effects of BDNF are mainly mediated by the TrkB receptor (Klein et al. 1991; Barbacid 1994; Huang and Reichardt 2001). Full-length TrkB receptors are ~ 145-kDa membrane bound glycoproteins (core protein ~ 93 kDa) that bind with high affinity to BDNF and a lesser extent to NT-3 (neurotrophin-3) and NT-4. TrkB receptors exist also as truncated forms that lack the majority of the intracellular domain while the extracellular domain is identical with the full-length receptors (Middlemas et al. 1991). These receptors act as BDNF scavengers or dominant-negative regulators of the full-length TrkB (Eide et al. 1996; Saarelainen et al. 2000), although the biological role of these receptors appears more complex (Fenner 2012). Upon binding to BDNF, TrkB receptors dimerize and transphosphorylate each other within the autophosphorylation loop (Y705/6) (Fig. 1). These posttranslational modifications further launch phosphorylation of other tyrosine residues that serve as docking sites for adaptor proteins that set off complex intracellular signaling cascades (Huang and Reichardt 2001). Phosphorylation of the Shc binding site (Y515) ultimately regulates protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) pathways that are important regulators of cell survival and neuronal differentiation. AKT regulates a number of apoptotic and survival mechanisms and is also an important regulator of mTor (mammalian target of rapamycin) and GSK3β (glycogen synthase 3β) (Kim et al. 2001; Beurel et al. 2015; Hermida et al. 2017). GSK3β is a promiscuous serine-threonine kinase that regulates a wide variety of cellular functions including metabolism and differentiation (Beurel et al. 2015; Hermida et al. 2017). Phosphorylated Y816 serves as a docking site for the phospholipase-Cγ1 (PLCγ1) that upregulates [Ca2+]i levels and thereby activation of, e.g., calcium/calmodulin kinase pathway. This pathway and MAPK ultimately regulate gene expression via transcription factor CREB (cAMP response element binding protein) (Finkbeiner et al. 1997). Activation of the PLCγ1 pathway has been shown to be particularly important for TrkB-induced synaptic plasticity (Minichiello et al. 1999, 2002). Direct application of BDNF (> 1–5 ng/ml) into cultured neurons readily phosphorylates Y705/6, Y515 and Y816 residues in TrkB (note: since Trk receptors share close similarity within their tyrosine kinase domains and the PLCγ1 binding sites, it is difficult to develop antibodies that specifically detect TrkB phosphorylation on these sites).

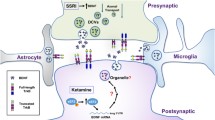
Effects of BDNF and antidepressants on TrkB receptor signaling. Upon BDNF binding, TrkB receptors dimerize at the cell surface leading to transphosphorylation of the autophosphorylation loops (Y705/6) and phosphorylation of tyrosine residues 515 and 816 that set fourth MAPK/Akt and PLCγ1 signaling cascades, respectively. Monoaminergic antidepressants and isoflurane anesthesia increase phosphorylation of Y705/6 and Y816 within TrkB while leaving Y515 unaffected. Unlike BDNF, these drugs appear to (trans)activate TrkB receptors (immaturely glycosylated) residing also in intracellular compartments. BDNF, brain-derived neurotrophic factor; MW, molecular weight; MAPK, mitogen-activated protein kinase; CREB, cAMP response element binding protein; Akt, protein kinase B; mTor, mammalian target of rapamycin; PLCγ1, phospholipase Cγ1; GSK3β, glycogen synthase kinase 3β; DAG, diacylglycerol; PKC, protein kinase C; IP3, inositol triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; TCA, tricyclic antidepressant; SSRI, serotonin-selective reuptake inhibitor
Despite the indisputable effects of BDNF on neuronal function and behavior in the brains of adult animals (incl. intracerebral applications of BDNF; see, e.g., Siuciak et al. 1994, 1996, 1997; Mamounas et al. 1995) BDNF-TrkB signaling seems to undergo significant developmental-dependent changes (Fig. 2). First, in contrast to adult animals, seizures produce negligible effects on hippocampal bdnf mRNA when delivered during early postnatal development (< P14) (Dugich-Djordjevic et al. 1992). Moreover, while BDNF strongly phosphorylates TrkB receptors in cultured neurons and in ex vivo preparations of an early postnatal brain, the responsiveness of TrkB to BDNF drastically reduces in a more mature brain. At around the third postnatal week and thereafter, an application of BDNF produces only minor effects on TrkB phosphorylation (Knüsel et al. 1994; Di Lieto et al. 2012). This intriguing phenomenon is unrelated to the gradual increase of dominant-negative truncated TrkB receptors during postnatal development (Di Lieto et al. 2012). Indeed, an essentially similar developmentally regulated reduced responsiveness of TrkA (no truncated receptors) to nerve growth factor (NGF) is observed (Knüsel et al. 1994). Moreover, basal TrkB phosphorylation levels are strongly reduced in the hippocampus of Bdnf-deficient mice during early development, while no baseline difference is seen in adult mice (Di Lieto et al. 2012). The neurobiological basis of these developmental-dependent changes in BDNF-TrkB signaling remains poorly known. For example, p75NTR and SorCS2 receptors, family members of vacuolar protein sorting 10 (VPS10) domain-containing receptor proteins, have been shown to facilitate the responsiveness of TrkB to BDNF (Berg et al. 1991; Glerup et al. 2016). In the absence of SorCS2, the ability of BDNF to induce TrkB signaling, LTP and neurotrophic effects is essentially abolished (Glerup et al. 2016).

Contrasting developmental-dependent effects of BDNF and antidepressants on TrkB. The ability of BDNF to activate TrkB gradually reduces during early postnatal development in rodents. At around 2 weeks of age, BDNF brings negligible effects on TrkB while systemic administrations of antidepressants begin to bring their effects on TrkB signaling. ADs, antidepressants; BDNF, brain-derived neurotrophic factor; P, postnatal day; p-TrkB, TrkB phosphorylation
The complexity of Trk(B) signaling is further highlighted by transactivation mechanism. At least glucocorticoids, zinc, H2O2, adenosine and pituitary adenylate cyclase–activating polypeptide (PACAP) can phosphorylate and activate Trk receptors independently of neurotrophins (Lee and Chao 2001; Lee et al. 2002; Huang et al. 2008; Jeanneteau et al. 2008; Huang and McNamara 2010, 2012). Notably, such transactivation seems to appear also in intracellular compartments, as evidenced by the phosphorylation of low molecular weight Trk receptors presumably corresponding to immaturely glycosylated forms of the receptor (Rajagopal et al. 2004). While most transactivation mechanisms have been studied in cultured cells, zinc is shown to activate TrkB also when administered systemically in adult mice (Huang et al. 2008). The effect of zinc and H2O2 on TrkB signaling also becomes evident rapidly while it takes a few hours for the other transactivators to bring similar effects in vitro. Src family kinases have been implicated in Trk transaction (Lee and Chao 2001; Huang and McNamara 2010) while other mechanisms may also be involved.
Pharmacologically diverse antidepressant drugs target TrkB receptors, but how?
Since the important scientific discoveries demonstrating the effects of stress and antidepressants on bdnf mRNA levels in the early 1990s (Smith et al. 1995; Nibuya et al. 1995), the role of BDNF has been thoroughly studied in the context of mood disorders and their treatments (Duman and Monteggia 2006; Castrén and Hen 2013). The opposing effects of stress (reduction) and classical antidepressant drugs (increase) on bdnf synthesis in the hippocampus and cortex have been repeatedly reported. Notably however, induction of bdnf synthesis is by no means restricted to pharmacological agents carrying antidepressant effects. The effects of classical antidepressant on bdnf mRNA levels emerge gradually within the time course of a few weeks, which corresponds with the delayed onset of therapeutic actions of these medications (Nibuya et al. 1995). Changes in bdnf mRNA have been, however, also reported already after a single treatment of monoaminergic antidepressants by some groups (e.g., Zetterström et al. 1999). Changes in BDNF protein have been reported less consistently, indicating that the total tissue levels of BDNF are relatively stable and BDNF is synthesized and released on demand in active synapses. Only minute amounts of BDNF are released upon activation, making the investigation of the neuronal release of BDNF challenging (Balkowiec and Katz 2002), especially in in vivo settings.
In an (initial; see below) attempt to indirectly investigate the effects of antidepressants on BDNF release, Dr. Castrén and colleagues—including myself—assayed TrkB phosphorylation levels in the brain lysates of mice subjected to antidepressant treatments (Fig. 1). Both imipramine (tricyclic) and fluoxetine (serotonin-selective reuptake inhibitor) induce TrkB phosphorylation and phosphorylation of CREB in the adult rodent cortex within an hour after a single systemic injection (Saarelainen et al. 2003). These findings were later confirmed and extended using a battery of pharmacologically distinct antidepressant drugs (Rantamäki et al. 2007) and with lithium (Rantamäki et al. 2006). Classical antidepressants activate TrkB in various brain areas, including the hippocampus, striatum, visual cortex and cerebellum (Saarelainen et al. 2003; Rantamäki et al. 2007, 2011) (Rantamäki, unpublished). Notably, antidepressants preferentially increase the phosphorylation of the autophosphorylation loop (Y705/6) and the PLCγ1 site while leaving the phosphorylation of the Shc binding site mostly unaffected (Saarelainen et al. 2003; Rantamäki et al. 2007, 2011; Di Lieto et al. 2012), although amitriptyline has been shown to phosphorylate all sites (Aonurm-Helm et al. 2015). Indeed, phosphorylation of AKT and MAPK remains unchanged (or even reduced) after acute antidepressant treatments (Rantamäki et al. 2007; Di Lieto et al. 2012).
Phospho-TrkB antibodies recognize ~ 100–110-kDa band in brain lysates obtained from antidepressant-treated mice (Saarelainen et al. 2003; Rantamäki et al. 2011). This protein may represent the immaturely glycosylated form of the receptor that is often observed when Trk receptors are transactivated independently by BDNF (see above) (Fig. 1). To support this idea, we have shown that imipramine activates TrkB receptors also in the hippocampus of forebrain-specific conditional BDNF knockout mice (Rantamäki et al. 2011). Moreover, the ability of antidepressants to activate TrkB is developmentally regulated (Di Lieto et al. 2012). A significant effect is seen in around a 2-week-old mouse and thereafter. This developmental time window corresponds with the differential responsiveness of TrkB to BDNF (discussed above) (Fig. 2). In fact, antidepressants have no effect on TrkB phosphorylation in culture neurons derived from embryonic rat brain or in cell lines (Rantamäki et al. 2011), although amitriptyline may again be an exception (Jang et al. 2009). All in all, the precise mechanism how antidepressants (trans)activate TrkB remains to be discovered but an intact and mature central nervous system seems to be required.
Differential effects of electroconvulsive shock and anesthesia on TrkB
The therapeutic effects of ECT become evident faster than those of conventional antidepressants (Segman et al. 1995), yet reduction of symptoms after a single ECT treatment is only seldom reported (Rich 1984; Thomas and Kellner 2003; Fligelman et al. 2016). An electric current leading into a short epileptiform electroencephalogram (EEG) activity is delivered during ECT under light anesthesia but how this “seizure” leads into a remedy remains a mystery.
Electroconvulsive shock (ECS; an animal model of ECT)—and seizures in general—increases bdnf mRNA levels (and other immediate early genes) in the adult rodent cortex and hippocampus. The effects appear faster and are in magnitude greater than those observed after classical antidepressants (Nibuya et al. 1995). Indeed, ECS, especially when delivered repeatedly during a course of several days or weeks, increases BDNF protein levels (Altar et al. 2003; Hansen et al. 2007; Segawa et al. 2013; Neyazi et al. 2018). To nail down the putative involvement of TrkB signaling, we investigated the effects of ECS on TrkB phosphorylation. Unexpectedly however, TrkB phosphorylation levels were reduced after a single ECS treatment (Hansen et al. 2007). Lack of effects on TrkB was also seen with repeated ECS treatment paradigm, even if BDNF protein levels were concomitantly increased in the same samples (Hansen et al. 2007). Since these early attempts, there has been little interest to investigate the effects of ECS on TrkB activation, although one recent study showed increased TrkB phosphorylation following repeated administration of ECS (Enomoto et al. 2017). Notably, brain samples were collected for analyses 4 h after the last ECS in this study. Pharmacologically induced seizures on the other hand have been more consistently shown to lead into increased TrkB phosphorylation in the adult hippocampus and cortex (Aloyz et al. 1999; Binder et al. 1999; He et al. 2002, 2004). Here too, the involvement of BDNF is however unclear as seizure-induced phosphorylation of TrkB in the hippocampus is readily also seen in conditional Bdnf−/− (synapsin-cre) mice (He et al. 2004).
A postictal emergence electroencephalogram (EEG) burst suppression pattern has been associated with the efficacy and onset-of-action of ECT (Sackeim et al. 1993; Nobler et al. 1993; Tadler and Mickey 2018). This encouraged clinical investigations to test whether deep burst-suppressing isoflurane anesthesia can ameliorate depressive symptoms with partially promising outcomes (Langer et al. 1985, 1995). This prompted us recently to investigate the effects of isoflurane on TrkB signaling in rodents. Isoflurane, at anesthetic doses, increased TrkB phosphorylation and phosphorylation of CREB within minutes in the adult prefrontal cortex and hippocampus while BDNF levels (mRNA and protein) remained unchanged (Antila et al. 2017; Theilmann et al. 2019). Remarkably, a single and brief isoflurane anesthesia produced antidepressant-like behavioral responses and facilitation of LTP several hours or a day later after the treatment (Antila et al. 2017). Antidepressant-like behavioral effects of isoflurane have been recently reported also by others (Brown et al. 2018). Similarly with the classical antidepressants, isoflurane regulates TrkB activation also in Bdnf-deficient mice (Antila et al. 2017) and the effects seem restricted to adult animals (Rantamäki et al., unpublished). Halothane, a closely related volatile anesthetic, is reported to have no antidepressant-like effects (Brown et al. 2018), although it does regulate TrkB signaling at anesthetic doses (Antila et al. 2017). It should be noted that of all the tested pharmacological manipulations so far, deep anesthesia is clearly one of the most potent and reliable means of inducing TrkB phosphorylation in the adult rodent brain (Theilmann et al. 2019). Moreover, we recently showed that medetomidine, a hypnotic-sedative drug, readily activates TrkB signaling although it did not bring antidepressant-like behavioral responses (Kohtala et al. 2018). Notably, as in the case of monoaminergic antidepressants and lithium, the effects of isoflurane on TrkB signaling follow closely the pharmacokinetic profile of the drug (Antila et al. 2017) (Fig. 3).

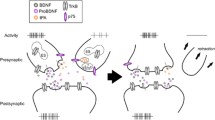
Drug-induced TrkB phosphorylation in vivo—under drug influence or upon drug withdrawal? Monoaminergic antidepressants, volatile anesthetic and medetomidine rapidly increase TrkB phosphorylation during the peak of pharmacological effect. Most clinically effective rapid-acting antidepressants (e.g., subanesthetic ketamine, nitrous oxide) are thought to regulate TrkB signaling after the peak of pharmacological effect. p-TrkB, TrkB phosphorylation; TCA, tricyclic antidepressant; SSRI, serotonin-selective reuptake inhibitor; ECT, electroconvulsive therapy
Rapid-acting antidepressants also act through TrkB: is the mechanism different from other antidepressants?
Ketamine is a widely used intravenous analgesic-anesthetic agent. At low subanesthetic doses (≤ 0.3 mg/kg IV), ketamine possesses marked analgesic properties with negligible effects on consciousness and cognition. Ketamine produces dose-dependent psychotomimetic effects, sedation and ultimately dissociative anesthesia (≥ 1.0 mg/kg, IV). Importantly, the antidepressant effects of ketamine become evident already at subanesthetic doses (Berman et al. 2000). A slow IV infusion of 0.5 mg/kg is routinely delivered in psychiatric practice but the optimal dose-window for its antidepressant effects remains to be determined and is likely specific for a given patient (Aan Het Rot et al. 2012). Anyhow, it is obvious that deep burst-suppressing EEG (c.f. isoflurane) is not essential for ketamine’s antidepressant effects.
The molecular mechanisms underlying the antidepressant effects of ketamine have been extensively studied in animal models. In most reports, ketamine has been injected intraperitoneally at a dose range of 3–10 mg/kg, while few studies indicate that ketamine might induce antidepressant-like behavioral responses also at higher doses and when administered intravenously (Browne and Lucki 2013). Somewhat similarly with ECS, subanesthetic treatment of ketamine increases glutamate release and cortical excitability. Here, the inhibition of NMDA-R and release of the tonic inhibition of GABAergic interneurons on glutamatergic neurons have been proposed as one putative mechanism (Duman and Aghajanian 2012). While also blocking the NMDA-R on glutamatergic neurons, released glutamate binds and activates preferentially postsynaptic AMPA-Rs (amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors). Indeed, AMPA-R blockade abolishes antidepressant effects of ketamine in animal models (Maeng et al. 2008). Ketamine and positive allosteric modulators of AMPA-Rs upregulate immediate early gene expression, including the bdnf. Moreover, ketamine has been shown to increase local translation of BDNF and activation of TrkB receptor in the rodent prefrontal cortex and hippocampus (Autry et al. 2011; Monteggia et al. 2013; Yang et al. 2015, 2016; Carreno et al. 2016; Sun et al. 2016; Dong et al. 2017; Ma et al. 2017) (Fig. 1). Ketamine also stimulates BDNF release in neuronal cultures (Lepack et al. 2016). To my knowledge however, the ability of ketamine to regulate TrkB phosphorylation in Bdnf-deficient mice has not been yet studied. Moreover, the dose-dependent effects of ketamine on TrkB have not been thoroughly examined in vivo or in vitro.
Among putative intracellular signaling events downstream of TrkB, activation of MAPK, activation of mTor (mammalian target of rapamycin) (and its target p70S6 kinase) and inhibition of GSK3β (glycogen synthase 3β) have been intimately connected with ketamine’s antidepressant effects in animal models (Li et al. 2010; Beurel et al. 2011). Ketamine regulates MAPK signaling even in cultured neurons (Lepack et al. 2016). Activation of mTor pathway is thought to underlie ketamine’s rapid changes on synaptogenesis (Li et al. 2010). These acute molecular changes are however not specific for ketamine. For example, isoflurane anesthesia produces very similar changes on mTor and GSK3β (Kohtala et al. 2016; Leikas et al. 2017; Antila et al. 2017), without significant effects on synaptogenesis in adult rodents (De Roo et al. 2009; Antila et al. 2017). Notably, isoflurane, as also several other general anesthetics (including ketamine), has been shown to increase synaptogenesis during early postnatal development during a time window when BDNF’s ability to activate TrkB is strongly reduced (De Roo et al. 2009; Briner et al. 2010, 2011).
Nitrous oxide (N2O), another NMDA-R blocker and a dissociative anesthetic, has been recently shown to produce rapid antidepressant effects in a subset of patients (Nagele et al. 2015). We revealed that N2O readily regulates Bdnf synthesis while TrkB signaling remains unchanged during gas administration (Kohtala et al. 2018). Importantly, TrkB signaling is set on only upon gas withdrawal (eliminated within minutes) during a brain state dominated by slow EEG oscillations (Kohtala et al. 2018). Subanesthetic ketamine and flurothyl (a volatile convulsant analogous to ECT) produce very similar EEG effects after the acute pharmacological effects on cortical excitation fade (Kohtala et al. 2018). The exact adaptive mechanism that is induced in response to N2O exposure (and other rapid-acting antidepressants) (Autry et al. 2011; Ly et al. 2018) responsible for the facilitation of TrkB signaling upon drug withdrawal (Fig. 3) remains under intense study in my lab.
Conclusions
Essentially, all treatments carrying antidepressant potential have the ability to rapidly—within an hour—regulate TrkB receptor signaling and this effect seems to be at the core of the neurotrophic and neuroplastic alterations set forth by these treatments. While this effect has not yet unequivocally demonstrated with ECT (or ECS), our recent study with the volatile convulsant flurothyl supports a notion that convulsive therapies also facilitate TrkB signaling but only after the epileptiform activity has dissipated when the brain goes into “a silent mode,” a phenomenon thought to predict the efficacy and onset of action of ECT. TrkB signaling is, however, facilitated also by a number of agents not carrying antidepressant effects (Autio et al. 2011; Koskimäki et al. 2014; Antila et al. 2017; Kohtala et al. 2018) pointing out that TrkB activation per se is not sufficient in antidepressant responses. Future efforts are thus needed to identify the specific neurobiological basis of how antidepressants, both conventional and rapid-acting and both clinically established and putative novel agents effective in animal models, target TrkB signaling (Figs. 1, 2 and 3).
References
Aan Het Rot M, Zarate CA, Charney DS, Mathew SJ (2012) Ketamine for depression: where do we go from here? Biol Psychiatry 72:537–547. https://doi.org/10.1016/j.biopsych.2012.05.003
Aid T, Kazantseva A, Piirsoo M et al (2007) Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 85:525–535. https://doi.org/10.1002/jnr.21139
Aloyz R, Fawcett JP, Kaplan DR et al (1999) Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem Cold Spring Harb N 6:216–231
Altar CA, Whitehead RE, Chen R et al (2003) Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry 54:703–709
Antila H, Ryazantseva M, Popova D et al (2017) Isoflurane produces antidepressant effects and induces TrkB signaling in rodents. Sci Rep 7:7811. https://doi.org/10.1038/s41598-017-08166-9
Aonurm-Helm A, Anier K, Zharkovsky T et al (2015) NCAM-deficient mice show prominent abnormalities in serotonergic and BDNF systems in brain - restoration by chronic amitriptyline. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol 25:2394–2403. https://doi.org/10.1016/j.euroneuro.2015.10.001
Autio H, Mätlik K, Rantamäki T et al (2011) Acetylcholinesterase inhibitors rapidly activate Trk neurotrophin receptors in the mouse hippocampus. Neuropharmacology 61:1291–1296. https://doi.org/10.1016/j.neuropharm.2011.07.033
Autry AE, Adachi M, Nosyreva E et al (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. https://doi.org/10.1038/nature10130
Balkowiec A, Katz DM (2002) Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22:10399–10407
Barbacid M (1994) The Trk family of neurotrophin receptors. J Neurobiol 25:1386–1403. https://doi.org/10.1002/neu.480251107
Barde YA, Edgar D, Thoenen H (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J 1:549–553
Berg MM, Sternberg DW, Hempstead BL, Chao MV (1991) The low-affinity p75 nerve growth factor (NGF) receptor mediates NGF-induced tyrosine phosphorylation. Proc Natl Acad Sci U S A 88:7106–7110
Berman RM, Cappiello A, Anand A et al (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354
Beurel E, Grieco SF, Jope RS (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–131. https://doi.org/10.1016/j.pharmthera.2014.11.016
Beurel E, Song L, Jope R (2011) Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry 16:1068–1070. https://doi.org/10.1038/mp.2011.47
Binder DK, Routbort MJ, McNamara JO (1999) Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci 19:4616–4626
Briner A, De Roo M, Dayer A et al (2010) Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology 112:546–556. https://doi.org/10.1097/ALN.0b013e3181cd7942
Briner A, Nikonenko I, De Roo M et al (2011) Developmental stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology 115:282–293. https://doi.org/10.1097/ALN.0b013e318221fbbd
Brown PL, Zanos P, Wang L et al (2018) Isoflurane but not halothane prevents and reverses helpless behavior: a role for EEG burst suppression? Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyy029
Browne CA, Lucki I (2013) Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front Pharmacol 4. https://doi.org/10.3389/fphar.2013.00161
Carreno FR, Donegan JJ, Boley AM et al (2016) Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol Psychiatry 21:1298–1308. https://doi.org/10.1038/mp.2015.176
Castrén E, Hen R (2013) Neuronal plasticity and antidepressant actions. Trends Neurosci 36:259–267. https://doi.org/10.1016/j.tins.2012.12.010
Castrén E, Pitkänen M, Sirviö J et al (1993) The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. Neuroreport 4:895–898
Castrén E, Rantamäki T (2010) The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol 70:289–297. https://doi.org/10.1002/dneu.20758
Castrén E, Zafra F, Thoenen H, Lindholm D (1992) Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci U S A 89:9444–9448
Chacón-Fernández P, Säuberli K, Colzani M et al (2016) Brain-derived neurotrophic factor in megakaryocytes. J Biol Chem 291:9872–9881. https://doi.org/10.1074/jbc.M116.720029
De Roo M, Klauser P, Briner A et al (2009) Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS One 4:e7043. https://doi.org/10.1371/journal.pone.0007043
Di Lieto A, Rantamäki T, Vesa L et al (2012) The responsiveness of TrkB to BDNF and antidepressant drugs is differentially regulated during mouse development. PLoS One 7. https://doi.org/10.1371/journal.pone.0032869
Dong C, Zhang J-C, Yao W et al (2017) Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: comparison with ketamine. Int J Neuropsychopharmacol 20:228–236. https://doi.org/10.1093/ijnp/pyw089
Dugich-Djordjevic MM, Tocco G, Willoughby DA et al (1992) BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron 8:1127–1138. https://doi.org/10.1016/0896-6273(92)90133-X
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72. https://doi.org/10.1126/science.1222939
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127. https://doi.org/10.1016/j.biopsych.2006.02.013
Eide FF, Vining ER, Eide BL et al (1996) Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci 16:3123–3129
Enomoto S, Shimizu K, Nibuya M et al (2017) Activated brain-derived neurotrophic factor/TrkB signaling in rat dorsal and ventral hippocampi following 10-day electroconvulsive seizure treatment. Neurosci Lett 660:45–50. https://doi.org/10.1016/j.neulet.2017.09.011
Fava M (2003) Diagnosis and definition of treatment-resistant depression. Biol Psychiatry 53:649–659
Fenner BM (2012) Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev 23:15–24. https://doi.org/10.1016/j.cytogfr.2012.01.002
Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19(5):1031–1047
Fligelman B, Pham T, Bryson EO et al (2016) Resolution of acute suicidality after a single right unilateral electroconvulsive therapy. J ECT 32:71–72. https://doi.org/10.1097/YCT.0000000000000258
Glerup S, Bolcho U, Mølgaard S et al (2016) SorCS2 is required for BDNF-dependent plasticity in the hippocampus. Mol Psychiatry 21:1740–1751. https://doi.org/10.1038/mp.2016.108
Hansen HH, Rantamäki TPJ, Larsen MH et al (2007) Rapid activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway by electroconvulsive shock in the rat prefrontal cortex is not associated with TrkB neurotrophin receptor activation. Cell Mol Neurobiol 27:585–594. https://doi.org/10.1007/s10571-007-9145-1
He X-P, Kotloski R, Nef S et al (2004) Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43:31–42. https://doi.org/10.1016/j.neuron.2004.06.019
He X-P, Minichiello L, Klein R, McNamara JO (2002) Immunohistochemical evidence of seizure-induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci 22:7502–7508
Hensch TK (2005) Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6:877–888. https://doi.org/10.1038/nrn1787
Hermida MA, Dinesh Kumar J, Leslie NR (2017) GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul 65:5–15. https://doi.org/10.1016/j.jbior.2017.06.003
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. https://doi.org/10.1146/annurev.neuro.24.1.677
Huang YZ, McNamara JO (2010) Mutual regulation of Src family kinases and the neurotrophin receptor TrkB. J Biol Chem 285:8207–8217. https://doi.org/10.1074/jbc.M109.091041
Huang YZ, McNamara JO (2012) Neuroprotective effects of reactive oxygen species mediated by BDNF-independent activation of TrkB. J Neurosci 32:15521–15532. https://doi.org/10.1523/JNEUROSCI.0755-12.2012
Huang YZ, Pan E, Xiong Z-Q, McNamara JO (2008) Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 57:546–558. https://doi.org/10.1016/j.neuron.2007.11.026
Huang ZJ, Kirkwood A, Pizzorusso T et al (1999) BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98:739–755
Isackson PJ, Huntsman MM, Murray KD, Gall CM (1991) BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron 6:937–948
Jang S-W, Liu X, Chan C-B et al (2009) Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem Biol 16:644–656. https://doi.org/10.1016/j.chembiol.2009.05.010
Jeanneteau F, Garabedian MJ, Chao MV (2008) Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A 105:4862–4867. https://doi.org/10.1073/pnas.0709102105
Kim AH, Khursigara G, Sun X et al (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 21:893–901. https://doi.org/10.1128/MCB.21.3.893-901.2001
Klein R, Nanduri V, Jing SA et al (1991) The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 66:395–403
Knüsel B, Rabin SJ, Hefti F, Kaplan DR (1994) Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci 14:1542–1554
Kohtala S, Theilmann W, Rosenholm M et al (2018) Cortical excitability and activation of TrkB signaling during rebound slow oscillations are critical for rapid antidepressant responses. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1364-6
Kohtala S, Theilmann W, Suomi T et al (2016) Brief isoflurane anesthesia produces prominent phosphoproteomic changes in the adult mouse hippocampus. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.6b00002
Koskimäki J, Matsui N, Umemori J et al (2014) Nimodipine activates TrkB neurotrophin receptors and induces neuroplastic and neuroprotective signaling events in the mouse hippocampus and prefrontal cortex. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-014-0110-5
Langer G, Karazman R, Neumark J et al (1995) Isoflurane narcotherapy in depressive patients refractory to conventional antidepressant drug treatment. A double-blind comparison with electroconvulsive treatment. Neuropsychobiology 31:182–194
Langer G, Neumark J, Koinig G, et al (1985) Rapid psychotherapeutic effects of anesthesia with isoflurane (ES narcotherapy) in treatment-refractory depressed patients. Neuropsychobiology 14:118–120. doi: 118216
Lee FS, Chao MV (2001) Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci 98:3555–3560. https://doi.org/10.1073/pnas.061020198
Lee FS, Rajagopal R, Chao MV (2002) Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev 13:11–17
Leikas JV, Kohtala S, Theilmann W et al (2017) Brief isoflurane anesthesia regulates striatal AKT-GSK3β signaling and ameliorates motor deficits in a rat model of early-stage Parkinson’s disease. J Neurochem. https://doi.org/10.1111/jnc.14066
Lepack AE, Bang E, Lee B et al (2016) Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 111:242–252. https://doi.org/10.1016/j.neuropharm.2016.09.011
Li N, Lee B, Liu R-J et al (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. https://doi.org/10.1126/science.1190287
Ly C, Greb AC, Cameron LP et al (2018) Psychedelics promote structural and functional neural plasticity. Cell Rep 23:3170–3182. https://doi.org/10.1016/j.celrep.2018.05.022
Ma Z, Zang T, Birnbaum SG et al (2017) TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat Commun 8:1668. https://doi.org/10.1038/s41467-017-01709-8
Maeng S, Zarate CA Jr, Du J et al (2008) Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63:349–352. https://doi.org/10.1016/j.biopsych.2007.05.028
Mamounas LA, Blue ME, Siuciak JA, Altar CA (1995) Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain. J Neurosci 15:7929–7939
Matsumoto T, Rauskolb S, Polack M et al (2008) Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci 11:131–133. https://doi.org/10.1038/nn2038
Middlemas DS, Lindberg RA, Hunter T (1991) trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol 11:143–153
Minichiello L, Calella AM, Medina DL et al (2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36:121–137
Minichiello L, Korte M, Wolfer D et al (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414
Monteggia LM, Gideons E, Kavalali ET (2013) The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol Psychiatry 73:1199–1203. https://doi.org/10.1016/j.biopsych.2012.09.006
Nagele P, Duma A, Kopec M et al (2015) Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry 78:10–18. https://doi.org/10.1016/j.biopsych.2014.11.016
Neeper SA, Gómez-Pinilla F, Choi J, Cotman C (1995) Exercise and brain neurotrophins. Nature 373:109. https://doi.org/10.1038/373109a0
Neyazi A, Theilmann W, Brandt C et al (2018) P11 promoter methylation predicts the antidepressant effect of electroconvulsive therapy. Transl Psychiatry 8:25. https://doi.org/10.1038/s41398-017-0077-3
Nibuya M, Morinobu S, Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15:7539–7547
Nobler MS, Sackeim HA, Solomou M et al (1993) EEG manifestations during ECT: effects of electrode placement and stimulus intensity. Biol Psychiatry 34:321–330
Pang PT, Teng HK, Zaitsev E et al (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306:487–491. https://doi.org/10.1126/science.1100135
Park H, Poo M (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. https://doi.org/10.1038/nrn3379
Payne NA, Prudic J (2009) Electroconvulsive therapy: part I. A perspective on the evolution and current practice of ECT. J Psychiatr Pract 15:346–368. https://doi.org/10.1097/01.pra.0000361277.65468.ef
Pruunsild P, Kazantseva1 A, Aid T et al (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90:397–406. https://doi.org/10.1016/j.ygeno.2007.05.004
Rajagopal R, Chen Z-Y, Lee FS, Chao MV (2004) Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci 24:6650–6658. https://doi.org/10.1523/JNEUROSCI.0010-04.2004
Rantamäki T, Castrén E (2008) Targeting TrkB neurotrophin receptor to treat depression. Expert Opin Ther Targets 12:705–715. https://doi.org/10.1517/14728222.12.6.705
Rantamäki T, Hendolin P, Kankaanpää A et al (2007) Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 32:2152–2162. https://doi.org/10.1038/sj.npp.1301345
Rantamäki T, Knuuttila JEA, Hokkanen M-E, Castrén E (2006) The effects of acute and long-term lithium treatments on trkB neurotrophin receptor activation in the mouse hippocampus and anterior cingulate cortex. Neuropharmacology 50:421–427. https://doi.org/10.1016/j.neuropharm.2005.10.001
Rantamäki T, Vesa L, Antila H et al (2011) Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. PLoS One 6:e20567. https://doi.org/10.1371/journal.pone.0020567
Rich CL (1984) Recovery from depression after one ECT. Am J Psychiatry 141:1010–1011
Rosenfeld RD, Zeni L, Haniu M et al (1995) Purification and identification of brain-derived neurotrophic factor from human serum. Protein Expr Purif 6:465–471
Saarelainen T, Hendolin P, Lucas G et al (2003) Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci 23:349–357
Saarelainen T, Pussinen R, Koponen E et al (2000) Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons have impaired long-term spatial memory but normal hippocampal LTP. Synap N Y N 38:102–104. https://doi.org/10.1002/1098-2396(200010)38:1<102::AID-SYN11>3.0.CO;2-K
Sackeim HA, Prudic J, Devanand DP et al (1993) Effects of stimulus intensity and electrode placement on the efficacy and cognitive effects of electroconvulsive therapy. N Engl J Med 328:839–846. https://doi.org/10.1056/NEJM199303253281204
Segawa M, Morinobu S, Matsumoto T et al (2013) Electroconvulsive seizure, but not imipramine, rapidly up-regulates pro-BDNF and t-PA, leading to mature BDNF production, in the rat hippocampus. Int J Neuropsychopharmacol 16:339–350. https://doi.org/10.1017/S1461145712000053
Segman RH, Shapira B, Gorfine M, Lerer B (1995) Onset and time course of antidepressant action: psychopharmacological implications of a controlled trial of electroconvulsive therapy. Psychopharmacology 119:440–448
Siuciak JA, Altar CA, Wiegand SJ, Lindsay RM (1994) Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res 633:326–330
Siuciak JA, Boylan C, Fritsche M et al (1996) BDNF increases monoaminergic activity in rat brain following intracerebroventricular or intraparenchymal administration. Brain Res 710:11–20
Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM (1997) Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav 56:131–137. https://doi.org/10.1016/S0091-3057(96)00169-4
Smith MA, Makino S, Kvetnansky R, Post RM (1995) Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15:1768–1777
Sun H-L, Zhou Z-Q, Zhang G-F et al (2016) Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl Psychiatry 6:e741. https://doi.org/10.1038/tp.2016.21
Tadler SC, Mickey BJ (2018) Emerging evidence for antidepressant actions of anesthetic agents. Curr Opin Anaesthesiol 31:439–445. https://doi.org/10.1097/ACO.0000000000000617
Teng HK, Teng KK, Lee R et al (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 25:5455–5463. https://doi.org/10.1523/JNEUROSCI.5123-04.2005
Theilmann W, Alitalo O, Yorke I, Rantamäki T (2019) Dose-dependent effects of isoflurane on TrkB and GSK3β signaling: importance of burst suppression pattern. Neurosci Lett 694:29–33
Thoenen H (1995) Neurotrophins and neuronal plasticity. Science 270:593–598
Thomas SG, Kellner CH (2003) Remission of major depression and obsessive-compulsive disorder after a single unilateral ECT. J ECT 19:50–51
Woo NH, Teng HK, Siao C-J et al (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8:1069–1077. https://doi.org/10.1038/nn1510
Yang B, Zhang J-C, Han M et al (2016) Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology 233:3647–3657. https://doi.org/10.1007/s00213-016-4399-2
Yang C, Shirayama Y, Zhang J– et al (2015) R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry 5:e632. https://doi.org/10.1038/tp.2015.136
Zetterström TS, Pei Q, Madhav TR et al (1999) Manipulations of brain 5-HT levels affect gene expression for BDNF in rat brain. Neuropharmacology 38:1063–1073
Acknowledgements
M. Sci. Okko Alitalo is thanked for helping with their preparation of the figures. I wish to thank all the members of my lab for the support.
Funding
Open access funding provided by the University of Helsinki including Helsinki University Central Hospital. This study has been supported by the Academy of Finland (grants 276333 and 305195).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rantamäki, T. TrkB neurotrophin receptor at the core of antidepressant effects, but how?. Cell Tissue Res 377, 115–124 (2019). https://doi.org/10.1007/s00441-018-02985-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-02985-6