
Abstract
In patients with progressive podocyte disease, such as focal segmental glomerulosclerosis (FSGS) and membranous nephropathy, upregulation of transforming growth factor-ß (TGF-ß) is observed in podocytes. Mechanical pressure or biomechanical strain in podocytopathies may cause overexpression of TGF-ß and angiotensin II (Ang II). Oxidative stress induced by Ang II may activate the latent TGF-ß, which then activates Smads and Ras/extracellular signal-regulated kinase (ERK) signaling pathways in podocytes. Enhanced TGF-ß activity in podocytes may lead to thickening of the glomerular basement membrane (GBM) by overproduction of GBM proteins and impaired GBM degradation in podocyte disease. It may also lead to podocyte apoptosis and detachment from the GBM, and epithelial-mesenchymal transition (EMT) of podocytes, initiating the development of glomerulosclerosis. Furthermore, activated TGF-ß/Smad signaling by podocytes may induce connective tissue growth factor and vascular endothelial growth factor overexpression, which could act as a paracrine effector mechanism on mesangial cells to stimulate mesangial matrix synthesis. In proliferative podocytopathies, such as cellular or collapsing FSGS, TGF-ß-induced ERK activation may play a role in podocyte proliferation, possibly via TGF-ß-induced EMT of podocytes. Collectively, these data bring new mechanistic insights into our understanding of the TGF-ß overexpression by podocytes in progressive podocyte disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glomerular podocytes are the target of injury in most glomerular diseases. Glomerulosclerosis is a hallmark of progressive glomerular damage, and is characterized by the collapse of the glomerular tuft and mesangial matrix accumulation. Transforming growth factor-ß (TGF-ß) is a key regulator of extracellular matrix (ECM) protein synthesis in renal cells. In progressive podocyte diseases, such as focal segmental glomerulosclerosis (FSGS) (Kim et al. 2003), membranous nephropathy (Kim et al. 1999; Shankland et al. 1996), diabetic nephropathy (Wahab et al. 2005), Alport renal disease (Sayers et al. 1999), and Denys-Drash syndrome (Patek et al. 2003), expression of TGF-ß mRNA and/or protein is increased in podocytes. In addition, mesangial matrix expansion occurs frequently in these diseases associated with glomerulosclerosis (Lee and Koh 1993; Lee and Lim 1995; Patek et al. 2003). TGF-ß, which is overexpressed by podocytes, may contribute to the glomerular basement membrane (GBM) thickening and mesangial matrix expansion in progressive podocyte disease (Lee 2011).
Besides ECM protein synthesis, TGF-ß has effects on proliferation, hypertrophy, and apoptosis in renal cells. Podocytes are growth-arrested terminally differentiated cells, and their loss following glomerular injury may contribute to the development of FSGS (Appel et al. 2009; Kriz and Le Hir 2005; Mundel and Shankland 2002). Yet damaged podocytes can proliferate leading to the development of cellular FSGS and even crescents (Bariety et al. 1998, 2005; Barisoni et al. 1999; Ding et al. 2006; Griffin et al. 2005; Moeller et al. 2004; Thorner et al. 2008), although Smeets et al. (2009) argued that proliferating cells in these lesions mainly originate from parietal epithelial cells. In this regard, TGF-ß, which is overexpressed by hyperplastic podocytes, may be involved in podocyte proliferation (Lee and Song 2010).
This review will discuss the recent findings on the mechanisms and consequences of TGF-ß overexpression by podocytes in chronic progressive podocyte disease.
Structure of podocytes
Podocytes are highly differentiated polarized epithelial cells. They have a main cell body which bulges into the urinary space. The long primary (cytoplasmic) processes extend from the cell body and divide into individual foot processes, which adhere to the outer surface of the GBM. Podocytes sustain the structural integrity of the GBM and synthesize most components of the GBM. The foot processes of neighboring podocytes regularly interdigitate, leaving between them filtration slits that are bridged by the slit diaphragm (Pavenstädt et al. 2003). There is a recent report documenting that the filtration slit is a heteroporous structure instead of the previously suggested zipper-like structure (Gagliardini et al. 2010).
Cell-cycle regulation in podocytes
Mature podocytes are terminally differentiated cells and are unable to proliferate in a wide variety of normal or disease conditions. Cell proliferation is controlled at the cell-cycle level by cell-cycle regulatory proteins. Activation of specific cyclin-dependent kinases (CDKs) by a partner cyclin in each phase of the cell cycle leads to cell proliferation. Cyclin–CDK complexes are negatively regulated by the CDK inhibitors p21, p27, and p57. The inability of podocytes to undergo proiferation in most adult diseases is most likely the consequences of a robust expression or even upregulation of the CDK inhibitors with disease progression.
Podocyte injury, however, may induce a loss of the strict cell-cycle control leading to podocyte proliferation. In the cellular lesion of FSGS, damaged podocytes seem to inhibit p27 and p57 protein expression but activate mitotic cell cyclins to promote podocyte proliferation (Wang et al. 2004). Despite prevailing thoughts that podocytes proliferate in human cellular FSGS (Bariety et al. 1998; Barisoni et al. 1999; Chun et al. 2004; Schwartz et al. 1999), Smeets et al. (2009) suggested that the lesion mainly originates from parietal epithelial cells in a murine model of cellular FSGS. In any case, podocyte proliferation is not regeneration but rather represents an ominous sign of the disease progression.
Components of the GBM
In the adult glomerulus, the podocyte continues to add and assemble matrix molecule to the GBM, maintaining a gel-like hydrated meshwork consisting of collagen IV, laminin, fibronectin and proteoglycan. Collagen type IV is the main component of the GBM, which includes six genetically distinct isoforms named α1(IV) to α6(IV). α3–α5(IV) chains originate solely from podocytes in both the developing and mature glomerulus (Abrahamson et al. 2009). In contrast, the α1/α2(IV) collagen network seems to originate mainly from glomerular endothelial cells (Lee et al. 1993) and is localized predominantly at the endothelial aspect of human GBM (Zhang and Lee 1997). Laminin is the most ample glycoprotein in the GBM. Laminin-11 (α5ß2γ1) continues to be deposited in the GBM whereas the fetal laminin chains (α1, α2 and ß1) gradually disappear from the GBM (Miner 2005).
The flexibility and dynamic of mature GBM require a constant turnover. Thus, podocytes not only produce GBM components but also secrete matrix-modifying enzymes, such as matrix metalloproteinase (MMP)-9 (Kang et al. 2010; Ma et al. 2010; McMillan et al. 1996) and MMP-2 (Hayashi et al. 2010; Ma et al. 2010).
Activation of latent TGF-ß
TGF-ß is secreted as latent complexes associated with a latency-associated peptide (LAP). TGF-ß/LAP complex is referred to as the small latent complex. Most cells secrete TGF-ß as part of a large latent complex, in which latent TGF-ß binding protein (LTBP) is linked to the small latent complex. The large latent complex is susceptible to proteolysis, within which LTBP is first cleaved, and then TGF-ß is released from LAP (Koli et al. 2001, 2008).
Under in vitro conditions, latent TGF-ß is activated by heating, acid or alkaline treatment, irradiation, reactive oxygen species (ROS), proteases including plasmin, cathepsin, calpain, MMP-2 and MMP-9, some integrins, or thrombospondin-1 (TSP-1) (for review, see Koli et al. 2001). Several activation mechanisms, such as proteolysis, TSP-1, ROS and some integrins, may exist in vivo (for review, see Lee and Song 2009).
Fibrogenic TGF-ß signaling cascade in podocytes
TGF-ß is known to stimulate the production of type IV collagen, fibronectin and laminin in podocytes (Nakamura et al. 1992), particularly α3(IV) collagen (Iglesias-de la Cruz et al. 2002). TGF-ß signals through sequential activation of two cell surface receptor serine-threonine kinases. Smad2 and Smad3 proteins are activated by TGF-ß receptor kinases. The phosphorylated Smads form complexes with Smad4, and then translocate to the nucleus, transducing signals to the target genes (Miyazono et al. 2000). In podocytes, TGF-ß1 phosphorylates Smad2 (Liu et al. 2005; Schiffer et al. 2004). Expression levels ofTGF-ß1, TGF-ß type II receptor and phosphorylated Smad2/Smad3 are increased in the podocytes covering the lesions of FSGS (Kim et al. 2003).
TGF-ß signaling intermediates controlling cell growth
Smad pathway
TGF-ß causes growth arrest in late G1 of the cell cycle through Smad2 and Smad3. In the nucleus, the Smad3–Smad4 complex can activate transcription of specific genes, such as p15 and p21 (Massague et al. 2000). In injured podocytes, TGF-ß/Smad signaling is activated, which may increase p15 and p21 expression (Lee and Song 2010).
TGF-ß can also induce its downstream inhibitory Smad7, which in turn inhibits Smad2/3 phosphorylation via the negative feedback mechanisms (Massague and Wotton 2000). TGF-ß1 induces Smad7 synthesis in cultured podocytes (Schiffer et al. 2002), in which TGF-ß1 and Smad7 each induce apoptosis (Schiffer et al. 2001).
Ras/mitogen-activated protein kinase (MAPK) pathway
TGF-ß is able to signal via Ras protein, which plays an essential role in eukaryotic cell growth. Ras is required for TGF-ß-mediated activation of extracellular signal-regulated kinase (ERK) (Hartsough et al. 1996). ERK activation mediates primarily mitogenic and/or anti-apoptotic signaling (Johnson and Lapadat 2002) and attenuates the nuclear accumulation of the Smads (Massague and Chen 2000).
In podocytes, TGF-ß activates ERK (Liu et al. 2005; Schiffer et al. 2004) and p38 MAPK (Schiffer et al. 2001, 2004). Activation of p38 MAPK is required for induction of apoptosis by TGF-ß in podocytes (Schiffer et al. 2001).
Phosphatidyl inositol-3-kinase (PI3K) pathway
TGF-ß also rapidly activates anti-apoptotic mediator PI3K/AKT in podocytes, the kinetic profiles of which are similar to ERK (Schiffer et al. 2004). Indeed, PI3K and ERK signals appear to be synergistically activated to mediate anti-apoptotic machinery (Davies et al. 2004). In addition, TGF-ß signaling through PI3K induces the expression of monocyte chemoattractant protein-1 (MCP-1) in podocytes (Lee et al. 2009), and MCP-1 is involved in podocyte proliferation (Burt et al. 2007).
Chronic progressive podocyte diseases with TGF-ß overexpression
Primary FSGS
FSGS is a clinicopathologic entity characterized by nephrotic syndrome and progression to end-stage renal disease. Intrarenal transcription of TGF-ß1 is increased in children with FSGS compared to those with minimal lesion, suggesting that TGF-ß1 gene transcription is indicative of progressive renal damage typical of FSGS (Strehlau et al. 2002). Expression of TGF-ß1 is increased in patients with primary FSGS, particularly in podocytes of sclerotic segments (Kim et al. 2003). Volume density of mesangial matrix is significantly greater in the FSGS patients than in minimal lesion cases. In patients with FSGS, the percent glomerulosclerosis correlates directly with mesangial volume per glomerulus (Lee and Lim 1995).
In rats with subtotal renal ablation, TGF-ß1 is upregulated by podocytes in response to enhanced transcapillary passage of plasma proteins, which precedes the development of glomerulosclerosis (Abbate et al. 2002).
Cellular lesion of FSGS
Up to 46% of patients with primary FSGS show cellular or collapsing lesion (Chun et al. 2004). The cellular lesion of FSGS comprises podocyte proliferation overlying the segmental scar. Collapsing glomerulopathy is characterized by segmental or global wrinkling of the GBM with podocyte proliferation, and may be distinguished from cellular FSGS in view of the different morphologic, etiologic and prognostic implications (Barisoni et al. 2009). Yet the two terms, cellular and collapsing FSGS, had often been used interchangeably or synonymously, because the glomerular pathology of both lesions is basically identical (Bariety et al. 2005; Chun et al. 2004; Schwartz et al. 1999). Evidence from renal allograft or repeat biopsies suggests that cellular or collapsing FSGS may be the early features of FSGS, which may evolve into the classic FSGS pattern in the course of disease progression (Bariety et al. 2001; Chun et al. 2004; Schwartz et al. 1999).
The lesions of FSGS following primary glomerular diseases may represent the nonspecific chronic scarred phase of the disease. Indeed, cellular FSGS is present in IgA nephropathy with TGF-ß overexpression by hyperplastic podocytes (Kim et al. 2002b). In this regard, damaged podocytes in various glomerular diseases can proliferate with expression of TGF-ß in the course of disease progression and exacerbation, leading to the development of cellular FSGS and even crescents (Lee and Song 2010).
Membranous nephropathy
In membranous nephropathy, the GBM is thickened due to the accumulation of GBM material between and around the subepithelial deposits, forming subepithelial projections or spikes. Subepithelial immune deposition, particularly complement membrane attack complex (C5b-9), promotes injury to the glomerular filtration barrier and proteinuria in passive Heymann nephritis (PHN), an experimental model of human membranous nephropathy (Couser and Nangaku 2006). Upregulation of TGF-ß1, α4(IV) and α1(IV) collagens, and laminin ß2 mRNAs by podocytes is shown in patients with membranous nephropathy (Kim et al. 1999). In addition, immunogold densities for polyclonal type IV collagen, α4(IV) collagen, laminin, and fibronectin are increased in the spikes (Zhang and Lee 1997). Expression of TGF-ß2 is also markedly increased in podocytes in experimental membranous nephropathy (Shankland et al. 1996).
Lesions of FSGS are observed in 43% of the membranous nephropathty patients, in whom the degree of mesangial expansion and GBM thickening is significantly greater than the remaining cases without FSGS (Lee and Koh 1993). In PHN, mesangial volume is significantly increased together with GBM thickening (Remuzzi et al. 1999).
Diabetic nephropathy
Thickening of the GBM and expansion of the mesangial matrix are hallmarks of diabetic nephropathy, which occur even within a few years after the onset of type 1 diabetes (Drummond and Mauer 2002). In insulin-dependent diabetes, the collagen α3(IV) through α5(IV) chains, collagen V, laminin, fibronectin, and serum proteins contribute to the thickened GBM (Miner 1999).
In human diabetic nodular glomerulosclerosis, podocytes covering the sclerotic segments show increased expression of TGF-ß1 mRNA and protein (Wahab et al. 2005). Increased expression of glomerular TGF-ß1 is observed mainly in podocytes of diabetic animals (Baba et al. 2005; Okada et al. 2006).
Alport renal disease
Alport syndrome is primary genetic disease of the basement membrane. In the kidney, this disorder is characterized by an absence of collagen α3α4α5(IV) in the GBM, progressive thickening and multilamination of the GBM, proteinuria, and renal failure. Collagen α1/α2(IV), however, is retained throughout the GBM, together with the deposition of the fetal laminin chains α1, α2 and ß1 (Abrahamson et al. 2003; Kashtan et al. 2001). In podocytes of α3(IV) collagen-knockout mice with Alport renal disease, mRNA expression of TGF-ß1, α1(IV) and α2(IV) collagen, fibronectin, and laminin ß1 chain is increased (Sayers et al. 1999). With disease progression, mesangial matrix and cells are increased, followed by the development of glomerulosclerosis (Gregory et al. 1996; Kim et al. 1995).
Denys-Drash syndrome
Wilms’ tumor suppressor gene, WT1, is essential for normal podocyte function. Mutations of the WT1 induce Denys-Drash syndrome (DDS) characterized by early onset nephrotic syndrome and diffuse mesangial matrix expansion. In DDS mice, the development of glomerulosclerosis is preceded by de novo TGF-ß1 expression in podocytes, while TGF-ß1 expression is absent in the mesangium (Patek et al. 2003). A gene mutation in DDS podocytes may not be sufficient to cause TGF-ß overexpression (Jin et al. 1999), but in the presence of a second injury, such as intraglomerular hypertension, TGF-ß seems to be overexpressed by podocytes (Patek et al. 2003). Recently, Sakairi et al. (2011) reported that TGF-ß1 downregulates WT1 expression in podocytes.
Altogether, TGF-ß is overexpressed by podocytes in progressive podocyte diseases, in which thickening of the GBM and expansion of the mesangial matrix are frequently present with the eventual development of glomerulosclerosis (Table 1).
Induction of TGF-ß by glomerular hypertension or biomechanical strain in the diseased glomeruli
Glomerular hemodynamic adaptive changes, including hyperfiltration and hyperperfusion, seem to promote progressive glomerulosclerosis in patients with reduced nephron mass and diabetes (Ziyadeh and Wolf 2008). The less cross-linked and possibly more elastic physical properties of the GBM in some diseased glomeruli may subject the podocytes to elevated biomechanical strain even under normal glomerular blood pressure. As the disease progresses and nephron mass is lost, glomerular hypertension develops, further exacerbating the biomechanical strain and the effector functions influenced by it (Meehan et al. 2009). In the remnant kidney model of glomerular capillary hypertension, TGF-ß1 (Abbate et al. 2002) and Ang II type I receptor (Durvasula et al. 2004) are upregulated by podocytes. In cultured podocytes, albumin load or mechanical strain increases the levels of TGF-ß1 and Ang II, as well as TGF-ß receptors (Abbate et al. 2002; Dessapt et al. 2009; Durvasula et al. 2004).
Together, an increase in glomerular capillary pressure may stimulate Ang II and TGF-ß1 expression in podocytes through mechanical force injury in progressive podocyte diseases (Fig. 1).

Effects of biomechanical strain on the induction of transforming growth factor-ß (TGF-ß) and angiotensin II (Ang II) in podocytes in patients with progressive podocyte disease. Ang II does not directly stimulate TGF-ß production in podocytes, yet activates NADPH oxidase to produce reactive oxygen species, leading to the activation of latent TGF-ß
Effects of Ang II on TGF-ß signaling in podocytes
Ang II is a major active product of the renin angiotensin system (RAS), and may enhance the generation of ROS through the activation of NADPH oxidases in podocytes. Unlike mesangial cells, podocytes do not overexpress TGF-ß1 when exposed to Ang II (Chen et al. 2005). Rather, Ang II-induced oxidative stress may activate latent TGF-ß and, subsequently, the TGF-ß signaling system in podocytes (Lee 2011) (Fig. 1).
Studies in animal models of chronic nephropathies have documented that RAS inhibitors significantly blunt the increased renal TGF-ß production. An angiotensin-converting enzyme (ACE) inhibitor prevents TGF-ß1 overexpression in podocytes and glomerulosclerosis in rats with reduced renal mass (Abbate et al. 2002). It also reduces the TGF-ß1, connective tissue growth factor (CTGF) and ECM protein overexpression in kidneys of Alport mice (Gross et al. 2003, 2004; Gross and Kashtan 2009), and limits mesangial expansion in PHN (Remuzzi et al. 1999). Combined anti-TGF-ß and ACE inhibition therapy abrogates the glomerulosclerosis of diabetic nephropathy in the rat (Benigni et al. 2003). In addition, administration of Ang II type I (AT1) receptor blocker to diabetic rats lowers glomerular expression of TGF-ß1 and vascular endothelial growth factor (VEGF) (Vieitez et al. 2008).
GBM thickening in relation to TGF-ß
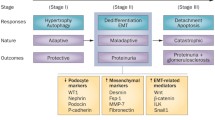
The GBM is significantly thickened in TGF-ß1 transgenic mice as compared with wild-type animals (Krag et al. 2007; Wogensen et al. 1999). Aberrant deposition of fetal laminin α1, α2 and ß1 chains and α1/α2(IV) collagen appears in the GBM of transgenic mice (Chai et al. 2003). In cases with membranous nephropathy and Alport syndrome, there is aberrant expression of α1/α2(IV) collagen, and laminin α1, α2 and ß1 in the thickened GBM (Abrahamson et al. 2003; Cosgrove et al. 2000; Fischer et al. 2000; Kashtan et al. 2001; Zhang and Lee 1997). TGF-ß1 increased α3(IV) collagen expression in cultured mouse podocytes, although it decreased the levels of α1(IV) and α5(IV) mRNA and/or protein (Iglesias-de la Cruz et al. 2002). Collectively, GBM thickening by abnormal deposition of ECM in podocytopathies could be due to the enhanced TGF-ß1 levels in podocytes (Fig. 2).

Hypothetical pathway for transforming growth factor-ß (TGF-ß)-induced glomerular basement membrane (GBM) thickening, mesangial matrix expansion, podocyte loss, podocyte proliferation, and epithelial-mesenchymal transition (EMT) by podocytes in progressive podocyte disease. CTGF connective tissue growth factor, ERK extracellular signal-regulated kinase, ILK integrin-linked kinase, PI3K phosphatidyl inositol-3-kinase, VEGF vascular endothelial growth factor
A further potential mechanism for GBM destruction and thickening involves the action of proteolytic enzymes, such as MMPs. MMP-9 expression is increased in podocytes in experimental membranous nephropathy (McMillan et al. 1996) and in human and experimental diabetic nephropathy (Li et al. 2008). Levels of MMPs are increased in the glomeruli of Alport mice and kidneys of patients with Alport syndrome (Zeisberg et al. 2006). The aberrant collagen α1/α2(IV) network deposited in the GBM contains fewer interchain crosslinks than wild-type GBM, and is more susceptible to proteolytic degradation by endogenously expressed MMPs (Kalluri et al. 1997; Zeisberg et al. 2006). Blocking the activity of specific MMPs has been shown to ameliorate the progression of glomerular pathology (Zeisberg et al. 2006). In cultured podocytes, TGF-ß1 stimulates the production of MMP-9 (Li et al. 2008; Liu et al. 2005), and many of the MMPs (MMP-3, -9, -10, and -14) are induced by mechanical strain (Meehan et al. 2009). Together, increased TGF-ß1 levels in podocytes may induce MMPs, resulting in proteolytic damage and thickening of the GBM in progressive podocyte disease.
Integrins are important cellular receptors for ECM. Type IV collagen receptors, integrin α2, are able to downregulate de novo collagen synthesis as long as the GBM is intact. Loss of integrin α2 in the nonlethal phenotype of the α2-deficient mice results in localized matrix overproduction in the GBM and increased TGF-ß and CTGF expression in the kidney (Girgert et al. 2010). Thus, altered cell-matrix interaction via α2 integrin may contribute to GBM thickening in association with TGF-ß.
Podocyte loss in relation to TGF-ß: the link to glomerulosclerosis
In podocyte diseases, enhanced TGF-ß activity in podocytes may lead to podocyte apoptosis and/or detachment with podocytopenia (Dessapt et al. 2009; Lee and Song 2010; Schiffer et al. 2001; Wolf et al. 2005) (Fig. 2). C5b-9 can induce apoptosis of podocytes in membranous nephropathy (Mundel and Shankland 2002), a process that may involve TGF-ß. Apoptosis is also observed in the crescentic lesion of DDS kidneys (Yang et al. 2004). In TGF-ß1 transgenic mice, podocytes undergo apoptosis at an early stage of glomerulosclerosis with overexpression of Smad7 (Schiffer et al. 2001). In CD2-associated protein-deficient mice, TGF-ß-induced podocyte apoptosis is an early pathomechanism developing FSGS (Schiffer et al. 2004).
Another mechanism of podocyte loss in podocyte diseases may relate to the detachment of podocytes from the GBM. α3ß1 integrin, an adhesion receptor for laminins and type IV collagen isoforms, is expressed primarily on podocytes (Kreidberg and Symons 2000). Downregulation of α3ß1 integrin is observed in the podocytes of patients with primary FSGS (Chen et al. 2006) and diabetes (Chen et al. 2000) associated with podocytopenia. TGF-ß1 suppresses the glomerular expression of α3 integrin in nephrotic rats (Kagami et al. 1993). In cultured podocytes, TGF-ß1 and mechanical stretch significantly reduce the α3ß1 integrin expression linked to decreased podocyte adhesion and increased apoptosis (Dessapt et al. 2009). Thus, TGF-ß1 may reduce podocyte adhesion to the GBM via downregulation of α3ß1 integrin, resulting in podocyte depletion in podocyte diseases (Fig. 2).
Because of their limited proliferative capacity, podocyte detachment from the GBM will lead to cell depletion or drop out into urinary space. The denuded GBM may adhere to the Bowman’s capsule with synechiae formation, initiating the development of FSGS (Kriz and Le Hir 2005). In a rat model of progressive glomerular injury, progressive reduction in the number of podocytes preceded the development of glomerulosclerosis (Macconi et al. 2006).
In summary, TGF-ß may induce podocyte apoptosis and detachment from the GBM in podocyte diseases leading to the development of glomerulosclerosis.
Epithelial-mesenchymal transition (EMT) of podocytes by TGF-ß
TGF-ß1 suppresses the slit-diaphragm-associated protein P-cadherin, zonula occludens-1, and nephrin in cultured podocytes mediated by Snail, a key transcription factor mediating EMT, leading to podocyte dedifferentiation (Li et al. 2008). In patients with FSGS and membranous nephropathy, nephrin mRNA expression by podocytes is significantly decreased as compared with minimal lesion cases (Kim et al. 2002a). EMT may be a potential pathway leading to podocyte dysfunction, proteinuria and glomerulosclerosis. TGF-ß1 induces integrin-linked kinase (ILK) in podocytes, and inhibition of ILK activity ameliorates podocyte Snail induction and EMT (Kang et al. 2010). Expression of ILK is also increased in cellular crescents of experimental glomerulonephritis (GN) (Shimizu et al. 2006), suggesting that TGF-ß1-induced ILK may lead to EMT of podocytes, contributing to the formation of cellular crescents. Together, more severe and/or longer podocyte injury induced by TGF-ß may lead to EMT via upregulation of ILK in progressive podocyte disease (Kang et al. 2010; Liu 2010) (Fig. 2).
Mesangial matrix expansion in podocyte diseases via activation of TGF-ß signaling
In patients with FSGS, membranous nephropathy and DDS, mesangial matrix expansion is frequently present in association with the development of glomerulosclerosis (Lee and Koh 1993; Lee and Lim 1995; Patek et al. 2003). Podocyte-specific injury in transgenic mice induced mesangial expansion and glomerulosclerosis (Matsusaka et al. 2005).
In Smad3-knockout diabetic mice, mesangial matrix expansion is prevented (Wang et al. 2007), as shown in the anti-TGF-ß-treated or TGF-ß type II receptor-deficient diabetic mice (Chen et al. 2003; Kim et al. 2004; Ziyadeh et al. 2000). Indeed, activation of the TGF-ß/Smad signaling in podocytes from the diseased glomeruli appears to lead to overproduction of ECM in the mesangial areas resulting in the formation of glomerulosclerosis (Lee and Song 2009; Kim et al. 2003; Patek et al. 2003) (Table 1).
Paracrine effector mechanism of CTGF and VEGF for TGF-ß to act on mesangial cells in podocytopathies
The podocyte TGF-ß, the active form of which has a very short half-life in plasma (Coffey et al. 1987), is unlikely to traverse the GBM to promote sclerosis in the adjacent mesangium. In this regard, some TGF-ß-induced humoral factors produced by podocytes seem to have fibrogenic effects on mesangial cells (Lee and Song 2009).
CTGF is a major autocrine growth factor induced by TGF-ß. TGF-ß1 induces CTGF mRNA and protein expression in podocytes (Ito et al. 2001). Expression of CTGF mRNA and/or protein in the mesangium and podocytes is upregulated in human chronic glomerular disease (Ito et al. 1998; Wahab et al. 2005). CTGF is increased particularly in the glomeruli of patients with mesangial matrix expansion (Suzuki et al. 2003). Furthermore, induction of diabetes in podocyte-specific CTGF-transgenic mice results in an increased mesangial CTGF expression with more severe mesangial expansion than diabetic wild-type mice (Yokoi et al. 2008). Treatment with the CTGF antisense oligonucleotides in diabetic mice significantly reduced mesangial matrix expansion as compared with those receiving mismatch oligonucleotides (Guha et al. 2007).
VEGF is a potent angiogenic molecule and is detected predominantly in podocytes (Bailey et al. 1999; Wendt et al. 2003). Yet glomeruli are not sites of angiogenesis, possibly because podocytes mainly express VEGF165b protein, which inhibits VEGF165-mediated angiogenesis (Cui et al. 2004). TGF-ß1 stimulates VEGF expression in podocytes (Iglesias-de la Cruz et al. 2002). VEGF may play an important role in TGF-ß1-induced glomerular fibrosis (Chen et al. 2004, 2005). Indeed, anti-VEGF attenuates mesangial matrix expansion in diabetic mice (Flyvbjerg et al. 2002).
Receptors for VEGF-A include VEGFR-1, VEGFR-2 and neuropilin-1. The dominant production of VEGF-A by podocytes and the localization of VEGFR-2 on glomerular endothelial cells suggest that VEGF-A moves across the GBM, opposing the ultrafiltration gradient to move water and solutes from the capillaries into the Bowman’s space (Satchell et al. 2006). In fact, about one-third of VEGF secreted from podocytes would reach the capillary lumen and accumulate there, supporting the view that VEGF can move against the flow of glomerular filtration (Katavetin and Katavetin 2008). It is not clear whether this is also the case for CTGF, yet the experiments performed by Yokoi et al. (2008) support that possibility.
To sum up, TGF-ß-induced CTGF and VEGF secretion by podocytes may act as a paracrine regulatory mechanism, necessary for the mesangial matrix accumulation (Fig. 2).
Role of TGF-ß/Ras/ERK signaling in podocyte proliferation in proliferative podocytopathies
In cellular crescents, TGF-ß overexpression (Shimizu et al. 2006) and ERK activation (Masaki et al. 2004) are observed. TGF-ß signaling appears to play a central role in the development of crescentic GN by inducing the activation of ERK (Song et al. 2007).
ERK activation is shown in hyperplastic podocytes from the human immunodeficiency virus (HIV)-associated nephropathy and/or idiopathic collapsing FSGS patients (He et al. 2004). Cyclin D1 is a key down-stream target of ERKs (Ammit and Panettieri 2001). In podocytes of HIV-transgenic mice, cyclin D1 protein is increased and the increase coincides with entry into the cell cycle (Pettermann et al. 2004).
Stabilization of hypoxia-inducible factor (HIF) in mice by selective deletion of the von Hippel-Lindau gene from podocytes leads to the development of crescentic GN with expression of HIF target gene Cxcr4 in podocytes (Ding et al. 2006). Cxcr4 is both required and sufficient for proliferation of podocytes in vivo. Overexpression of HIF-2 is also shown in hyperplastic podocytes from the patients with HIV-associated nephropathy and HIV-1-transgenic mice (Korgaonkar et al. 2008). Activation of HIF-1 occurs via PI3K and MAPK signaling pathways (Hellwig-Bürgel et al. 2005). Together, Ras/ERK and PI3K activation in podocytes could stimulate cyclin D1 expression resulting in podocyte proliferation in proliferative podocytopathies.
In cultured podocytes, TGF-ß1 does not stimulate cell proliferation (Xavier et al. 2009). Yet it may induce the proliferation of injured podocytes possibly via TGF-ß-induced EMT, since TGF-ß stimulates proliferation of mesenchymal-type cells (Fukuda et al. 2009). In this regard, TGF-ß1-induced Ras/ERK/PI3K activation may play a role in podocyte proliferation in proliferative podocytopathies (Fig. 2).
Conclusions
Biomechanical strain in progressive podocyte diseases may upregulate Ang II and TGF-ß expression in podocytes. Oxidative stress induced by Ang II may activate the latent TGF-ß in podocytes. Enhanced TGF-ß activity by podocytes may induce GBM thickening by overproduction of ECM proteins and impaired GBM degradation in podocyte diseases. It may also lead to podocyte apoptosis and detachment from the GBM together with EMT, initiating the development of glomerulosclerosis. Furthermore, activated TGF-ß/Smad signaling by podocytes may induce CTGF and VEGF overexpression, which may act as a paracrine effector mechanism on mesangial cells to stimulate mesangial matrix synthesis. In proliferative podocytopathies, TGF-ß-induced ERK activation may play a role in podocyte proliferation, possibly via TGF-ß-induced EMT of podocytes. Together, this review provides new mechanistic insights into the TGF-ß overexpression by podocytes in progressive podocyte disease. Better understanding of the activation of TGF-ß signaling by podocytes and its downstream effectors may provide novel tools for the prevention of glomerulosclerosis.
References
Abbate M, Zoja C, Morigi M, Rottoli D, Angioletti S, Tomasoni S, Zanchi C, Longaretti L, Donadelli R, Remuzzi G (2002) Transforming growth factor-ß1 is up-regulated by podocytes in response to excess intraglomerular passage of proteins: a central pathway in progressive glomerulosclerosis. Am J Pathol 161:2179–2193
Abrahamson D, Prettyman A, Robert B, St John PL (2003) Laminin-1 reexpression in Alport mouse glomerular basement membranes. Kidney Int 63:826–834
Abrahamson DR, Hudson BG, Stroganova L, Borza D-B, St. John PL (2009) Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol 20:1471–1479
Ammit AI, Panettieri RA Jr (2001) The circle of life: cell cycle regulation in airway smooth muscle. J Appl Physiol 91:1431–1437
Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ (2009) Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20:333–343
Baba M, Wada J, Eguchi J, Hashimoto I, Okada T, Yasuhara A, Shikata K, Kanwar YS, Makino H (2005) Galectin-9 inhibits glomerular hypertrophy in db/db diabetic mice via cell-cycle-dependent mechanisms. J Am Soc Nephrol 16:3222–3234
Bailey E, Bottomley MJ, Westwell S, Pringle JH, Furness PN, Feehally J, Brenchley PEC, Harper SJ (1999) Vascular endothelial growth factor mRNA expression in minimal change, membranous, and diabetic nephropathy demonstrated by nonisotopic in situ hybridization. J Clin Pathol 52:735–738
Bariety J, Nochy D, Mandet C, Jacquot C, Glotz D, Meyrier A (1998) Podocytes undergo phenotypic changes and express macrophagic-associated markers in idiopathic collapsing glomerulopathy. Kidney Int 53:918–928
Bariety J, Bruneval P, Hill G, Irinopoulou T, Mandet C, Meyrier A (2001) Posttransplantation relapse of FSGS is characterized by glomerular epithelial cell transdifferentiation. J Am Soc Nephrol 12:261–274
Bariety J, Bruneval P, Meyrier A, Mandet C, Hill G, Jacquot C (2005) Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int 68:1109–1119
Barisoni L, Kriz W, Mundel P, D’Agati V (1999) The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10:51–61
Barisoni L, Schnaper HW, Kopp JB (2009) Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med 133:201–216
Benigni A, Zoja C, Corna D, Zatelli C, Conti S, Campana M, Gagliardini E, Rottoli D, Zanchi C, Abbate M, Ledbetter S, Remuzzi G (2003) Add-on anti-TGF-ß antibody to ACE inhibitor arrests progressive diabetic nephropathy in the rat. J Am Soc Nephrol 14:1816–1824
Burt D, Salvidio G, Tarabra E, Barutta F, Pinach S, Dentelli P, Camussi G, Perin PC, Gruden G (2007) The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am J Pathol 171:1789–1799
Chai Q, Krag S, Miner JH, Nyengaard JR, Chai S, Wogensen L (2003) TGF-ß1 induces aberrant laminin chain and collagen type IV isotype expression in the glomerular basement membrane. Nephron Exp Nephrol 94:e123–e136
Chen CA, Hwang JC, Guh JY, Chang JM, Lai YH, Chen HC (2006) Reduced podocyte expression of α3ß1 integrins and podocyte depletion in patients with primary focal segmental glomerulosclerosis and chronic PAN-treated rats. J Lab Clin Med 147:74–82
Chen HC, Chen CA, Guh JY, Chang JM, Shin SJ, Lai YH (2000) Altering expression of α3ß1 integrin on podocytes of human and rats with diabetes. Life Sci 67:2345–2353
Chen S, Iglesias-de la Cruz MC, Jim B, Hong SW, Isono M, Ziyadeh FN (2003) Reversibility of established diabetic glomerulopathy by anti-TGF-ß antibodies in db/db mice. Biochem Biophys Res Commun 300:16–22
Chen S, Kasama Y, Lee JS, Jim B, Marin M, Ziyadeh FN (2004) Podocyte-derived vascular endothelial growth factor mediates the stimulation of α3(IV) collagen production by transforming growth factor-ß1 in mouse podocytes. Diabetes 53:2939–2949
Chen S, Lee JS, Iglesias-de la Cruz MC, Wang A, Izquierdo-Lahuerta A, Gandhi NK, Danesh FR, Wolf G, Ziyadeh FN (2005) Angiotensin II stimulates α3(IV) collagen production in mouse podocytes via TGF-ß and VEGF signaling: implications for diabetic glomerulopathy. Nephrol Dial Transplant 20:1320–1328
Chun MJ, Kobert SM, Schwartz MM, Lewis EJ (2004) Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol 15:2169–2177
Coffey RJ Jr, Kost LJ, Lyons RM, Moses HL, LaRusso NE (1987) Hepatic processing of transforming growth factor ß in the rat. Uptake, metabolism, and biliary excretion. J Clin Invest 80:750–757
Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R (2000) Integrin α1ß1 and transforming growth factor-ß1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol 157:1649–1659
Couser WG, Nangaku M (2006) Cellular and molecular biology of membranous nephropathy. J Nephrol 19:699–705
Cui T-G, Foster RR, Saleem M, Mathieson PW, Gillatt DA, Bates DO, Harper SJ (2004) Differentiated human podocytes endogenously express an inhibitory isoform of vascular endothelial growth factor (VEGF165b) mRNA and protein. Am J Physiol Renal Physiol 286:F767–F773
Davies CC, Mason J, Wakelam MJO, Young LS, Eliopoulos AG (2004) Inhibition of phosphatidylinositol 3-kinase- and ERK MAPK-regulated protein synthesis reveals the pro-apoptotic properties of CD40 ligation in carcinoma cells. J Biol Chem 279:1010–1019
Dessapt C, Baradez MO, Hayward A, Dei Cas A, Thomas SM, Viverti G, Gnudi L (2009) Mechanical forces and TGFß1 reduce podocyte adhesion through α3ß1 integrin downregulation. Nephrol Dial Transplant 24:2645–2655
Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, Marsden PA, Pippin J, Shankland S, Rastaldi MP, Cohen CD, Kretzler M, Quaggin SE (2006) Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med 12:1081–1087
Drummond K, Mauer M (2002) The early natural history of nephropathy in type 1 diabetes. II. Early renal structural changes in type 1 diabetes. Diabetes 51:1580–1587
Durvasula RV, Pettermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ (2004) Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 65:30–39
Fischer E, Mougenot B, Callard P, Ronco P, Rossert J (2000) Abnormal expression of glomerular basement membrane laminins in membranous glomerulonephritis. Nephrol Dial Transplant 15:1956–1964
Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R (2002) Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 51:3090–3094
Fukuda N, Tahira Y, Matsuda H, Matsumoto K (2009) Transforming growth factor-ß as a treatment target in renal diseases. J Nephrol 22:708–715
Gagliardini E, Conti S, Benigni A, Remuzzi G, Remuzzi A (2010) Imaging of the porous ultrastructure of the glomerular epithelial filtration slit. J Am Soc Nephrol 21:2080–2089
Girgert R, Martin M, Kruegel J, Miosge N, Temme J, Eckes B, Müller G-A, Gross O (2010) Integrin α2-deficient mice provide insights into specific functions of collagen receptors in the kidney. Fibrogenesis Tissue Repair 3:19
Gregory MC, Terreros DA, Barker DF, Fain PN, Denison JC, Atkin CL (1996) Alport syndrome-clinical phenotypes, incidence, and pathology. Contrib Nephrol 117:1–28
Griffin S, Krofft RD, Pippin JW, Shankland SJ (2005) Limitation of podocyte proliferation improves renal function in experimental crescentic glomerulonephritis. Kidney Int 67:977–986
Gross O, Kashtan CE (2009) Treatment of Alport syndrome: beyond animal models. Kidney Int 76:599–603
Gross O, Beirowski B, Koepke M-L, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M (2003) Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int 63:438–446
Gross O, Schulze-Lohoff E, Koepke M-L, Beirowski B, Addicks K, Bloch W, Smyth N, Weber M (2004) Antifibrotic, nephroprotective potential of ACE-inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol Dial Transplant 19:1716–1723
Guha M, Xu Z-G, Tung D, Lanting L, Natarajan R (2007) Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J 21:3355–3368
Hartsough MT, Frey RS, Zipfel PA, Buard A, Cook SJ, McCormick F, Mulder KM (1996) Altered transforming growth factor-ß signaling in epithelial cells when ras activation is blocked. J Biol Chem 271:22368–22375
Hayashi K, Sasamura H, Ishiguro K, Sakamaki Y, Azegami T, Itoh H (2010) Regression of glomerulosclerosis in response to transient treatment with angiotensin II blockers is attenuated by blockade of matrix metalloproteinase-2. Kidney Int 78:69–78
He JC, Husain M, Sunamoto M, D’Agati VD, Klotman ME, Iyengar R, Klotman PE (2004) Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1, 2 pathways. J Clin Invest 114:643–651
Hellwig-Bürgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W (2005) Hypoxia-inducible factor-1: a novel transcription factor in immune reactions. J Interferon Cytokine Res 25:297–310
Iglesias-de la Cruz MC, Ziyadeh FN, Isono M, Kouahou M, Han DC, Kalluri R, Mundel P, Chen S (2002) Effects of high glucose and TGF-ß1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int 62:901–913
Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R (1998) Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 53:853–861
Ito Y, Goldschmeding R, Bende RJ, Claessen N, Chand MA, Kleij L, Rabelink TJ, Weening JJ, Aten J (2001) Kinectics of connective tissue growth factor expression during experimental proliferative glomerulonephritis. J Am Soc Nephrol 12:472–484
Jin DK, Kang SI, Kim SL, Bang EH, Hwang HZ, Tadokoro K, Yamada M, Kohsaka T (1999) Transcriptional regulation of PDGF-A and TGF-ß by +KTS WT1 deletion mutants and a mutant mimicking Denys-Drash syndrome. Ren Fail 21:685–694
Johnson GL, Lapadat R (2002) Mitogen-activated kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911–1912
Kagami S, Border WA, Ruoslahti E, Noble NA (1993) Coordinated expression of ß1 integrins and TGF-ß-induced matrix proteins in glomerulonephritis. Lab Invest 69:68–76
Kalluri R, Shield CF III, Todd P, Hudson BG, Nielson EG (1997) Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membrane to endoproteolysis. J Clin Invest 99:2470–2478
Kang YS, Li Y, Dai C, Kiss LP, Wu C, Liu Y (2010) Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int 78:363–373
Kashtan C, Kim Y, Lees GE, Thorner PS, Virtanen I, Miner JH (2001) Abnormal glomerular basement membrane laminins in murine, canine and human Alport syndrome: aberrant laminin α2 deposition is species independent. J Am Soc Nephrol 12:252–260
Katavetin P, Katavetin P (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 359:205–206
Kim BK, Hong HK, Kim JH, Lee HS (2002a) Differential expression of nephrin in acquired human proteinuric diseases. Am J Kidney Dis 40:964–973
Kim HW, Moon KC, Park SY, Hong HK, Lee HS (2002b) Differential expression of platelet-derived growth factor and transforming growth factor-ß in relation to progression of IgA nephropathy. Nephrology 7:S131–S139
Kim HW, Kim BC, Song CY, Kim JH, Hong HK, Lee HS (2004) Heterozygous mice for TGF-ßIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int 66:1859–1865
Kim JH, Kim BK, Moon KC, Hong HK, Lee HS (2003) Activation of the TGF-ß/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int 64:1715–1721
Kim KH, Kim Y, Gubler MC, Steffes MW, Lane PH, Kashtan CE, Crosson JT, Mauer SM (1995) Structural-functional relationships in Alport syndrome. J Am Soc Nephrol 5:1659–1668
Kim TS, Kim JY, Hong HK, Lee HS (1999) mRNA expression of glomerular basement membrane proteins and TGF-ß1 in human membranous nephropathy. J Pathol 189:425–430
Koli K, Saharinen J, Hyytiäinen M, Penttinen C, Keski-Oja J (2001) Latency, activation, and binding proteins of TGF-ß. Microsc Res Tech 52:354–362
Koli K, Myllärniemi M, Keski-Oja J, Kinnula VL (2008) Transforming growth factor-ß activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid Redox Signal 10:333–342
Korgaonkar SN, Feng X, Ross MD, Lu T-C, D’Agati V, Iyengar R, Klotman PE, He JC (2008) HIV-1 upregulates VEGF in podocytes. J Am Soc Nephrol 19:877–883
Krag S, Nyengaard JR, Wogensen L (2007) Combined effects of moderately elevated blood glucose and locally produced TGF-ß1 on glomerular morphololgy and renal collagen production. Nephrol Dial Transplant 22:2485–2496
Kreidberg JA, Symons JM (2000) Integrins in kidney development, function, and disease. Am J Physiol Renal Physiol 279:F233–F242
Kriz W, Le Hir M (2005) Pathways to nephron loss starting from glomerular diseases – insights from animal models. Kidney Int 67:404–419
Lee EY, Chung CH, Khoury CC, Yeo TK, Pyagay PE, Wang A, Chen S (2009) The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-ß, increases podocyte motility and albumin permeability. Am J Physiol Renal Physiol 297:F85–F94
Lee HS (2011) Pathogenic role of TGF-ß in the progression of podocyte diseases. Histol Histopathol 26:107–116
Lee HS, Koh HI (1993) Nature of progressive glomerulosclerosis in human membranous nephropathy. Clin Nephrol 39:7–16
Lee HS, Lim SD (1995) The significance of glomerular hypertrophy in focal segmental glomerulosclerosis. Clin Nephrol 44:349–355
Lee HS, Song CY (2009) Differential role of mesangial cells and podocytes in TGF-ß-induced mesangial matrix synthesis in chronic glomerular disease. Histol Histopathol 24:901–908
Lee HS, Song CY (2010) Effects of TGF-ß on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press Res 33:24–29
Lee LK, Pollock AS, Lovett DH (1993) Asymmetric origins of the mature glomerular basement membrane. J Cell Physiol 157:169–177
Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y (2008) Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol 172:299–308
Liu S, Liang Y, Huang H, Wang L, Li Y, Li J, Li X, Wang H (2005) ERK-dependent signaling pathway and transcriptional factor Ets-1 regulate matrix metalloproteinase-9 production in transforming growth factor-ß1 stimulated glomerular podocytes. Cell Physiol Biochem 16:207–216
Liu Y (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 21:212–222
Ma H, Togawa A, Soda K, Zhang J, Lee S, Ma M, Yu Z, Ardito T, Czyzyk J, Diggs L, Joly D, Hatakeyama S, Kawahara E, Holzman L, Guan JL, Ishibe S (2010) Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J Am Soc Nephrol 21:1145–1156
Macconi D, Bonomelli M, Benigni A, Plati T, Sangalli F, Longaretti L, Conti S, Kawachi H, Hill P, Remuzzi G, Remuzzi A (2006) Pathophysiologic implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol 168:42–54
Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Nikolic-Paterson DJ (2004) Activation of the extracellular-signal regulated protein kinase pathway in human glomerulopathies. J Am Soc Nephrol 15:1835–1843
Massague J, Chen YG (2000) Controlling TGF-ß signaling. Genes Dev 14:627–644
Massague J, Wotton D (2000) Transcriptional control by the TGF-ß/Smad signaling system. EMBO J 19:1745–1759
Massague J, Blain SW, Lo RS (2000) TGF-ß signaling in growth control, cancer, and heritable disorders. Cell 103:295–309
Matsusaka T, Xin J, Niwa S, Kobayashi K, Akatsuka A, Hashizume H, Wang QC, Pastan I, Fogo AB, Ichikawa I (2005) Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol 16:1013–1023
McMillan JI, Riordan JW, Couser WG, Pollock AS, Lovett DH (1996) Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J Clin Invest 97:1094–1101
Meehan DT, Delimont D, Cheung L, Zallocchi M, Sansom SC, Holzclaw JD, Rao V, Cosgrove D (2009) Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int 76:968–976
Miner JH (1999) Renal basement membrane components. Kidney Int 56:2016–2024
Miner JH (2005) Building the glomerulus: a matricentric view. J Am Soc Nephrol 16:857–861
Miyazono K, ten Duke P, Heldin C-H (2000) TGF-ß signaling by Smad proteins. Adv Immunol 75:115–157
Moeller MJ, Soofi A, Hartmann I, Le Hir M, Wiggins R, Kriz W, Holzman LB (2004) Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J Am Soc Nephrol 15:61–67
Mundel P, Shankland SJ (2002) Podocyte biology and response to injury. J Am Soc Nephrol 13:3005–3015
Nakamura T, Miller D, Ruoslahti E, Border WA (1992) Production of extracellular matrix by glomerular epithelial cells is regulated by transforming growth factor-ß1. Kidney Int 41:1213–1221
Okada T, Wada J, Hida K, Eguchi J, Hashimoto I, Baba M, Yasuhara A, Shikata K, Makino H (2006) Thiazolidinediones ameliorate diabetic nephropathy via cell cycle-dependent mechanisms. Diabetes 55:1666–1677
Patek CE, Fleming S, Miles CG, Bellamy CO, Ladomery M, Spraggon L, Mullins J, Hastie ND, Hooper ML (2003) Murine Denys-Drash syndrome: evidence of podocyte dedifferentiation and systemic mediation of glomerulosclerosis. Hum Mol Genet 12:2379–2394
Pavenstädt H, Kriz W, Kretzler M (2003) Cell biology of the glomerular podocyte. Physiol Rev 83:253–307
Pettermann A, Hiromura K, Pippin J, Blonski M, Couser WG, Kopp J, Mundel P, Shankland SJ (2004) Differential expression of D-type cyclins in podocytes in vitro and in vivo. Am J Pathol 164:1417–1424
Remuzzi A, Monaci N, Bonassi ME, Corna D, Zoja C, Mohammed EI, Remuzzi G (1999) Angiotensin-converting enzyme inhibition prevents loss of glomerular hydraulic permeability in passive Heymann nephritis. Lab Invest 79:1501–1510
Sakairi T, Abe Y, Kopp JB (2011) TGF-ß1 reduces Wilms’ tumor suppressor gene expression in podocytes. Nephrol Dial Transplant (in press)
Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O’Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW (2006) Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int 69:1633–1640
Sayers R, Kalluri R, Rodgers KD, Shield CF III, Meehan DT, Cosgrove D (1999) Role for transforming growth factor-ß1 in Alport renal disease progression. Kidney Int 56:1662–1673
Schiffer M, Bitzer M, Roberts ISD, Kopp JB, ten Dijke P, Mundel P, Böttinger EP (2001) Apoptosis in podocytes induced by TGF-ß and Smad7. J Clin Invest 108:807–816
Schiffer M, Schiffer LE, Gupta A, Shaw AS, Roberts ISD, Mundel P, Böttinger EP (2002) Inhibitory Smads and TGF-ß signaling in glomerular cells. J Am Soc Nephrol 13:2657–2666
Schiffer M, Mundel P, Shaw AS, Böttinger EP (2004) A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-ß-induced apoptosis. J Biol Chem 279:37004–37012
Schwartz MM, Evans J, Bain R, Korbet SM (1999) Focal segmental glomerulosclerosis: prognostic implications of the cellular lesions. J Am Soc Nephrol 10:1900–1907
Shankland SJ, Pippin J, Pichler RH, Gordon KL, Friedman S, Gold LI, Johnson RJ, Couser WG (1996) Differential expression of transforming growth factor-ß isoforms and receptors in experimental membranous nephropathy. Kidney Int 50:116–124
Shimizu M, Kondo S, Urushihara M, Takamatsu M, Kanemoto K, Nagata M, Kagami S (2006) Role of integrin-linked kinase in epithelial mesenchymal transition in crescent formation of experimental glomerulonephritis. Nephrol Dial Transplant 21:2380–2390
Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JFM, Floege J, Moeller MJ (2009) Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 20:2604–2615
Song CY, Kim BC, Hong HK, Lee HS (2007) TGF-ß type II receptor deficiency prevents renal injury via decrease in ERK activity in crescentic glomerulonephritis. Kidney Int 71:882–888
Strehlau J, Schachter AD, Pavlakis M, Singh A, Tejani A, Strom TB (2002) Activated intrarenal transcription of CTL-effectors and TGF-ß1 in children with focal segmental glomerulosclerosis. Kidney Int 61:90–95
Suzuki D, Toyoda M, Umezono T, Uehara G, Zhang S-Y, Sakai T, Nishina M, Suga T, Endoh M, Yagame M, Sakai H (2003) Glomerular expression of connective tissue growth factor mRNA in various renal diseases. Nephrology 8:92–97
Thorner PS, Ho M, Eremina V, Sado Y, Quaggin S (2008) Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol 19:495–502
Vieitez P, Gomez O, Uceda ER, Vera ME, Molina-Holgado E (2008) Systemic and local effects of angiotensin II blockade in experimental diabetic nephropathy. J Renin Angiotensin Aldosterone Syst 9:96–102
Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, Schaefer R, Mason RM (2005) Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 48:2650–2660
Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S (2007) Interference with TGF-ß signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol 293:F1657–F1665
Wang S, Kim JH, Moon KC, Hong HK, Lee HS (2004) Cell cycle mechanisms involved in podocyte proliferation in cellular lesion of focal segmental glomerulosclerosis. Am J Kidney Dis 43:19–27
Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D’Agati VD, Schmidt AM (2003) RAGE drives the develolpment of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162:1123–1137
Wogensen L, Nielsen CB, Hjorth P, Rasmussen LM, Nielsen AH, Gross K, Sarvetnick N, Ledet T (1999) Under control of the Ren-1c promoter, locally produced transforming growth factor-ß1 induces accumulation of glomerular extracellular matrix in transgenic mice. Diabetes 48:182–192
Wolf G, Chen S, Ziyadeh FN (2005) From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1626–1634
Xavier S, Niranjan T, Krick S, Zhang T, Ju W, Shaw AS, Schiffer M, Böttinger EP (2009) TßR1 independently activates Smad- and CD2AP-dependent pathways in podocytes. J Am Soc Nephrol 20:2127–2137
Yang AH, Chen JY, Chen BF (2004) The dysregulated glomerular cell growth in Denys-Drash syndrome. Virchows Arch 445:305–314
Yokoi H, Mukoyama M, Mori K, Kasahara M, Suganami T, Sawai K, Yoshioka T, Saito Y, Ogawa Y, Kuwabara T, Sugawara A, Nakao K (2008) Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int 73:446–455
Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF III, Werb Z, Kalluri R (2006) Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med 3:e100
Zhang YZ, Lee HS (1997) Quantitative changes in the glomerular basement membrane components in human membranous nephropathy. J Pathol 183:8–15
Ziyadeh FN, Wolf G (2008) Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev 4:39–45
Ziyadeh FN, Hoffman BB, Han DC, Iglesias-de la Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-ß antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97:8015–8020
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lee, H.S. Mechanisms and consequences of TGF-ß overexpression by podocytes in progressive podocyte disease. Cell Tissue Res 347, 129–140 (2012). https://doi.org/10.1007/s00441-011-1169-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1169-7