
Abstract
The importance of TNF-α signals mediated by tumor necrosis factor receptor type 1 (TNFR1) in inflammation and fibrosis induced by carbon tetrachloride (CCl4), and in post-injury liver regeneration including a GFP/CCl4 model developed as a liver repair model by bone marrow cell (BMC) infusion, was investigated. In mice in which TNFR1 was suppressed by antagonist administration or by knockout, liver fibrosis induced by CCl4 was significantly decreased. In these mice, intrahepatic macrophage infiltration and TGF-β1 expression were reduced and stellate cell activity was decreased; however, expression of MMP-9 was also decreased. With GFP-positive BMC (TNFR1 wild-type, WT) infusion in these mice, fibrosis proliferation, including host endogenous intrahepatic macrophage infiltration, TGF-β1 expression and stellate cell activity, increased significantly. There was no significant increase of MMP-9 expression. In this study, TNFR1 in hosts had a promoting effect on CCl4-induced hepatotoxicity and fibrosis, whereas BMC infusion in TNFR1 knockout mice enhanced host-derived intrahepatic inflammation and fibrosis proliferation. These findings differed from those in WT recipient mice, in which improvement in inflammation and fibrosis with BMC infusion had previously been reported. TNFR1-mediated signaling might be important to induce the improvement of liver fibrosis by bone marrow cell infusion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The mechanism of hepatotoxicity induced by many factors, including CCl4, has been studied for a long time (Drill 1952) and besides direct cytotoxicity due to these factors, enhanced hepatotoxicity due to inflammatory cell infiltration and stellate cell activation induced by these factors also occurs. TNF-α is an important inflammatory cytokine that induces hepatotoxicity; however, it also initiates liver regeneration after injury. This has been examined in various previous hepatotoxicity, hepatic resection and hepatic failure models. In addition, in recent stem cell studies, TNF-α signals have been found to be essential in the differentiation/proliferation process from stem cells to functional cells. For effective expression of TNF-α signals, of the various receptors, TNFR1 (p55) is important. In studies using TNFR1 knockout (KO) mice, while inhibition of hepatotoxicity has been shown, decreased liver regeneration and hepatocyte proliferation have also been confirmed (Gardner et al. 2003; Simeonova et al. 2001; Sudo et al. 2005; Yamada and Fausto 1998).
In our laboratory, using a GFP/CCl4 model, BMC infusion has been shown to improve fibrosis and liver function in cirrhosis (Sakaida et al. 2004, 2008; Terai et al. 2002, 2005). In the liver of mice not treated with CCl4, without hepatic toxicity, repopulation of GFP-positive BMCs into the liver has not been observed, thus highlighting the need for chronic inflammatory signals in cell repopulation and effective expression. In addition, after BMC infusion, alterations in TNFR1 expression and associated increases in various cytokines such as fibroblast growth factor 2 (FGF2) have been recognized (Omori et al. 2004). This also suggested the importance of inflammatory signals, particularly TNF-α signals, in liver function improvement. Therefore, to investigate the importance of TNF-α signals mediated by TNFR1 in liver repair by BMC infusion in recipients, this study using TNFR1 antagonist-administered mice and TNFR1 KO mice as recipients was conducted.
Materials and methods
Mice
C57BL/6 TNFR1 KO mice were kindly provided by Hiroshi Nishina (Department of Developmental and Regenerative Biology, Medical Research Institute, Tokyo Medical and Dental University). TNFR1 KO mouse typing was performed by PCR analysis using primer typing box [p55R636 and HSV-TK (to confirm induction of silencing gene factor)]. Those primers were p55R636 (5′-GGCTGCAGTCCACGCACTGG-3′), and HSV-TK (5′-ATTCGCCAATGACAAGACGCTGG-3′) (data not shown) (Pfeffer et al. 1993). C57BL/6 female mice were purchased from Chiyoda SLC (Tokyo, Japan). GFP-transgenic mice (TgN(β-act-EGFP)Osb) were kindly provided by Masaru Okabe (Genome Research Center, Osaka University, Osaka, Japan). These mice were maintained in specific pathogen-free housing at the Animal Experiment Facility of Yamaguchi University School of Medicine and cared for in accordance with the animal ethics requirements at Yamaguchi University School of Medicine.
Experimental protocol
First, to confirm that signals mediated by TNFR1 are important in intrahepatic inflammatory responses and fibrosis induced by CCl4, an experiment using TNFR1 antagonist (anti-mouse TNFR1 antibody, MAB430; R&D Systems, Minneapolis, MN, USA) was conducted in wild-type (WT) mice. In WT mice, CCl4 was administered for 4 weeks to create a liver cirrhosis model; then, by caudal vein injection of 100 μg/body of antagonist, signals mediated by TNFR1 were suppressed in recipients; and thereafter, CCl4 was continued for 1 week. A model was also created with infusion of GFP-positive BMCs after 1 h, when in vivo activity of the antagonist reaches a peak. There were 3 groups: WT (Control) (wild-type, without BMC infusion); WT + A (wild-type, with TNFR1 antagonist only, without BMC infusion); and GFP/WT + A (wild-type, with TNFR1 antagonist and with BMC infusion) (Fig. 1).

Experimental protocols. Protocol I In each group of mice, CCl4 (1.0 ml/kg body) was administered twice a week for 5 weeks to create a liver cirrhosis model. In WT + A mice, 100 μg/body of TNFR1 antagonist was injected via tail vein before 1 week of sacrifice. GFP/WT + A mice were treated with GFP positive BMC (1 × 105 cells) infusion via tail vein 1 hour after TNFR1 antagonist injection. Protocol II WT (Control) mice and KO (Control) mice were treated with CCl4 (same as above) for 8 weeks. In GFP/WT mice and GFP/KO mice, after 4 weeks of CCl4 administration, BMCs (1 × 105 cells) were injected, followed by 4 weeks of CCl4 treatment
As will be described later, by blocking TNFR1, suppression of fibrosis and suppression of inflammatory cell infiltration were confirmed. Therefore, as a more highly specific model, a model was created by the following protocol with TNFR1 KO mice as BMC infusion recipients.
Six-week-old female C57BL/6 mice and female isogenic TNFR1 KO mice were treated with CCl4 (1.0 ml/kg body diluted 1:3 in corn oil) twice a week for 8 weeks. In the other group, after 4 weeks of CCl4 administration in each group (C57BL/6 wild-type and TNFR1 KO), bone marrow cells (BMC) (1 × 105 cells) from GFP transgenic mice were injected via the tail vein as previously described (Terai et al. 2003).
After 8 weeks, 36 h after the last CCl4 injection, the mice were sacrificed to examine the blood data and liver tissue specimens. The liver was fixed in 4% buffered paraformaldehyde for 24–48 h and paraffin embedded. Blood samples were obtained by cardiac puncture and drawn into a glass tube containing 7.5% EDTA (pH 7.4). After centrifugal separation, the plasma was stored at 4°C.
There was a total of 4 groups in this study: WT (Control), wild-type without BMC infusion; KO (Control), TNFR1 KO without BMC infusion; GFP/WT, wild-type with GFP-positive BMC infusion; and GFP/KO, TNFR1 KO with GFP-positive BMC infusion (Fig. 1).
Quantitative analysis of liver fibrosis and immunohistochemistry
The liver fibrosis area was quantified with Sirius-red staining using an Olympus Provis microscope equipped with a CCD camera (Olympus, Tokyo, Japan). The red area, considered the fibrotic area, was assessed by computer-assisted image analysis with MetaMorph software (Universal Imaging, Downingtown, PA, USA) at a magnification of ×40. The mean value of 10 randomly selected areas per sample was used as the expressed percent area of fibrosis.
Immunohistochemistry of TGF-β1, alpha smooth muscle actin (α-SMA), matrix metalloproteinase (MMP)-9 and F4/80
Three-μm-thick liver sections were mounted on microscope slides, routinely dewaxed and rehydrated and pretreated with Vector Antigen Unmasking Solutions (Citrate-based, Cat. No. H-3300). For the immunohistochemical analysis, the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) was used for GFP (anti-GFP, rabbit IgG fraction, A11122; Invitrogen, Carlsbad, CA, USA), TGF-β1[TGF-β1(V), SC-146; Santa Cruz Biotechnology], alpha-smooth muscle actin (α-SMA) (alpha smooth muscle actin antibody, ab6594; Abcam, Cambridge, MA, USA), matrix metalloproteinase (MMP)-9 (anti-mouse MMP-9 antibody, AF909; R&D Systems) and F4/80 [F4/80 antibody(BM8), ab16911; Abcam] staining by the avidin-biotin-peroxidase complex method.
Additionally, double immunofluorescent staining was performed to study co-expression of GFP and F4/80 in bone marrow cell-infused mice. The mixture of the first antibodies was GFP and F4/80 noted above. The secondary antibodies, goat anti-rabbit IgG (H + L), Alexa Fluor 488 (A11034,;Invitrogen) (Green) and goat anti-rat IgG (H + L), Alexa Fluor 568 (A11077; Invitrogen) (Red) were each applied at a concentration of 1:400 in PBS for 60 min at room temperature. Before attaching the coverslip, DAPI (D212; Dojindo Laboratories, Kumamoto, Japan) was applied for counterstaining to visualize all nuclei in the tissue sections. The sections were viewed and photographed with the CCD camera noted above.
Real-time quantitative PCR analysis
Total RNA was isolated from the livers of the mice treated at 4 weeks after the BMC infusion or control CCl4 treatment. The messenger RNA (mRNA) expressions of TGF-β1 and MMP-9 were evaluated using real-time quantitative PCR. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany). For cDNA synthesis, AMV reverse transcription reagents were used according to the manufacturer’s instructions (Roche Diagnostic, Pleasanton, CA, USA). Real-time PCR was performed with SYBR Green Master Mix (Roche Diagnostic). The primers used for TGF-β1 were 5′-GAAGCCATCCGTGGCCAGAT-3′ (forward) and 5′-GACGTCAAAAGACAGCACT-3′ (reverse), for MMP-9 were 5′-GGAACTCACACGACATCTTCCA-3′ (forward) and 5′-GAAACTCACACGCCAGAAGAATTT-3′ (reverse) and collagen type 1 alpha were 5′-CGGGCAGGACTTGGGTA-3′ (forward) and 5′-CGGAATCTGAATGGTCTGACT-3′ (reverse). The PCR primers used for mouse glyceraldehyde-3-phospatase dehydrogenase (GAPDH), which was used as an internal control, were: 5′-GTCTTCACCACCATGGAGAAGGC-3′, 5′-ATG CCAGTGAGCTTCCCGTTCAGC-3′. The cycle for PCR was as follows: 1 cycle of 95°C for 20 s; 40 cycles of 3 s at 95°C and 30 s at 60°C; and 1 cycle of 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Reactions were performed in a Step One Plus™ real-time PCR system (Applied Biosystems, California, CA, USA) and amounts of all mRNAs were quantified using StepOne™ version 2.1 software (Applied Biosystems).
Western blot analysis
The liver samples (approximately 40 mg) were homogenized in 1 ml of cell lysis buffer (Cell Signal Technology, Beverly, MA, USA) and 1 mM phenylmethanesulfonyl fluoride (PMSF) and complete mini-centrifuged (Roche Diagnostic). The supernatant represented the whole protein. Then, 40 μg of the protein sample were mixed with the same volume of loading buffer (5% 2-mercaptoethanol and 95% Laemmli Sample Buffer; Bio-Rad Laboratories, Hercules, CA, USA), heated for 4 min at 97°C and separated by 10% SDS-PAGE. The separated bands were transferred to Immobilon-P transfer membrane (Millipore, Billerica, MA, USA), followed by blocking of the membranes for 1 h with blocking buffer (0.1% Tween-20; Wako Pure Chemical Industries, Osaka, Japan), 0.2% I-Block™ reagent (Tropix, Bedford, MA, USA) and 1 mM Tris-HCl buffer (pH 7.5) (Invitrogen). The membranes were then washed with washing buffer (0.1% Tween-20, 1 mM Tris-HCl buffer; pH 7.5) and incubated overnight at 4°C with the primary antibodies against MMP-9 (R&D Sytems), TGF-β1 (Santa Cruz Biotechnology) and β-actin (Abcam) in blocking buffer. Then, after being washed, the membranes were incubated for 1 h at room temperature with the appropriate secondary antibodies. Reactive bands were identified using ECL (Amersham Biosciences, Piscataway, NJ, USA) and autoradiography, according to the manufacturer’s instructions.
Statistical analysis
Statistical significance was determined using 2-tailed Student's t test. Results are presented as the mean ± standard deviation and differences of p < 0.05 were considered significant.
Results
Alterations of liver fibrosis and intrahepatic inflammatory invasion by TNFR1 antagonist and BMC infusion
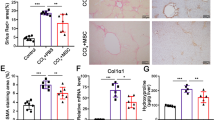
At 1 week after TNFR1 antagonist administration in wild-type mice, liver fibrosis was significantly suppressed [WT (Control): WT + A, p < 0.01] in the antagonist-treated group compared to the untreated group. In addition, F4/80-positive intrahepatic macrophage infiltration tended to be suppressed [WT (Control): WT + A, p = 0.07] and stellate cell activity, shown by α-SMA staining, was significantly suppressed [WT (Control): WT + A, p < 0.01]. However, in the group with GFP-positive BMC infusion in addition to the antagonist, liver fibrosis did not improve with BMC infusion (WT + A: GFP/WT + A, p = 0.44) Conversely, both macrophage infiltration (WT + A: GFP/WT + A, p = 0.02) and stellate cell activity (WT + A: GFP/WT + A, p < 0.01) increased significantly (Fig. 2a–l).

Suppression of liver fibrosis, hepatic stellate cell activation and F4/80 positive cell invasion using TNFR1 antagonist. a–c Sirius red staining of liver sections (original magnifications ×40). d Statistical analysis of fibrotic area by the staining. Scale bars standard deviations. WT liver (a) had severe fibrosis but WT + A liver (b) and GFP/WT + A liver (c) did not. There was no significant improvement induced by BMC infusion with TNFR1 antagonist. e–g Immunohistochemistry of F4/80 (original magnifications ×100). h Statistical analysis of F4/80 positive area. Comparing WT (e) with WT + A (f), TNFR1 antagonist suppressed the invasion of F4/80 positive cells into the liver but in GFP/WT + A (g), the invasion increased. i–k Immunohistochemistry of α-SMA (original magnifications ×40). l Statistical analysis of α-SMA positive area. Comparing WT (i) with WT + A (j), TNFR1 antagonist suppressed the expression of α-SMA but in GFP/WT + A (k), the bone marrow cell infusion raised the activation regardless of TNFR1 antagonist administration
Liver fibrosis did not improve in the KO recipient model
The migration of GFP positive cells was confirmed using immunohistochemistry of GFP (Fig. 3a, b). On Sirius red staining, liver fibrosis was significantly suppressed in KO (Control) mice compared to WT (Control) mice [WT (Control): KO (Control), p < 0.05]. WT mice tended to have strong fibrosis but similar to the GFP/CCl4 model that we previously reported (Terai, et al. 2003), with BMC infusion, fibrosis improved significantly [WT (Control): GFP/WT, p < 0.01]. However, in TNFR1 KO mice, in whom fibrosis was suppressed, BMC infusion did not improve liver fibrosis [KO (Control): GFP/KO, p = 0.91] (Fig. 3c–g).

Comparison of liver fibrosis using TNFR1 KO mice. a, b anti-GFP immunohistochemistry (original magnifications, ×200). Both in wild type mouse live (a) and in TNFR1 KO mouse liver (b), the migrations of GFP positive bone marrow cells were confirmed. c–f Sirius red staining of liver sections (original magnifications, ×40). g Statistical analysis of fibrotic area. Scale bars standard deviations. Compared with WT (Control) (c), GFP/WT (d) had marked fibrolysis. KO (Control) (e) had significantly suppressed fibrosis but there was no marked improvement of fibrosis in GFP/KO (f). h Real-time RT-PCR analysis of expression of collagen type 1 alpha-1, indicating the fibrotic activity, showed similar results. i–l Immunohistochemistry of MMP-9 in liver sections (original magnifications, ×200). Liver section of WT (Control) (i) [magnified figure (×400) (j)] had certain degree of expression, but in KO (Control) liver (k) and in GFP/KO liver (l), the expression was significantly low level. m Real-time RT-PCR analyses. n Western blot analyses of MMP-9; both showed suppression in TNFR1 KO mice, but no significant increase in GFP/KO mice was seen, and the suppression remained
In addition, on real-time RT-PCR for collagen I-α1, in WT mice, fibrosis proliferation was increased and in KO mice, it was suppressed [WT (Control): KO (Control), p < 0.05]. In WT mice, as in our previous GFP/CCl4 model, after BMC infusion, collagen I-α1 mRNA expression was decreased [WT (Control): GFP/WT, p < 0.05]. On the other hand, there were no changes in KO mice with BMC infusion [KO (Control): GFP/KO, p = 0.67] (Fig. 3h).
Fibrolytic reactions induced by BMC infusion
Expression of MMP-9, important in liver fibrosis improvement, corresponded with sites of inflammation and fibrosis in each model and based on western blot and real-time RT-PCR analysis, MMP-9 expression was significantly suppressed in KO mice compared to wild-type mice [WT (Control): KO(Control), p < 0.01]. However, in KO mice, even with GFP-positive BMC infusion, there was no significant increase, statistically proven in Real-time RT-PCR analysis [KO (Control): GFP/KO, p = 0.41]. This showed that, in KO mice, although MMP-9 expression is suppressed, BMC infusion in these mice did not clearly increase MMP-9 (Fig. 3i-n).
Alterations of F4/80 positive cells into TNFR1 KO mice
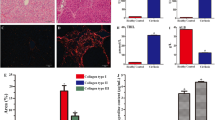
In KO (Control) mice, compared to WT (Control) mice, intrahepatic infiltration of F4/80-positive cells (macrophages) induced by CCl4 was significantly suppressed [WT (Control): KO (Control), p < 0.01] but with BMC infusion, this infiltration was significantly increased [KO (Control): GFP/KO, p < 0.01] (Fig. 4a–e). Upon evaluation of macrophage origin, in double immunofluorescent staining (GFP: emitting green light, and F4/80: emitting red light), no cells showed co-positivity with GFP and F4/80. It was indicated that the increased F4/80-positive macrophages were derived from differentiation and proliferation of recipient pre-existing cells (Fig. 4f).

Alteration of F4/80-positive cell infiltration, TGF-β1 expression, and hepatic stellate cell activation. a–d Immunohistochemical analysis of F4/80 (original magnifications, ×100). e Statistical analysis of F4/80 positive area. Scale bars standard deviations. Compared with WT (Control) (a) [magnified figure (×200) (b)], the invasion of F4/80 positive cells was markedly suppressed in KO (Control) (c) and significantly increased in GFP/KO (d). f Double fluorescent immunohistochemistry of GFP and F4/80 (original magnifications, ×400). GFP was marked with FITC (green) and F4/80 with Cy3 (red). This showed the mismatch between the GFP-positive area and that of F4/80. g–j Immunohistochemical analyses of expression (original magnifications, ×100). WT (Control) (g) [magnified figure (×200) (h)] had much expression, but in KO (Control) (i), it was suppressed. Adversely, GFP/KO (j) had more TGF-β1 expression than KO (Control). k Real-time RT-PCR analyses of TGF-β1. l Western blot analyses using TGF-β1 antibody. Both showed the increase of TGF-β1 expression in GFP/KO, suppressed in KO (Control). m–o Immunohistochemical analyses of α-SMA (original magnifications ×40). p Statistical analysis of α-SMA positive area. Scale bars standard deviations. Compared with WT (Control) (m), KO (Control) (n) had less hepatic stellate cell activation, but the activation was markedly increased in GFP/KO (o)
Upregulation of TGF-β1 signals and hepatic stellate cell activation
Upon evaluation by immunostaining, TGF-β1 expression was decreased in KO (Control) mice but after BMC infusion, it was significantly increased (Fig. 4g–j). On real-time RT-PCR analysis as well, in KO mice, TGF-β1 increased after BMC infusion [KO (Control): GFP/KO, p < 0.05] (Fig. 4k). On western blot analysis, the findings were similar (Fig. 4l). With alterations in TGF-β1 expression, stellate cell activity, evaluated by α-SMA staining, was suppressed in KO (Control) mice but with BMC infusion, in GFP/KO mice, it was significantly increased [KO (Control): GFP/KO, p < 0.05] (Fig. 4m–p).
Discussion
TNF-α has long been known as an inflammatory cytokine whose action is mediated mainly by TNFR1. In addition to TNFR1, several other receptors exist, including TNFR2, each with a variety of actions, including cytotoxic effects, cell proliferation and protective effects. TNFR1 itself is known to be involved in various actions, including induction of hepatocyte apoptosis, extracellular matrix proliferation via downstream signals, anti-apoptosis effects and hepatocyte proliferation. In a previous partial hepatectomy model, the importance of TNF-α in liver regeneration was demonstrated (Yamada et al. 1997) but on the other hand, in studies using hepatotoxic factors such as CCl4 and LPS, the involvement of TNF-α–TNFR1 pathways in the enhancement of liver cell injury and fibrosis has often been reported (Simeonova et al. 2001; Sudo et al. 2005; Horn et al. 2000). For applications of these findings to clinical liver disease, studies using TNFR1 knockout models, anti-TNF-α neutralizing antibodies and anti-TNFR1 antagonists have been conducted. Reductions in liver injury and liver fibrosis have been reported (Haak-Frendscho et al. 1994; Inui et al. 2005; Shibata et al. 2008a).
In the GFP/CCl4 model, we reported that BMC infusion has liver repair effects and improves liver fibrosis (Terai et al. 2003). In reports to date from our laboratory, we have shown that inflammatory cytokines induced by CCl4, primarily TNF-α, are important in BMC survival and liver repair (Omori et al. 2004). In addition, at other facilities, in vitro models have shown the importance of TNF-α signals in the differentiation of bone marrow stem cells to functional cells. There are various theories about BMC kinetics when infused in vivo (Higashiyama et al. 2007; Dalakas et al. 2009; Terai and Sakaida 2008; Khurana and Mukhopadhyay 2007) but much remains unknown.
In our study, to first confirm the effects of TNFR1, a TNFR1 antagonist was used. Because TNFR1 antagonists differ from anti-TNF-α antibodies, only block TNFR1, and do not affect other signal pathways such as TNFR2 (Shibata et al. 2008b), administration of a sufficient dose of antagonist in wild-type mice can reproduce a TNFR1 KO mouse system. A dose of 100 μg/body is sufficient to enable inhibition of intrahepatic inflammatory responses and liver fibrosis (Shibata et al. 2008a). Furthermore, the in vivo half-life of an intravenously administered TNFR1 antagonist is 5–6 days (Haak-Frendscho et al. 1994), so the blocking effect of TNFR1, up to the time of tissue sampling after 1 week, as used in the present protocol, probably continued.
In mice administered with the TNFR1 antagonist, intrahepatic macrophage infiltration was suppressed and stellate cell activation was decreased. Studies in liver injury suppression models, by inhibition of TNFα-TNFR1 signals, not only with TNFR1 antagonists but also with anti-TNF-α antibodies, have been reported by several facilities (Sudo et al. 2005; Yamada and Fausto 1998; Shibata et al. 2008a; Kitamura et al. 2002; Bahcecioglu et al. 2008). In the present study as well, in a model of advanced liver fibrosis induced by CCl4 administration in wild-type mice, the TNFR1 antagonist suppressed intrahepatic inflammatory cell infiltration and improved liver fibrosis. However, in this model, with combined GFP-positive BMC infusion, intrahepatic inflammatory cell infiltration and stellate cell activation, which were suppressed by the antagonist, worsened with BMC infusion (attenuation of the suppression effect).
Next, to further clarify this phenomenon, TNFR1 KO mice were used as recipients to investigate whether there were differences in liver repair with BMC infusion based on the presence or absence of TNFR1. In GFP/KO mice, using TNFR1 KO mice as recipients, no liver repair effects with BMC infusion were observed. In TNFR1 KO mice, compared to wild-type mice, although CCl4-induced hepatotoxicity and liver fibrosis were suppressed, GFP-positive BMC infusion showed no synergism in liver repair or anti-hepatotoxic effects.
The importance of an increase in hepatic expression of MMP-9 by BMC infusion in liver fibrosis improvement has been reported by many facilities, including our laboratory (Sakaida et al. 2004, 2008; Higashiyama et al. 2007; Alwayn et al. 2008). In the present study, MMP-9 expression was markedly suppressed in TNFR1 KO mice compared to wild-type mice. MMP-9 expression has been shown to increase by TNF-α signals mediated via TNFR1 (Robinson et al. 2002; Vaday et al. 2001). In TNFR1 KO mice, the fact that this was suppressed is a consistent finding. In our laboratory, GFP-positive BMC infusion in wild-type mice was reported to enhance hepatic expression of MMP-9 (Sakaida et al. 2004). Within this series of cascades, inflammatory signals such as TNFR1 have been shown to be important (Omori et al. 2004). On the other hand, in GFP/KO mice, in which GFP-positive BMCs were infused in TNFR1 KO mice, there was no significant increase in MMP-9 expression and, moreover, the amount of expression compared to wild-type mice was significantly decreased, with no demonstrable effect on liver fibrolysis. With regard to the origin of MMP-9, infused BMCs have received attention but signals mediated by TNFR1 in pre-existing hepatocytes and host-derived hepatic macrophages are also important (Haruyama et al. 2000). These are suppressed in KO mice; furthermore, an increase in cytokine groups, such as increased TGF-β1 expression, which conversely suppresses MMP-9 expression, also has an influence (Vaday et al. 2001).
In TNFR1 KO mice, macrophage infiltration, increased TGF-β1 and enhanced α-SMA activity were noted. But no cells showing co-positivity with GFP staining were confirmed in F4/80 immunohistochemistry. We think the increase in macrophage came from recipient macrophage. In addition, as a result, other inflammatory signals such as TGF-β1 increased, followed by enhanced stellate cell activity and increased liver fibrosis (Parsons et al. 2007; Moreira 2007). DNA-Chip analysis also showed that the macrophage-related gene such as “monocyte-macrophage differentiation, MMD” was activated in TNFR1 KO mice by bone marrow cell infusion (data not shown). Several reports have evaluated the relationship between infiltrating macrophages in the liver with hepatotoxicity, liver fibrosis and liver regeneration (Liu et al. 2010; Duffield et al. 2005; Tacke et al. 2009). An association between intrahepatic macrophages with liver fibrolysis has also been reported (Fallowfield et al. 2007). Based on the present study, after BMC infusion in TNFR1 KO recipients, macrophages infiltrating the liver enhanced the above-mentioned inflammatory responses and increased liver fibrosis. In conclusion, proper TNFR1 signaling might be a key factor to induce the improvement of liver fibrosis by bone marrow cell infusion. Further studies will be needed to understand the precise mechanism.
References
Alwayn I, Verbesey J, Kim S et al (2008) A critical role for matrix metalloproteinases in liver regeneration. J Surg Res 145:192–198
Bahcecioglu I, Koca S, Poyrazoglu O et al (2008) Hepatoprotective effect of infliximab, an anti-TNF-alpha agent, on carbon tetrachloride-induced hepatic fibrosis. Inflammation 31:215–221
Dalakas E, Newsome P, Boyle S, et al. (2009) Bone marrow stem cells contribute to alcohol liver fibrosis in humans. Stem Cells Dev
Drill V (1952) Hepatotoxic agents; mechanism of action and dietary interrelationship. Pharmacol Rev 4:1–42
Duffield J, Forbes S, Constandinou C et al (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65
Fallowfield JA, Mizuno M, Kendall TJ et al (2007) Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 178:5288–5295
Gardner C, Laskin J, Dambach D et al (2003) Exaggerated hepatotoxicity of acetaminophen in mice lacking tumor necrosis factor receptor-1. Potential role of inflammatory mediators. Toxicol Appl Pharmacol 192:119–130
Haak-Frendscho M, Marsters S, Mordenti J et al (1994) Inhibition of TNF by a TNF receptor immunoadhesin. Comparison to an anti-TNF monoclonal antibody. J Immunol 152:1347–1353
Haruyama T, Ajioka I, Akaike T et al (2000) Regulation and significance of hepatocyte-derived matrix metalloproteinases in liver remodeling. Biochem Biophys Res Commun 272:681–686
Higashiyama R, Inagaki Y, Hong Y et al (2007) Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 45:213–222
Horn T, O'Brien T, Schook L, Rutherford M (2000) Acute hepatotoxicant exposure induces TNFR-mediated hepatic injury and cytokine/apoptotic gene expression. Toxicol Sci 54:262–273
Inui T, Nakashima H, Habu Y et al (2005) Neutralization of tumor necrosis factor abrogates hepatic failure induced by alpha-galactosylceramide without attenuating its antitumor effect in aged mice. J Hepatol 43:670–678
Khurana S, Mukhopadhyay A (2007) Characterization of the potential subpopulation of bone marrow cells involved in the repair of injured liver tissue. Stem Cells 25:1439–1447
Kitamura K, Nakamoto Y, Akiyama M et al (2002) Pathogenic roles of tumor necrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liver fibrosis. Lab Invest 82:571–583
Liu C, Tao Q, Sun M et al (2010) Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Invest 90:1805–1816
Moreira R (2007) Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med 131:1728–1734
Omori K, Terai S, Ishikawa T et al (2004) Molecular signature associated with plasticity of bone marrow cell under persistent liver damage by self-organizing-map-based gene expression. FEBS Lett 578:10–20
Parsons CJ, Takashima M, Rippe RA (2007) Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol 22(Suppl 1):S79–S84
Pfeffer K, Matsuyama T, Kunding TM et al (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457–467
Robinson S, Scott K, Balkwill F (2002) Chemokine stimulation of monocyte matrix metalloproteinase-9 requires endogenous TNF-alpha. Eur J Immunol 32:404–412
Sakaida I (2008) Autologous bone marrow cell infusion therapy for liver cirrhosis. J Gastroenterol Hepatol 23:1349–1353
Sakaida I, Terai S, Yamamoto N et al (2004) Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology 40:1304–1311
Shibata H, Yoshioka Y, Ohkawa A et al (2008a) The therapeutic effect of TNFR1-selective antagonistic mutant TNF-alpha in murine hepatitis models. Cytokine 44:229–233
Shibata H, Yoshioka Y, Ohkawa A et al (2008b) Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J Biol Chem 283:998–1007
Simeonova P, Gallucci R, Hulderman T et al (2001) The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol Appl Pharmacol 177:112–120
Sudo K, Yamada Y, Moriwaki H, Saito K et al (2005) Lack of tumor necrosis factor receptor type 1 inhibits liver fibrosis induced by carbon tetrachloride in mice. Cytokine 29:236–244
Tacke F, Luedde T, Trautwein C (2009) Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol 36:4–12
Terai S, Sakaida I (2008) Current status of autologous bone marrow cell infusion therapy for liver cirrhosis patients. Hepatol Res 38:S72–S75
Terai S, Yamamoto N, Omori K et al (2002) A new cell therapy using bone marrow cells to repair damaged liver. J Gastroenterol 37(Suppl 14):162–163
Terai S, Sakaida I, Yamamoto N et al (2003) An in vivo model for monitoring trans-differentiation of bone marrow cells into functional hepatocytes. J Biochem 134:551–558
Terai S, Sakaida I, Nishina H et al (2005) Lesson from the GFP/CCl4 model–translational research project: the development of cell therapy using autologous bone marrow cells in patients with liver cirrhosis. J Hepatobiliary Pancreat Surg 12:203–207
Vaday G, Schor H, Rahat M et al (2001) Transforming growth factor-beta suppresses tumor necrosis factor alpha-induced matrix metalloproteinase-9 expression in monocytes. J Leukoc Biol 69:613–621
Yamada Y, Fausto N (1998) Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am J Pathol 152:1577–1589
Yamada Y, Kirillova I, Peschon J et al (1997) Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 94:1441–1446
Acknowledgements
Ms. Mariko Yamada, Ms. Isako Fujimoto and Ms. Yoko Fukuzumi helped us with the cell culture and immunohistochemical analysis. This study was supported by Grants-in-Aid for scientific research from the Japan Society for the Promotion of Science (No. 22390150) and the Ministry of Health, Labour and Welfare, the Knowledge Cluster Initiative and Japan Science and Technology agency.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License that permits any noncommercial use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hisanaga, T., Terai, S., Iwamoto, T. et al. TNFR1-mediated signaling is important to induce the improvement of liver fibrosis by bone marrow cell infusion. Cell Tissue Res 346, 79–88 (2011). https://doi.org/10.1007/s00441-011-1236-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1236-0