
Abstract
The Long Life Family Study (LLFS) enrolled 4953 participants in 539 pedigrees displaying exceptional longevity. To identify genetic mechanisms that affect cardiovascular risks in the LLFS population, we developed a multi-omics integration pipeline and applied it to 11 traits associated with cardiovascular risks. Using our pipeline, we aggregated gene-level statistics from rare-variant analysis, GWAS, and gene expression-trait association by Correlated Meta-Analysis (CMA). Across all traits, CMA identified 64 significant genes after Bonferroni correction (p ≤ 2.8 × 10–7), 29 of which replicated in the Framingham Heart Study (FHS) cohort. Notably, 20 of the 29 replicated genes do not have a previously known trait-associated variant in the GWAS Catalog within 50 kb. Thirteen modules in Protein–Protein Interaction (PPI) networks are significantly enriched in genes with low meta-analysis p-values for at least one trait, three of which are replicated in the FHS cohort. The functional annotation of genes in these modules showed a significant over-representation of trait-related biological processes including sterol transport, protein-lipid complex remodeling, and immune response regulation. Among major findings, our results suggest a role of triglyceride-associated and mast-cell functional genes FCER1A, MS4A2, GATA2, HDC, and HRH4 in atherosclerosis risks. Our findings also suggest that lower expression of ATG2A, a gene we found to be associated with BMI, may be both a cause and consequence of obesity. Finally, our results suggest that ENPP3 may play an intermediary role in triglyceride-induced inflammation. Our pipeline is freely available and implemented in the Nextflow workflow language, making it easily runnable on any compute platform (https://nf-co.re/omicsgenetraitassociation).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Long Life Family Study (LLFS) is a multi-center, longitudinal family study that enrolled families enriched for exceptional longevity to discover genetic, behavioral, and environmental factors contributing to healthy aging and long life. LLFS enrolled 4953 participants in 539 families, including probands, offspring, grandchildren, and spouses. Participants are primarily of European ancestry (99%). The data it has generated include microarray genotypes, whole genome sequences, gene expression from whole blood, and biomarkers of health and aging. Healthy aging and long life are heritable traits (Brooks-Wilson 2013; Perls and Terry 2003) and the LLFS cohort is exceptional in both (Wojczynski et al. 2022). The LLFS probands and offspring were less likely to have diabetes, chronic pulmonary disease, and peripheral artery disease than participants in the Cardiovascular Health Study (CHS) and Framingham Heart Study (FHS) in the same age group (Newman et al. 2011). High-density cholesterol levels were higher, and pulse pressure and triglycerides were lower in the LLFS cohort than in CHS and FHS (Newman et al. 2011). In this work, we look for genes that affect cardiovascular health in the LLFS population and the biological processes through which they work. We focus on 11 traits associated with cardiovascular risks spanning four categories: pulmonary (forced expiratory volume, forced vital capacity, and the ratio of the two), lipids (high-density lipoprotein, low-density lipoprotein, triglycerides, total cholesterol), anthropometric (BMI, BMI-adjusted waist), and cardiovascular (pulse, ankle-brachial index) (Barter et al. 2007; Miller et al. 2011; Flint et al. 2010; Ramalho and Shah 2021; Korhonen et al. 2009).
Genome-wide association studies (GWAS) have identified many loci for cardiovascular-related domains, including pulmonary function (Wyss et al. 2018; Shrine et al. 2019), lipids (Graham et al. 2021), obesity and body fat distribution (Locke et al. 2015; Shungin et al. 2015), and blood pressure and ankle-brachial index (Murabito et al. 2012; Evangelou et al. 2018). However, GWAS has some well-known limitations. Testing millions of individual variants requires extremely small p-values and hence very large cohorts. When GWAS does identify statistically significant variants, it is difficult to determine which are causal and which are merely tagging a causal variant in linkage disequilibrium (Donnelly 2008). If a causal non-coding variant is found, it is often unclear which gene it acts through. We set out to address these challenges. To reduce the multiple testing burden, we aggregated variant-level GWAS p-values for common variants (minor allele frequency (MAF) > 5%) to obtain gene-level p-values, used a SKAT-based (Wu et al. 2011) analysis method (Li et al. 2020) to calculate gene-level p-values for rare variants (MAF < 5%) (Wu et al. 2011; Li et al. 2020; Auer and Lettre 2015) and calculated gene-level p-values for association between measured gene expression levels and traits (Transcriptome-wide association studies (TWAS); throughout this paper, TWAS refers to association with measured gene expression levels, not predicted levels). We combined the gene-level p-values from TWAS, GWAS, and rare variant analysis (RVA) using a meta-analysis approach that accounts for expected correlations among these (Province and Borecki 2013). Aggregating variants to the gene level creates strong evidence about which gene is implicated, which can be difficult when focusing on individual variants. By incorporating evidence from TWAS, we reduce the chance that a significant gene is simply tagging a nearby gene in LD (since LD does not induce correlation in the expression levels of nearby genes). TWAS alone has a different problem—gene expression may be associated with a trait because it is affected by the trait, rather than affecting the trait, or by a confounding factor affecting both trait and gene expression. However, when there is supporting evidence from genetic variants, that is less likely.
To further investigate a gene’s potential for causally affecting a trait, we started with the hypothesis that, among genes statistically associated with a trait, the most likely to be causal are those that interact with other statistically associated genes (1) through a common molecular system, and (2) serve a common biological function. To identify genes that interact with other statistically associated genes through a common molecular system, we searched for network modules in protein–protein interaction networks whose genes, as a group, are significantly enriched for genes with suggestive/significant p-values from correlated meta-analysis. To identify common biological functions served by module genes, we looked for GO biological process terms significantly overrepresented among genes in the enriched modules (Wang et al. 2017).
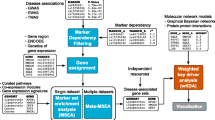
This paper makes three contributions. First, it presents 64 genes from meta-analysis that are genome-wide significant for at least one of 11 traits associated with cardiovascular risks, of which 29 are replicated in the FHS population. Second, it presents 13 protein–protein interaction network modules significantly enriched in genes with comparatively low meta-analysis p-values for at least one of the traits. Three such modules are replicated in the FHS population. Third, it presents software that researchers can use to conduct similar analyses. The software is packaged as a Nextflow pipeline, which containerizes each analysis step, simplifies the maintenance of software dependencies, and enables deployment across multiple computing environments, including cloud computing provided by data repositories (Ewels et al. 2020). The software pipeline and complete documentation can be found at https://nf-co.re/omicsgenetraitassociation/. Figure 1 depicts the pipeline.

Pipeline diagram. A Inputs to GWAS, TWAS, and RVA. B GWAS output is fed into PASCAL, which calculates gene-level p-values. TWAS outputs gene-level p-values. STAAR splits variants into ten functional categories and outputs 10 p-values per gene. C Correlated meta-analysis (CMA) is run 10 times. Each run uses outputs from PASCAL and TWAS together with one variant category of STAAR, outputting 10 p-values. D For each gene, the minimum p-value from 10 CMA runs is fed into module enrichment analysis, which is also performed by PASCAL. PASCAL outputs enriched modules and their p-values. Gene ontology over-representation analysis identifies biological processes with significant over-representation among genes in each module
Material and methods
Participants
The recruitment procedure, eligibility criteria, and enrollment of the LLFS participants have been previously described (Newman et al. 2011). We used data from the first clinical exam, which started in 2006 and recruited 4953 individuals from 539 families. Across 11 studied traits, the participants ranged from n = 2528 to 4166 for GWAS, n = 595 to 1200 for gene expression level–trait association, and n = 2528 to 4166 for rare-variant analysis. Descriptive statistics for all the traits and covariates can be found in File S3. The number of participants in each analysis depended on the number of participants with data for the trait, microarray genotypes for GWAS, whole genome sequencing for rare-variant analysis, and RNA-Seq for TWAS.
Cardiovascular-related traits
We used trait values from the first clinical exam. BMI was calculated as weight (kg)/height (m)2, and waist as the average of three abdominal circumference measurements in cm. Pulse was calculated as the average of three measurements of the sitting pulse. FEV1 and FVC were measured in a portable spirometer (EasyOne, NDD Medical Technologies, Andover, MA), as previously reported (Newman et al. 2011). High-density lipoprotein (HDL), low-density lipoprotein (LDL), triglyceride (TG), and total cholesterol (TC) were assessed and analyzed by the LLFS central laboratory based at the University of Minnesota, as previously reported (Newman et al. 2011). Participants were excluded if fasting < 8 h for LDL, TG, and TC. Ankle-brachial index (ABI) was derived as the average of the right and left ankle-arm blood pressure ratios. We excluded participants with non-compressible arteries (ABI > = 1.4). For all analyses, each of the traits was adjusted for age, sex, field center, and square of the age. Waist and pulse were additionally adjusted for BMI. FEV1, FVC, and FEV1/FVC were also adjusted for height and smoking. LDL and TC were also adjusted for statin use, and TG was log-transformed. All traits were also adjusted for the top 10 genetic principal components stepwise. The trait residuals unexplained by the covariates were used for GWAS, TWAS, and RVA. After covariate adjustments, all trait residuals were inverse normal transformed.
GWAS and gene level aggregation of GWAS results
GWAS SNP-chip data for the LLFS participants were produced using Illumina 2.5 million HumanOmni array. Genotypes were called using Bead Studio. SNPs were removed if their call rate was less than 98%, if their allele frequency in the LLFS population was < 1% or > 99%, if they had an allelic mismatch with 1000 Genomes Project (1000Gp3v5), or if they displayed excess heterozygosity relative to Hardy Weinburg Equilibrium (p < 1E–6). A single-SNP association test was done for all SNPs passing the quality filter by using a linear mixed model. Family relatedness was accounted for using a pedigree-based kinship matrix, and an additive genetic model was assumed. The SNP-level summary statistics from GWAS for SNPs with minor allele frequency > = 5% were input to PASCAL (Lamparter et al. 2016). The SNPs were assigned to a gene if they lay within 50 kb of the gene body. PASCAL uses the sum of chi-squared statistics to calculate a gene-level p-value. Document S1 describes the GWAS and gene level aggregation process for the FHS population.
Gene-expression to trait association (TWAS)
The RNA extraction and sequencing were carried out by the McDonnell Genome Institute at Washington University (MGI). Total RNA was extracted from PAXgene™ Blood RNA tubes using the Qiagen PreAnalytiX PAXgene Blood miRNA Kit (Qiagen, Valencia, CA). The Qiagen QIAcube extraction robot performed the extraction according to the company's protocol. The RNA-Seq data were processed with the nf-core/RNASeq pipeline version 3.3 using STAR/RSEM and otherwise default settings (https://zenodo.org/records/5146005). RNA-Seq on whole blood samples from the LLFS participants in the first clinical visit was used for the analysis. Genes with less than three counts per million in greater than 98.5% of samples were filtered out from the analysis. Samples with greater than 8% of reads in intergenic regions were also filtered out. The resulting set were transformed using DESeq2’s (Love et al. 2014) variance stabilizing transform (VST) function. The VST transformed gene expression levels were adjusted for base covariates: age, age squared, sex, field center, percent of reads mapping to intergenic sequence, and the counts of red blood cells, white blood cells, platelets, monocytes, and neutrophils. The gene expression level was also adjusted stepwise for the RNA-seq batch and the top 10 principal components of gene expression. For each trait, the adjusted gene expression residuals were used as predictors, and the trait residuals after adjustment were used as a response variable in a linear mixed model implemented in MMAP (O’Connell 2017). A kinship matrix generated by MMAP from the LLFS pedigree was used to account for family relatedness. For traits with genomic inflation factor (GIF) > 1.1, the p-values were adjusted using BACON (van Iterson et al. 2017). The same RNA-Seq processing steps were implemented for replication in the FHS dataset.
Rare-variant analysis (RVA) using STAAR
LLFS Whole Genome Sequence (WGS) was produced by MGI using 150 bp Illumina reads. Variant calls with read depth less than 20 or greater than 300 were set to missing. Variants with call rate < 90% and those with excess heterozygosity (p < 1E–6) were excluded from the analysis. Missing genotype calls in the LLFS cohort were filled in using the call with the highest phred-scale likelihood from GATK. Bi-allelic SNVs with MAF < 5% and passing the above quality filters were input to STAAR (Li et al. 2020) for variant set association tests using SKAT (Wu et al. 2011). We also employed burden testing (Morgenthaler and Thilly 2007; Li and Leal 2008; Madsen and Browning 2009; Morris and Zeggini 2010) and Aggregate Cauchy Association Test (ACAT) (Liu et al. 2019; Liu and Xie 2020) as implemented in the STAAR framework. However, the resulting p-value distributions from these tests displayed a U-shaped pattern, deviating from the expected uniform distribution under the null hypothesis so we did not use them.
For each gene, variants are split into 10 functional categories, and an omnibus association test is performed for each category of each gene separately, resulting in 10 p-values per gene. Within each category, variants are weighted by functional annotations from the FAVOR database (Zhou et al. 2023), which is curated by the TOPMed Consortium. The annotations describe various aspects of variant function, and include conservation scores, epigenetic measurements, protein function scores and metrics for relative pathogenicity. The individual annotation scores for each variant are integrated into a single variant-specific weight by calculating the first principal component of the standardized annotation scores, and then transforming the principal component into a Phred-scale score (Li et al. 2020). The 10 functional categories include synonymous, missense, putative loss of function (plof), promoter CAGE, promoter DHS, enhancer CAGE, enhancer DHS, upstream, downstream, and untranslated region (UTR) (Li et al. 2020; Zhou et al. 2023). A minimum of 2 variants is required in a category to perform a SKAT test. Document S1 describes the WGS data processing steps for the FHS population.
Correlated meta-analysis (CMA)
CMA (Province and Borecki 2013) combined gene p-values from GWAS (after aggregation by PASCAL), TWAS, and RVA while preventing Type I errors by accounting for dependencies between individual analyses under the null as described (Province and Borecki 2013; Feitosa et al. 2022, 2024). CMA uses the multivariate normal distribution to integrate GWAS, TWAS, and RVA p-values. The individual p-values are first converted to corresponding z-scores which are assumed to have a multivariate normal distribution. The combined p-values are calculated using the standardized z-scores. This calculation is based on the sum of the z-scores and its standard deviation. The standard deviation is determined based on an empirically estimated covariance matrix, which represents the correlation between the individual studies. GWAS, TWAS, and RVA were performed on overlapping individuals from LLFS’s first clinical visit. Furthermore, genetic variants affect gene expression. Therefore, each pair of inputs to CMA may be correlated. Since STAAR outputs 10 p-values per gene, one for each category, we ran CMA 10 times resulting in 10 p-values per gene. To account for multiple testing using Bonferroni correction and identify significant genes following CMA, the number of tests is aggregated across all 10 CMA runs. If a gene meets the Bonferroni significance threshold for any category after adjusting for the total number of tests, it is considered significant after CMA. The significance threshold used for GWAS, TWAS, RVA, and CMA for LLFS can be found in Table S4.
Module enrichment analysis and Gene Ontology (GO) over-representation analysis
We started with modules (highly connected subnetworks) from two protein–protein interaction (PPI) networks, the STRING functional PPI network (Szklarczyk et al. 2015) and the InWeb physical PPI network (Li et al. 2017) which were identified by the best-performing module identification methods from a DREAM challenge (Choobdar et al. 2019): random walk algorithm R1 for STRING and modularity optimization algorithm M2 for InWeb. These modules and the gene-level p-values were input to PASCAL’s module enrichment algorithm (Lamparter et al. 2016). Genes with p-values from only one of the three sources (GWAS, TWAS, or RVA) were removed from the modules. The module enrichment p-values from PASCAL were corrected for the total number of modules tested using Bonferroni correction. GO over-representation analysis was done on the set of genes in each enriched module by using WebGestaltR package (version: 0.4.6) with the following configuration: (organism: hsapiens, method: ORA, enrichDatabase: GO Biological Process, FDRMethod: BH, FDRThreshold = 0.05) (Wang et al. 2017). The affinity propagation feature in WebGestaltR was used to eliminate GO biological processes with highly overlapping member genes.
Framingham Heart Study (FHS) replication
FHS is a multi-generational study to identify genetic and environmental factors affecting cardiovascular and other diseases (Splansky et al. 2007; Kannel et al. 1979). We used the data on the FHS participants from grandchildren and offspring spouse generation who attended examination 2 for replication purposes (Splansky et al. 2007; Kannel et al. 1979). Across 11 studied traits, the participants ranged from n = 2512–3341 for GWAS, n = 1080–1380 for TWAS, and n = 921–1233 for rare-variant analysis. Descriptive statistics for all the traits and covariates can be found in File S3. We use the same pipeline described in Fig. 1 to replicate the LLFS results in the FHS population. Replication analysis was done on genes that were significant in LLFS by CMA or by any of the CMA inputs: TWAS, GWAS, or RVA. For each trait, a gene is replicated if it meets the Bonferroni significance threshold, which is adjusted for the number of genes that were significant in the LLFS population in GWAS and TWAS, or for the number of gene-category pairs of significant genes in CMA and STAAR. A module is replicated if it is significantly enriched after applying Bonferroni correction based on the number of significantly enriched modules across all traits in the LLFS population.
GWAS Catalog search
We used the NHGRI-EBI GWAS Catalog database (version: v1.0.2-associations_e109) (Sollis et al. 2023) to check if the gene-trait associations with suggestive/significant signals from GWAS, TWAS, RVA, and CMA have a previously known trait-associated genome-wide significant variant within 50 kb of the gene body. Genes matching this criterion are designated as “previously associated in GWAS Catalog” throughout the paper. It is important to note that the presence of previously known trait-associated variants in a 50 kb region around the trait-associated gene’s body does not necessarily establish a causal role for the gene on the trait. However, we use this broad criterion to ensure that we classify genes with any hint of previous implication as “previously associated,” minimizing the risk of incorrectly classifying them as novel findings.
Results
Figure 1 shows the flowchart of the multi-omics integration pipeline we used to identify genes and biological processes affecting 11 cardiovascular-related traits. We implemented it as a Nextflow workflow, which containerizes each process (Ewels et al. 2020). This greatly simplifies the maintenance of software dependencies and enables easy deployment across various computing environments. The complete pipeline documentation can be found at https://nf-co.re/omicsgenetraitassociation/.
Gene-level aggregation of GWAS
File S3 shows the characteristics of study participants for covariates and 11 cardiovascular-related traits for GWAS, TWAS, and RVA. We employed GWAS on all traits. Genomic inflation factors (GIFs) for all traits (Table S1) indicate no systematic inflation, technical bias, or population stratification. We then aggregated GWAS summary statistics to the gene level using PASCAL (Lamparter et al. 2016). After aggregation, GIFs range from 1.07 to 1.21 (GIFs: Table S2). After Bonferroni correction, 30 gene-trait associations were genome-wide significant across five traits – low-density lipoprotein (LDL, 9 genes), total cholesterol (TC, 7 genes), High-density lipoprotein (HDL, 4 genes), waist (1 gene), and triglycerides (TG, 9 genes). 26 of these gene-trait pairs are previously associated in GWAS Catalog (Sollis et al. 2023). We replicated 9/30 genome-wide significant gene-trait associations in the FHS population using aggregated GWAS (Table S3). One of those genes for TG, BUD13-DT (p = 2.25 × 10−8), is not previously associated in the GWAS Catalog. However, BUD13-DT is a divergent transcript and shares 82 of the 83 genetic variants that are aggregated to the gene level with BUD13. BUD13 is previously associated in the GWAS Catalog.
Transcriptome-wide association study (TWAS)
We conducted TWAS on the 11 traits. After using BACON (van Iterson et al. 2017) to correct for inflation when GIF > 1.10, the GIFs range from 1.01 to 1.16 (GIFs: Table S2). After Bonferroni correction, 77 gene-trait associations were genome-wide significant across five traits—TC (5), BMI (21), HDL (21), FVC (1), and TG (29) (Table S5). 9 of the 77 genes are previously associated in the GWAS Catalog and 57 of the 77 associations were replicated in the FHS population (Tables S5, S10). The direction of the effect matches between the LLFS and the FHS populations for all 57 FHS-replicated associations. Of 21 genes significant for HDL, 18 were also significant for TG. Consistent with the inverse relationship between HDL and TG traits, the HDL and TG β-values had opposite signs for all 18 genes. 50 of the 57 replicated gene-trait associations are not previously associated with the corresponding traits in the GWAS Catalog (Sollis et al. 2023). Among 9 genes previously associated in GWAS Catalog, 7 were replicated in FHS – HCAR3 (BMI p = 1.12 × 10−8), HCK (BMI p = 3.91 × 10−7), SLC45A3 (HDL p = 1.42 × 10−15), LINC02458 (HDL p = 7.17 × 10−15), ABCG1 (HDL p = 1.98 × 10−9), ENPP3 (HDL p = 9.68 × 10−9), and ABCA1 (TG p = 5.22 × 10−9). These previously associated genes have GWAS Catalog reported trait-associated variant(s) within the 50 kb region of the gene body. The genome-wide significance of these previously associated genes after TWAS and their replication in the FHS population suggests a potential role as mediators linking trait-associated variants to traits. For 3 of the 7 replicated TWAS genes with trait-associated variants within 50 Kb, the variants are assigned to other, closer genes in the GWAS Catalog. Our analysis suggests the following reassignments: rs3747973 from NUCKS1 and Metazoa_SRP to SLC45A3, rs2245133 from MED23 to ENPP3, rs2245611 from HCAR1 and DENR to HCAR3, and rs6489191 from KNTC1 and HCAR2 to HCAR3 (Sollis et al. 2023).
Rare variant analysis (RVA)
We applied RVA on the same 11 traits using the STAAR package (Li et al. 2020). STAAR splits variants into 10 functional categories and performs 10 variant set tests for each gene. The GIFs of all 110 STAAR-category-trait combinations (10 categories by 11 traits) range from 0.77 to 1.20 (GIFs: Table S2). After Bonferroni correction, we identified 194 unique gene-trait associations at the genome-wide significant levels for ABI (13), LDL (2), TC (1), BMI (16), FEV1 (24), FEV1/FVC (8), HDL (7), FVC (49), TG (2), pulse (3), and waist (69) (Table S6). 22/194 are previously associated genes, and 5/194 associations were replicated in the FHS population (Table S6, Table S10). OR52A1 (p = 2.56 × 10−8) is genome-wide significant for ABI, was replicated in FHS, and not previously associated in the GWAS Catalog (Sollis et al. 2023). The low replication rate in FHS may stem from LLFS’s unique cohort enriched for exceptional longevity. Rare variants unique to LLFS could drive the phenotype under study. One example is NABP1 (p = 2.12 × 10−8), which is genome-wide significant for HDL in the upstream category. Two rare variants upstream of this gene (rs10931513, rs10177406) have minor allele counts of 5 and are present in the same group of individuals. GWAS on these variants for HDL shows that each one individually has a suggestive p-value (betas = 2.02, p < 8 × 10−6). Other genes that are significant in LLFS but not replicated in FHS warrant further investigation.
Correlated meta-analysis (CMA)
After aggregating gene-level p-values from PASCAL, TWAS, and each category of RVA (Province and Borecki 2013), we obtained 10 category-specific p-values for each gene. The GIFs across all 110 category-trait combinations ranged from 0.98 to 1.29 (GIFs: Table S2). After Bonferroni correction, we identified 64 significant genes across 9 traits—LDL (6), TC (1), BMI (4), FEV1 (3), FEV1/FVC (1), HDL (15), FVC (8), TG (23), and waist (3), of which 21 are previously associated genes (Table S7). Twenty-nine of 64 gene-trait associations were replicated in the FHS population, of which 9 genes are previously associated in the GWAS Catalog (Sollis et al. 2023) (Table S7, Table S10). We identified 20 genes that were not previously associated and were replicated in the FHS population (Table 1), including 14 for TG, 5 for HDL, and 1 for BMI. 7 of 29 FHS-replicated genes have significant or suggestive evidence from more than one source. The remaining genes are CMA-significant due to a highly significant p-value in one analysis. File S4 shows the distribution of p-values for one of the traits (TC) after GWAS, TWAS, RVA, and CMA.
CMA accounts for the correlation between p-values from PASCAL, TWAS, and RVA outputs, but we observed minimal correlation between the TWAS output and PASCAL or RVA output. The absolute median tetrachoric correlation across all trait-category pairs ranges from 0.001 to 0.009 for [TWAS, RVA] and from 0.004 to 0.017 for [TWAS, PASCAL]. The absolute median correlation across trait-category pairs is slightly higher between RVA and PASCAL, ranging from 0.02 to 0.05 (Table S8).
The p-values from CMA were inputted to PASCAL’s module enrichment analysis method, which identifies modules whose genes, as a group, have significantly lower p-values than would be expected by chance after Benjamini-Hochberg correction (Lamparter et al. 2016). We used modules from the InWeb (physical) (Li et al. 2017) and STRING (functional) (Szklarczyk et al. 2015) protein–protein interaction (PPI) networks. We identified 13 enriched modules across 7 traits – LDL (1), TC (1), BMI (3), FEV1/FVC (1), HDL (2), FVC (2), and TG (3) (Table 2). Of the 13, 6 modules are in the physical network and 7 are in the functional one. Three of 13 modules were replicated in FHS (see Methods). One replicated STRING module (cma-STRING-104) is enriched for genes with suggestive/significant p-values for both HDL and for TG. It contains three genome-wide significant TG genes – APOA5 (p = 1.56 × 10−15), APOC3 (p = 2.08 × 10−11), and APOA4 (p = 1.56 × 10−15), of which APOA5 and APOC3 were replicated in FHS. The module also contains two genome-wide significant HDL genes—APOC3 (p = 1.64 × 10−8) and CETP (p = 1.61 × 10−18), of which CETP was replicated (Fig. 2a). Functional annotation of genes in this module showed a significant over-representation of multiple biological processes. Notably, the top 5 most significant biological processes are lipid-related – sterol transport, glycerolipid catabolism, protein-lipid complex remodeling, phospholipid transport, and plasma lipoprotein particle assembly (Table S9). Another FHS-replicated STRING module for TG contains two replicated genes for TG that are not previously associated in the GWAS Catalog – MS4A2 (p = 3.14 × 10−34) and FCER1A (p = 1.71 × 10−18) along with SYK (not suggestive or significant) and CBL (suggestive). However, the expression level of MS4A2 and FCER1A has been associated with TG in two previous studies (Dekkers et al. 2023; Inouye et al. 2010). The over-represented biological processes for this module include immune-related processes such as positive regulation of immune response, mast-cell degranulation, and T-cell-activation (Fig. 2b). The most significant biological processes for 7 other significantly enriched lipid-related modules – LDL (1), TC (1), HDL (1), BMI (3), and TG (1), are primarily lipid-related or immune-related processes (Table 2). The genes in enriched modules and over-represented biological processes for enriched modules can be found in File S1.

Sub-modules within enriched modules. Genes with P < 10−4 are annotated as suggestive. Module enrichment analysis identifies trait-related genes missed by association analysis. A Module cma-STRING-104 is enriched for both TG and HDL. APOB is not significant for TG but directly interacts with genes with suggestive or significant p-values for TG. APOB and MSR1 participate in macrophage-derived foam cell differentiation with two genome-wide significant genes (CETP and ABCG1). B SYK is not genome-wide significant after CMA. SYK interacts with significant/suggestive genes for TG, FCER1A and MS4A2, and participates in mast-cell degranulation
Discussion
The value of correlated meta-analysis
Using correlated meta-analysis (CMA), we developed a strategy to integrate evidence from GWAS, TWAS, and rare-variant analysis (RVA). The summary statistics from all four analyses can be found in File S2. After CMA, we identified 64 genome-wide significant genes across 9 cardiovascular-related traits. Of 29 CMA-significant and FHS-replicated genes, TOMM40 for TC, ATG2A for BMI, and APOC3 for TG are more significant after CMA than in PASCAL, TWAS, and RVA alone (Table 1). TOMM40 for LDL, CETP for HDL, and APOC3, LPL, and SLC45A3 for TG have suggestive or genome-wide significant p-values from more than one input analysis (Table 1). The rest of the genes have strong evidence from TWAS. Modestly significant genes with support from only one of GWAS, TWAS, or RVA were filtered out by CMA.
Prior work using meta-analysis has primarily focused on integrating evidence from multiple GWAS on different cohorts (Thompson et al. 2011; Zeng et al. 2022; Kavvoura and Ioannidis 2008) or identifying shared/pleiotropic genetic effects across multiple traits (Feitosa 2022; Feistosa et al.2024; Zhang et al. 2014). Our approach integrates evidence from GWAS, TWAS, and RVA. Wang et al. (2020) performed a meta-analysis similar to ours by integrating methylation data (EWAS), TWAS, and GWAS gene-level statistics. However, the TWAS and EWAS statistics came from a single cohort without replication and the meta-analysis also did not account for the correlation between EWAS, TWAS, and GWAS statistics from the same cohort. These are expected to be correlated because genetic variants and methylation both affect gene expression (Wang et al. 2020).
Within the individual analyses, the replication rate in the FHS population for RVA (5/194) is lower than for GWAS (9/30) or TWAS (57/77). The low replication rate for RVA is expected because the FHS sub-population with whole genome sequencing and measured traits and covariates is much smaller than the LLFS cohort (File S3). For the most significant functional categories of the 194 RVA-significant genes in LLFS, only ~ 4% (59 / 1530) of the variants are present and analyzed in FHS.
ATG2A and its link to obesity
Autophagy-Related Protein 2 Homolog A (ATG2A) is a genome-wide significant gene (p = 6.11 × 10−8) for BMI after CMA and is replicated in FHS (p = 1.5 × 10−4). ATG2A is not previously associated in the GWAS Catalog. The ATG2A protein plays a role in autophagosome formation, regulation of lipid droplet morphology, and lipid-droplet dispersion during autophagy (Velikkakath et al. 2012; Valverde et al. 2019). In vitro experiments have shown that low expression of ATG2A can disrupt normal autophagy. Velikkakath et al. reported that silencing ATG2A/ATG2B via siRNA in HeLa cells leads to the aggregation of large lipid droplets. ATG2A/ATG2B double knockout in HEK293 cells led to an incomplete autophagy process (Valverde et al. 2019). The association between the expression level of ATG2A and BMI is genome-wide significant with a negative beta coefficient, which means higher expression of ATG2A is associated with lower BMI (beta = -0.7, P = 1.8 × 10−7). Obesity increases the inhibition of autophagy (Namkoong et al. 2018), so the lower expression of ATG2A, a pro-autophagy gene, may be a consequence of high BMI. On the other hand, increasing autophagy by genetic or pharmacological mechanisms protects mice from obesity and sequelae such as insulin resistance and fatty liver (Namkoong et al. 2018), so higher expression of ATG2A may protect against obesity and consequent cardiovascular risk (Ortega et al. 2016). Indeed, autophagy regulation has been proposed as a therapy to reduce the risk of obesity-associated cardiovascular diseases (Castaneda et al. 2019). Thus, ATG2A may participate in a positive feedback loop in which lower expression of ATG2A is both a cause and a consequence of obesity.
ENPP3: a potential mediator of TG-induced inflammation
ENPP3 is genome-wide significant (p = 4.52 × 10−13) for TG after CMA and replicated in FHS (p = 1.41 × 10−11). It encodes ecto-nucleotide pyrophosphatase-phosphodiesterase 3, one of several enzymes that hydrolyze extracellular ATP and thereby tamp down chronic inflammation (Virgilio et al. 2020). Extracellular ATP is a powerful “alarmin” that signals cellular damage, activates immune cells, and causes inflammation (Virgilio et al. 2020), a key element of atherosclerosis (Peng and Wu 2022). Enpp3–/– mouse cells exhibit lower ATP hydrolysis compared to WT cells (Tsai et al. 2015). The expression level of ENPP3 is genome-wide significant for TG with a negative coefficient (beta = – 1.04, P = 1.75 × 10−26) and the direction of effect is the same in FHS. The expression level of ENPP3 has been previously associated with TG in two prior studies with the same direction of effect (Dekkers et al. 2023; Inouye et al. 2010). A 2023 bidirectional Mendelian randomization study found a significant effect of TG on ENPP3 expression but no evidence for reverse causation (Dekkers et al. 2023). Thus, reduced expression of ENPP3 and subsequent increase in extracellular ATP concentration may be one of the mechanisms by which high TG induces inflammation and promotes atherosclerosis (Peng and Wu 2022).
Role of mast cell functional genes in atherosclerosis risks
FCER1A, MS4A2, GATA2, HDC, and HRH4 are genome-wide significant for TG in LLFS CMA and replicated in FHS. None of them has a TG-associated variant within 50 kb in the GWAS Catalog. All 5 genes play a role in either mast-cell activation, mast-cell proliferation, or secretion of pro-inflammatory markers (Turner and Kinet 1999; Galli and Tsai 2012; Wu et al. 2022; Shi et al. 2015; Krystel-Whittemore et al. 2015; Wang et al. 2011; Hofstra et al. 2003; Li et al. 2015). Active mast cells affect atherosclerosis risks. In mice, local activation of adventitial mast cells during atherogenesis increases plaque size, macrophage apoptosis, vascular leakage, and intraplaque hemorrhage (Bot et al. 2007). FCER1A and HDC have also been experimentally linked to atherosclerosis. Homozygous deletion of FCER1A reduced atherosclerosis in Apoe–/– mice (Shi et al. 2015). Similarly, Hdc–/– mice exhibited reduced atherosclerotic lesions in an Apoe–/– background (Wang et al. 2011). Using Mendelian randomization, Dekkers et al. found a significant effect of TG on all 5 genes but no evidence for reverse causation (Dekkers et al. 2023). This is consistent with the fact that elevated TG causes inflammation (Bernardi et al. 2018) and that these genes are pro-inflammatory. Surprisingly, the association between TG and the expression of these pro-inflammatory genes is not positive, as would be expected based on the inflammatory effect of high TG levels. In fact, we see a significant negative association for all 5 genes (Table 1). The expression level of these genes has been previously associated with TG in two different studies with the same direction of effect (Dekkers et al. 2023; Inouye et al. 2010). One explanation for the lack of a positive correlation between TG and these pro-inflammatory genes is that we have measured gene expression in whole blood, whereas inflammation associated with atherosclerosis occurs in plaques. However, the existence of such a strong and consistent negative correlation between TG and the expression of these genes is an intriguing mystery that demands further experimental investigation.
Module and GO enrichment analysis identified an additional gene, SYK, which may affect atherosclerosis risk via a similar mechanism. SYK lies in an enriched TG-module (cma-STRING-193) in which genes involved in mast-cell degranulation are significantly overrepresented (Fig. 2b). SYK directly interacts with FCER1A and MS4A2, two genes with known mast cell functions (Turner and Kinet 1999; Galli and Tsai 2012; Wu et al. 2022). An experimental study has shown that treating mice with SYK inhibitors significantly reduced atherosclerosis lesions in atherosclerosis-prone mice (Hilgendorf et al. 2011). This suggests that combined module and GO analysis can identify important trait-related genes that are not genome-wide significant.
A flexible and easy-to-use pipeline
We introduced a multi-omics integration pipeline (Fig. 1) and provided a NextFlow implementation that is easily run on a wide variety of platforms, from laptops to large compute clusters (https://nf-co.re/omicsgenetraitassociation/). While we used our multi-omics integration approach to aggregate signals from GWAS, TWAS, or RVA, our pipeline can also take in gene-level summary statistics from epigenome-wide association studies (EWAS) (Rakyan et al. 2011). While we used modules from the STRING and InWeb PPI networks, our pipeline can also take in modules from other networks, such as those linking transcription factors to their target genes. This flexibility makes the pipeline useful for a wide range of research problems.
In the future, we plan to enhance the pipeline to address some limitations. Currently, we aggregate variant-level statistics from GWAS to the gene level based on proximity to the gene. This could be improved by aggregating variants in the genes’ regulatory regions using publicly available resources on regulatory regions and their target genes (Gao and Qian 2020; Fishilevich et al. 2017). The current meta-analysis approach does not offer weighted aggregation of different input sources. This could be improved by providing options to use various meta-analysis tools. The current implementation offers only STAAR for rare variant analysis. This could be improved by offering other, less complex options.
Data and code availability
The code used to do all association analyses is available at https://nf-co.re/omicsgenetraitassociation/. The summary results from GWAS, TWAS, RVA, and CMA on all 11 traits for the LLFS cohort are available in File S2. The input datasets have not been deposited in public repositories due to data use constraints.
References
Auer PL, Lettre G (2015) Rare variant association studies: considerations, challenges and opportunities. Genome Med 7(1):16
Barter P et al (2007) HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 357(13):1301–1310
Bernardi S et al. The complex interplay between lipids, immune system and interleukins in cardio-metabolic diseases. Int J Mol Sci. 2018;19(12).
Bot I et al (2007) Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 115(19):2516–2525
Brooks-Wilson AR (2013) Genetics of healthy aging and longevity. Hum Genet 132(12):1323–1338
Castaneda D et al (2019) Targeting autophagy in obesity-associated heart disease. Obesity (Silver Spring) 27(7):1050–1058
Choobdar S et al (2019) Assessment of network module identification across complex diseases. Nat Methods 16(9):843–852
Dekkers KF et al. Lipid-induced transcriptomic changes in blood link to lipid metabolism and allergic response. Nature Commun. 2023;14(1).
Di Virgilio F, Sarti AC, Coutinho-Silva R (2020) Purinergic signaling, DAMPs, and inflammation. Am J Physiol Cell Physiol 318(5):C832–C835
Donnelly P (2008) Progress and challenges in genome-wide association studies in humans. Nature 456(7223):728–731
Evangelou E et al (2018) Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 50(10):1412–1425
Ewels PA et al (2020) The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 38(3):276–278
Feitosa MF et al. Genetic pleiotropy between pulmonary function and age-related traits: The Long Life Family Study. J Gerontol A Biol Sci Med Sci. 2022.
Feitosa MF, Lin SJ, Acharya S, Thyagarajan B, Wojczynski MK, Kuipers AL, Kulminski A, Christensen K, Zmuda JM, Brent MR, Michael A (2024) Province Discovery of genomic and transcriptomic pleiotropy between kidney function and soluble receptor for advanced glycation end products using correlated meta-analyses: the long life family study. Aging Cell. https://doi.org/10.1111/acel.14261
Fishilevich S et al (2017) GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database J Biol Databases Curation 2017:1–17
Flint AJ et al (2010) Body mass index, waist circumference, and risk of coronary heart disease: a prospective study among men and women. Obes Res Clin Pract 4(3):e163-246
Galli SJ, Tsai M (2012) IgE and mast cells in allergic disease. Nat Med 18(5):693–704
Gao T, Qian J (2020) EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res 48(D1):D58–D64
Graham SE et al (2021) The power of genetic diversity in genome-wide association studies of lipids. Nature 600(7890):675–679
Hilgendorf I et al (2011) The oral spleen tyrosine kinase inhibitor fostamatinib attenuates inflammation and atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 31(9):1991–1999
Hofstra CL et al (2003) Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther 305(3):1212–1221
Inouye M et al (2010) An immune response network associated with blood lipid levels. PLoS Genet 6(9):e1001113
Kannel WB et al (1979) An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 110(3):281–290
Kavvoura FK, Ioannidis JPA (2008) Methods for meta-analysis in genetic association studies: a review of their potential and pitfalls. Hum Genet 123(1):1–14
Korhonen PE et al (2009) Ankle-brachial index is lower in hypertensive than in normotensive individuals in a cardiovascular risk population. J Hypertens 27(10):2036–2043
Krystel-Whittemore M, Dileepan KN, Wood JG (2015) Mast cell: a multi-functional master cell. Front Immunol 6:620
Lamparter D et al. Fast and rigorous computation of gene and pathway scores from SNP-based summary statistics. PLoS Comput Biol. 2016;12(1).
Li B, Leal SM (2008) Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83(3):311–321
Li Y et al (2015) The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol 194(9):4328–4338
Li T et al (2017) A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 14(1):61–64
Li X et al (2020) Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat Genet 52(9):969–983
Liu Y, Xie J (2020) Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc 115(529):393–402
Liu Y et al (2019) ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am J Hum Genet 104(3):410–421
Locke AE et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518(7538):197–206
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21
Madsen BE, Browning SR (2009) A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 5(2):e1000384
Miller M et al (2011) Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 123(20):2292–2333
Morgenthaler S, Thilly WG (2007) A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat Res 615(1–2):28–56
Morris AP, Zeggini E (2010) An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 34(2):188–193
Murabito JM et al (2012) Association between chromosome 9p21 variants and the ankle-brachial index identified by a meta-analysis of 21 genome-wide association studies. Circ Cardiovasc Genet 5(1):100–112
Namkoong S et al (2018) Autophagy dysregulation and obesity-associated pathologies. Mol Cells 41(1):3–10
Newman AB et al (2011) Health and function of participants in the Long Life Family Study: a comparison with other cohorts. Aging (albany NY) 3(1):63–76
O’Connell J. Mixed Model Analysis for Pedigrees and Populations (MMAP) [Github] 2017 08/01/2022]. https://mmap.github.io/.
Ortega FB, Lavie CJ, Blair SN (2016) Obesity and cardiovascular disease. Circ Res 118(11):1752–1770
Peng X, Wu H (2022) Inflammatory links between hypertriglyceridemia and atherogenesis. Curr Atheroscler Rep 24(5):297–306
Perls T, Terry D (2003) Understanding the determinants of exceptional longevity. Ann Intern Med 139(5 Pt 2):445–449
Province MA, Borecki IB. A correlated meta-analysis strategy for data mining "OMIC" scans. In: Pacific Symposium on Biocomputing. 2013.
Rakyan VK et al (2011) Epigenome-wide association studies for common human diseases. Nat Rev Genet 12(8):529–541
Ramalho SHR, Shah AM (2021) Lung function and cardiovascular disease: a link. Trends Cardiovasc Med 31(2):93–98
Shi GP, Bot I, Kovanen PT (2015) Mast cells in human and experimental cardiometabolic diseases. Nat Rev Cardiol 12(11):643–658
Shrine N et al (2019) New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 51(3):481–493
Shungin D et al (2015) New genetic loci link adipose and insulin biology to body fat distribution. Nature 518(7538):187–196
Sollis E et al (2023) The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res 51(D1):D977–D985
Splansky GL et al (2007) The third generation cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 165(11):1328–1335
Szklarczyk D et al (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447–D452
Thompson JR, Attia J, Minelli C (2011) The meta-analysis of genome-wide association studies. Brief Bioinform 12(3):259–269
Tsai SH et al (2015) The ectoenzyme E-NPP3 negatively regulates ATP-dependent chronic allergic responses by basophils and mast cells. Immunity 42(2):279–293
Turner H, Kinet JP (1999) Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature 402(6760 Suppl):B24-30
Valverde DP et al (2019) ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol 218(6):1787–1798
van Iterson M et al., Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18(1).
Velikkakath AK et al (2012) Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 23(5):896–909
Wang KY et al (2011) Histamine deficiency decreases atherosclerosis and inflammatory response in apolipoprotein E knockout mice independently of serum cholesterol level. Arterioscler Thromb Vasc Biol 31(4):800–807
Wang J et al (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45(W1):W130–W137
Wang B et al (2020) Integrative omics approach to identifying genes associated with atrial fibrillation. Circ Res 126(3):350–360
Wojczynski MK et al (2022) NIA long life family study: objectives, design, and heritability of cross-sectional and longitudinal phenotypes. J Gerontol A Biol Sci Med Sci 77(4):717–727
Wu MC et al (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 89(1):82–93
Wu C et al (2022) Single-cell transcriptomics reveals the identity and regulators of human mast cell progenitors. Blood Adv 6(15):4439–4449
Wyss AB et al (2018) Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat Commun 9(1):2976
Zeng H et al (2022) Meta-analysis of genome-wide association studies uncovers shared candidate genes across breeds for pig fatness trait. BMC Genom 23(1):786
Zhang Q, Feitosa M, Borecki IB (2014) Estimating and testing pleiotropy of single genetic variant for two quantitative traits. Genet Epidemiol 38(6):523–530
Zhou H et al (2023) FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res 51(D1):D1300–D1311
Acknowledgements
We are grateful to the entire Long Life Family Study consortium, its participants, and its investigators, without whom this work would not have been possible. We would particularly like to thank Dr. Bharat Thyagarajan, Dr. Allison Kuipers, and Hannah Campbell for consultations on the 11 cardiovascular risk traits analyzed here. This work was supported by grant AG063893 from the National Institute on Aging. The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195, HHSN268201500001I and 75N92019D00031). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI.
Funding
This work was funded by grant AG063893 from the US National Institute on Aging.
Author information
Authors and Affiliations
Contributions
S.A., M.R.B, and M.A.P conceived the project. S.A. and M.R.B. worked on the methodology and drafted the manuscript. S.A. also performed data curation, formal project analysis, and downstream evaluation. S.L. performed STAAR analysis, processed whole genome sequencing data in LLFS and FHS, prepared Figs. 1 and 2, Document 10, and drafted the Rare-variant analysis part of the Methods section. W.J. developed the Nextflow pipeline omicsgenetraitassociation (https://nf-co.re/omicsgenetraitassociation/) and performed correlated meta-analysis (CMA). Y.S.K. performed module enrichment and GO over-representation analysis and drafted the respective methods section. V.A.M. performed GWAS with WGS data on CMA-significant genes across traits. M.F., M.W., S.L., J.A.A, and K.S. contributed to data processing of SNP-Chip genotypes and cardiovascular traits. J.O.C. assisted with TWAS runs in MMAP.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acharya, S., Liao, S., Jung, W.J. et al. A methodology for gene level omics-WAS integration identifies genes influencing traits associated with cardiovascular risks: the Long Life Family Study. Hum. Genet. (2024). https://doi.org/10.1007/s00439-024-02701-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00439-024-02701-1