
Abstract
Developmental language disorder (DLD) overlaps clinically, genetically, and pathologically with other neurodevelopmental disorders (NDD), corroborating the concept of the NDD continuum. There is a lack of studies to understand the whole genetic spectrum in individuals with DLD. Previously, we recruited 61 probands with severe DLD from 59 families and examined 59 of them and their families using microarray genotyping with a 6.8% diagnostic yield. Herein, we investigated 53 of those probands using whole exome sequencing (WES). Additionally, we used polygenic risk scores (PRS) to understand the within family enrichment of neurodevelopmental difficulties and examine the associations between the results of language-related tests in the probands and language-related PRS. We identified clinically significant variants in four probands, resulting in a 7.5% (4/53) molecular diagnostic yield. Those variants were in PAK2, MED13, PLCB4, and TNRC6B. We also prioritized additional variants for future studies for their role in DLD, including high-impact variants in PARD3 and DIP2C. PRS did not explain the aggregation of neurodevelopmental difficulties in these families. We did not detect significant associations between the language-related tests and language-related PRS. Our results support using WES as the first-tier genetic test for DLD as it can identify monogenic DLD forms. Large-scale sequencing studies for DLD are needed to identify new genes and investigate the polygenic contribution to the condition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Developmental language disorder (DLD) and other neurodevelopmental disorders (NDD) share many symptoms and signs (Nitin et al. 2022; Plug et al. 2021). DLD is a neurodevelopmental communication disorder characterized by persistent deficits in receptive and expressive language not attributable to other biomedical conditions and results in functional, psychological, social, and/or occupational limitations (American Psychiatric Association 2022; Bishop et al. 2016). DLD can co-occur with other NDD, complicating their clinical and etiological disentanglement (Nitin et al. 2022). DLD remains underdiagnosed and understudied despite its ~7% prevalence (Calder et al. 2022; McGregor 2020; Tomblin et al. 1997). Growing evidence suggests that genetic factors have a large impact on the etiology of DLD (Mountford et al. 2022). Some environmental risk factors, such as maternal smoking during pregnancy, have also been suggested (Calder et al. 2022).
Monogenic/genomic DLD forms, although rare, are apt to provide insight into the biological pathways associated with DLD. In this study, we used the term genomic DLD when referring to DLD caused by copy-number variants (CNVs). An increasing number of high-impact copy CNVs and rare variants have been identified in a proportion of DLD individuals (Chen et al. 2017a, b; Eising et al. 2019; Kalnak et al. 2018; Mountford et al. 2022; Plug et al. 2021; Villanueva et al. 2015). The recent advances in sequencing technologies and the availability of population-level variations data have enabled genetic diagnosis in DLD and the discovery of new genes implicated in the condition (Mountford et al. 2022).
Disentangling the polygenic contribution in DLD is lagging compared to the monogenic forms. A recent genome-wide association study (GWAS) identified a robust single-nucleotide polymorphism (SNP) heritability of five reading- and/or language-related traits, not including DLD, that explained 13–26% of trait variability (Eising et al. 2022).
Previously, we reported an enrichment of neurodevelopmental difficulties in families of 59 probands with severe DLD (Kalnak et al. 2012, 2014). In a subsequent study, we investigated those 59 probands and their families using array genotyping (Kalnak et al. 2018). We reported clinically significant CNVs in four probands, resulting in a 6.8% molecular diagnostic yield. Although we identified rare CNVs in some siblings and parents, a significant association with CNV burden did not explain the aggregation of NDD within the families (Kalnak et al. 2018). In the current study, we investigated 53 out of the 59 probands using whole exome sequencing (WES) to identify rare monogenic conditions. We report the high-impact variants identified in genes previously known to be associated with monogenic DLD/DLD-related disorders, highlighting the diagnostic utility of WES in DLD. Furthermore, we prioritized candidate genes for future studies. We also explored the possible association between the performance of individuals with DLD on selected language-related tests and their corresponding polygenic scores, and the potential of PRS to explain the enrichment of neurodevelopmental difficulties in the families.
Methods
Recruitment of participants
Sixty-one DLD probands aged 8–12 years were recruited from 14 school language units for children with severe DLD as described before (Kalnak et al. 2012, 2014, 2018). Admission to these schools requires pre-assessments by a speech-language pathologist, a psychologist, and a teacher, and is usually performed at the age of 5 or 6 years. To be admitted, each child had to have DLD as the primary diagnosis, i.e., excluding autism spectrum disorder (ASD) and intellectual disability (ID). Children with dyslexia or attention-deficit hyperactivity disorder (ADHD) could be admitted if they co-occurred with DLD. Children whose DLD status was confirmed by the school to still be the primary diagnosis at the time of participation were included.
Inclusion and exclusion criteria
As described previously (Kalnak et al. 2012, 2014, 2018; Kalnak and Sahlén 2022), our inclusion criteria were: (1) DLD at the time of school admission and not questioned by the school up to the time of recruitment to our study at the age of 8–12 years. (2) Normal nonverbal IQ at admission to school (3) Normal vision and hearing. (4) Monolingual Swedish speaking. We included only monolingual Swedish-speaking children to minimize the risk of bilingual effects on the language tests which were all administered in Swedish. Adopted children were excluded from the study. We also excluded probands without available high-quality DNA from this current study.
Interviews and assessments
The probands were assessed with a broad linguistic and cognitive test battery by a speech-language pathologist, as described previously (Kalnak et al. 2018). Herein, we used the results from the nonword repetition task (NWR), the Raven’s colored progressive matrices (CPM9), and the test of word-reading efficiency (TOWRE). NWR standard deviation units (z-scores) were calculated based on whole-word accuracy (NWR_binary; the binary outcome of either a positive or a negative response), numbers of syllabi (NWR_length), and percentage of correct consonants (NWR_PCC). TOWRE z-scores were based on the number of accurate decoding over time of words (TOWRE_words) and nonwords (TOWRE_nonwords). We also compared the performance of the probands with a monogenic diagnosis identified in the current or the previous study (Kalnak et al. 2018) on CPM9, NWR_binary, NWR_length, NWR_PCC, TOWRE_words, and TOWRE_nonwords.
To investigate the polygenic contribution of other neurodevelopmental diagnoses and traits/difficulties within these families, we also categorized each participant, including parents and siblings, whether they were reported with any kind of neurodevelopmental (1) diagnoses, such as language disorder, reading impairments, autism spectrum disorder, stuttering, ID, ADHD, cleft palate, congenital impairment of hearing or vision, or (2) difficulties with language, learning, social communication, and attention. These were based on family history interviews with the parents (Kalnak et al. 2012).
Sample collection and DNA extraction
Saliva samples were collected from the participants using the Oragene DNA OG-500 collection tubes (DNA Genotek, Inc., Ottawa, Ontario, Canada). DNA was extracted according to the prepIT.L2P manual protocol provided by the manufacturer. DNA quality was checked using agarose gel electrophoresis and NanoDrop spectrophotometer.
Whole exome sequencing and variant prioritization
Fifty-three out of the 59 participants with available DNA were examined using WES. Four samples were excluded due to degraded DNA and two failed during library preparation. WES was performed on NovaSeq 6000 using paired end reads at the Clinical Genomics Stockholm core facility, Karolinska Institutet, and Science for Life Laboratory, Stockholm, Sweden. Library preparation and WES data processing up to the generation of variant call format files (VCF) were described previously (Li et al. 2022). In short, Twist Human Core Exome v1.3 Enrichment Kit (Twist Bioscience) was used for library preparation with some modifications: xGen Dual Index UMI adapters [6-nucleotide unique molecular identifiers (UMI), 0.6 mM, Integrated DNA Technologies] were used for the ligation, and xGen Library Amp Primer (2 mM, Integrated DNA Technologies) was used for PCR amplification (10 cycles). Target enrichment was performed in a multiplex fashion with a library amount of 187.5 ng (8-plex). The libraries were hybridized to Exome probes v1.3 (Twist Bioscience), xGen Universal Blockers—TS Mix (Integrated DNA Technologies), and COT Human DNA (Life Technologies) for 16–19 h. The post-capture PCR was performed with xGen Library Amp Primer (0.5 mM, Integrated DNA Technologies) for eight cycles. Quality control was performed with the Qubit dsDNA HS assay (Invitrogen) and TapeStation HS D1000 assay (Agilent). Demultiplexing was done using Casava v2.20. The depth and coverage were assessed relative to the capture kit and the exome-coding regions of hg19 using Samtools v1.17. The average coverage of the hg19 whole exome was 79% (the range was 76–81%), 75% (70–77%), and 73% (65–74%) at 10×, 20×, and 30×, respectively. The average coverage of the capture kit was 99% (87–99.7%; only two samples were below 99%, we did not identify a candidate variant in those two samples) at 30× and the mean depth was 188.
The reads were aligned against the GRCh37 reference genome using Burrows Wheel Aligner (Li and Durbin 2009). We used VarAFT to annotate the VCF files and prioritize/filter the variants (Desvignes et al. 2018). First, we selected exonic and probable splice-site variants that were rare in gnomAD v2.1 general and European populations (minor allele frequency < 0.001 for recessive model and <0.000001 for dominant/de novo model), predicted as damaging/possibly damaging/probably damaging by in silico tools (Sift (Ng and Henikoff 2003), Polyphen2 HumDiv, and Polyphen2 HumVar (Adzhubei et al. 2010)), and located in brain-expressed genes. In the subsequent filters, we selected only high-impact variants in genes that fitted the postulated inheritance models in each family and were considered essential for brain function.
We regarded a variant as a high-impact variant if it was nonsense, start-loss, ±2 splice-site, or a frameshift variant; or if it was classified by ClinVar as pathogenic/likely pathogenic variant. Genes were considered essential for brain function if when mutated cause a central nervous system disease (CNS; searched in OMIM and PubMed databases) and/or if they were constrained genes (damaging variants of them are removed by natural selection). Additionally, genes not reported before to cause a CNS disease had to have evidence from the scientific literature supporting their role in the CNS. We considered dominant genes constrained if their gnomAD v2.1 probability of being loss-of-function intolerant (pLI) was ≥0.9. We considered recessive genes constrained if the number of homozygotes with loss-of-function variants in gnomAD v2.1 general population was zero. Furthermore, we searched the scientific literature using PubMed database by entering the name/s of the gene in the search field.
Supplementary Fig. S1 shows the filters we used for the dominant/de novo model and the average number of variants that remained after each filter. We used similar filters for recessive models (homozygous and compound heterozygous), with 0.001 as a frequency cutoff.
Candidate variants were sent for Sanger sequencing for segregation analysis. Primers were designed using Primer3Plus (Untergasser et al. 2007). Sanger sequencing was performed at Eurofins Genomics Europe Sequencing GmbH, Köln, Germany, and viewed using SnapGene Viewer. We submitted all the candidate novel genes to the GeneMatcher platform (Sobreira et al. 2015). Variants in genes known to cause NDD were classified according to the American College of Medical Genetics (ACMG) criteria (Richards et al. 2015) and amending recommendations for PVS1 and PP3/BP4 criteria (Abou Tayoun et al. 2018; Pejaver et al. 2022). For each prioritized variant, we further checked if it meets the amended recommendation for PVS1 using AutoPVS1 (Xiang et al. 2020). We confirmed trio status of the parents and the proband from genome-wide SNP array data, when available, using “--kinship” command of KING v2.2.9 to aid ACMG variants classification.
GnomAD, PubMed, ClinVar, and GeneMatcher platforms/databases were accessed November 22nd, 2023.
Genotyping and imputation
Genotyping using Genome-Wide SNP Array 6.0 (Affymetrix, Santa Clara, California) of the 59 families has been described earlier (Kalnak et al. 2018). Calling of the genotypes was performed using Affymetrix Analysis Power Tools (APT) software version APT-2.11.6. The average per sample call rate was >97%. APT output genotyping file was converted to VCF using affy2vcf plugin of BCFtools (Li 2011). Quality control was performed using PLINK 1.9 (Chang et al. 2015). Variants or individuals with missingness > 2% (tightened to > 1% before PRS calculation) were filtered. Variants not in Hardy–Weinberg equilibrium (p < 1 × 10−6) were also filtered. Individuals with heterozygosity rate >±3 standard deviations from the samples’ heterozygosity rate mean were removed. Discrepancies between reported and biological sex were checked using the “–check-sex” PLINK command (X chromosome homozygosity estimate for males > 0.8 and for females < 0.2). Checking population stratification was performed using the multidimensional scaling (MDS) approach anchored to phase 3 of the 1000 genome project as described by Marees et al. (Marees et al. 2018). We checked for any strand issues, any problematic SNPs, and references correspondence before anchoring. MDS component scores were generated using “--cluster --mds-plot” PLINK command. We visualized the first two principal components to check for outliers (Supplementary Fig. S2). Minor allele frequency was set at 5%. Out of 180 individuals and 907,343 variants, 165 individuals and 490,971 variants passed our quality control filters with a total genotyping rate of 99.7%. Six individuals were filtered due to high missingness and nine due to excess heterozygosity. Phasing and imputation were performed based on phase 3 of the 1000 genome project using the default parameters of SHAPEIT4 (Delaneau et al. 2019) and IMPUTE5 (Rubinacci et al. 2020), respectively.
We previously described calling CNVs from this same Genome-Wide SNP array data of the 59 families using three algorithms: PennCNV-Affy (August 2016 version), Chromosome Analysis Suite (ChAS) software v.3.1 (Affymetrix), and Partek Genomics Suite software version 6.6 (Partek Inc., St. Louis, Missouri) (Kalnak et al. 2018). In short, the CNV calls were mapped using GRCh37/hg19 coordinates and annotated using ANNOVAR. Only CNVs called by at least two of the algorithms with ≥50% reciprocal overlap were considered, excluding those that spanned ≥50% with centromeres or telomeres, <10 consecutive probes and were <10 kb in size (Kalnak et al. 2018).
Polygenic risk scores’ calculations and analysis
We selected five GWASs on European-ancestry individuals as base data sources to calculate PRS for specific developmental traits to test possible polygenic overlap with DLD. These traits included ASD (46,350 individuals) (Grove et al. 2019), ADHD (51,568 individuals) (Demontis et al. 2023), educational attainment (765,283 individuals) (Okbay et al. 2022), cognitive performance (257,828 individuals) (Lee et al. 2018), reading- and language-related skills, and performance IQ (Eising et al. 2022). The included reading- and language-related skills were word reading (27,180 individuals), nonword reading (16,746 individuals), nonword repetition (12,828 individuals), phoneme awareness (12,411 individuals), and spelling (17,278 individuals). PRS was calculated using the default parameters of PRS-CS (Ge et al. 2019) (PARAM_A = 1, PARAM_B = 0.5, PARAM_PHI = 0.01, MCMC_ITERATIONS = 1000, MCMC_BURNIN = 500, and MCMC_THINNING_FACTOR = 5). Polygenic transmission disequilibrium was tested in 30 individuals from 20 families using ptdt tool (Weiner et al. 2017). The first ten principal components of ancestry (PC1–10) were included as covariates in the association analysis between the selected language-related clinical measures and PRS. We calculated the first ten principal components using “--pca” PLINK command.
Statistical analysis
We compared the performance of the probands with a monogenic/genomic diagnosis identified in the current and the previous study (Kalnak et al. 2018) to the probands without any genetic diagnosis on CPM9, NWR_binary, NWR_length, NWR_PCC, TOWRE_words, and TOWRE_nonwords tasks using an unpaired student t test.
We fitted a generalized estimating equation (GEE) logistic regression model in R version 4.1.1 using the geeglm() function from the geepack package (Halekoh et al. 2006) to test associations between the PRS for ASD, ADHD, educational attainment, and cognitive performance and the neurodevelopmental phenotypes in the families. We added sex and the first ten principal components (PC1–10) as predictor variables, assuming an independent correlation structure and controlling for the family status.
The associations between six quantitative language and reading skill measures tested in the DLD probands and the corresponding PRS values were done using a linear regression with lm() function in R. The models were adjusted for age, sex, diagnosis with a monogenic condition/genomic syndrome, and PC1–10. CPM9 was tested against cognitive performance PRS and performance IQ PRS, each in a separate model. NWR scores were tested against nonword repetition PRS, and TOWRE scores were tested against word reading and nonword reading PRS, in separate models.
Results
Diagnostic yield of WES and inheritance patterns
Among the 53 probands, we identified four pathogenic/likely pathogenic variants in genes previously known to cause DLD or other related NDD in four probands/families, achieving a molecular diagnostic success rate of 7.5% (4/53) (Table 1). Three of the variants were de novo variants and one was inherited from an apparently affected father (Fig. 1). The four variants were pathogenic/likely pathogenic according to the ACMG criteria (Richards et al. 2015) and the amendments for PVS1 and/or PP3/BP4 criteria (Abou Tayoun et al. 2018; Pejaver et al. 2022). A variant of uncertain significance (NM_001005271.3:c.2327A>G (p.Asp776Gly)) in CHD3 was detected in family F60 (Supplementary materials) and was not included as a positive diagnosis when we calculated the molecular diagnostic success rate. KING kinship inference was as expected in the families.

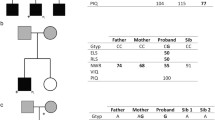
Segregation analysis of the variants identified in genes known to cause developmental language disorders. A–D Thin arrows point to the proband in each family, thick red arrows point to culprit variants, and black arrows point to wild-type variants, circles depict females, squares depict males, filled circles and squares depict patients, and unfilled circles and squares depict controls. DLD, developmental language disorders. We previously identified a 3.5 Mb deletion of Xp22.31-p22.33 in F34.6, highlighted with “*” sign
Pathogenic/likely pathogenic variants in genes known to cause DLD/NDD in human
In family F2, we identified the de novo variant NM_002577.4(PAK2):c.1051G>T (p.Glu351Ter) in the proband F2.1 (Fig. 1A). Heterozygous mutations in PAK2 were only reported before in a Chinese ASD individual (Wang et al. 2018) and in two New Zealand siblings with a variable combination of retinal detachment, vitreous abnormalities, severe developmental delay, incomprehensible speech, ASD, interstitial parenchymal pulmonary changes on chest X-rays, and enamel hypoplasia (OMIM 618458) (Antonarakis et al. 2021).
In family F10, we identified the de novo nonsense variant affecting MED13 [NM_005121.3(MED13):c.4225C>T (p.Arg1409Ter)] (Fig. 1B). Heterozygous mutations in MED13 cause intellectual developmental disorder type 61 (OMIM 618009) (Snijders Blok et al. 2018). The clinical presentations of individuals with pathogenic MED13 variants include variable combinations of NDD, dysmorphic features, and epilepsy among others (Snijders Blok et al. 2018; Trivisano et al. 2022).
In family F34, we identified the missense variant NM_001377142.1(PLCB4):c.1898G>A (p.Arg633His) (rs397514481) in the proband and her mother (Fig. 1C). The variant was absent from the gnomAD v4.0 database, had a CADD score of 35, was classified as pathogenic in ClinVar database by six submitters (VCV000031639.6, accessed in 28/02/2023), and was validated by functional studies (Kanai et al. 2022). Recessive and dominant mutations in PLCB4 cause auriculocondylar syndrome 2 (OMIM 614669) (Gordon et al. 2013; Rieder et al. 2012). Dominant mutations, including rs397514481, cause the disease through dominant-negative effects on the endothelin receptor type A-Gq/11 pathway important for forming lower jaw and middle ear structures during embryonic development (Kanai et al. 2022). Auriculocondylar syndrome 2 is characterized by craniofacial anomalies and other less frequent features, including speech delay (Li et al. 2023). Variable expressivity and incomplete penetrance have also been observed in auriculocondylar syndrome 2 (Vegas et al. 2022). While the proband only had severe DLD, his mother had reading and writing difficulties, language impairment, and learning difficulties. The proband had a sister reported to have social communication difficulties, DLD, and, as we reported before, a 3.5 Mb deletion of Xp22.31–p22.33. This deletion was absent in the proband (Kalnak et al. 2018).
We identified a de novo variant in TNRC6B [NM_001162501.2(TNRC6B):c.830_836del (p.Asn277MetfsTer3)] in the proband F35.1 (Fig. 1D). Heterozygous mutations in TNRC6B cause a syndrome of global developmental delay with speech and behavioral abnormalities (OMIM 619243) (Eising et al. 2019). Speech delay was documented in 94% of individuals with pathogenic TNRC6B variants, followed by other forms of developmental delay, intellectual disability, ASD, ADHD, behavioral abnormalities, musculoskeletal abnormalities, and dysmorphic features (Granadillo et al. 2020).
Variants in novel genes and genes with nonconclusive associations with DLD/NDD
We identified four high-impact variants in four candidate novel genes (ZBTB38, AQR, APBA1, and TXLNA) not implicated before in NDD or genetic diseases in four probands/families (Table 2; Fig. 2B–D, F). Two of those variants were dominantly inherited from apparently affected parents, one variant was likely a de novo variant, and one, APBA1, was compound heterozygous with a paternal deletion (Fig. 2). The deletion in the father was identified in our earlier study showing a ~13 kb (chr9:72035199–72048477) heterozygous deletion including APBA1 exons 12 and 13 and present in both siblings. We also identified two high-impact variants in two genes (DIP2C and PARD3) with yet nonconclusive associations with NDD (Table 2; Fig. 2A, E).

Segregation analysis of the variants identified in candidate novel genes and genes with no strong evidence that they cause neurodevelopmental disorders. A–F Thin arrows point to the proband in each family, thick red arrows point to culprit variants, black arrows point to wild-type variants, circles depict females, squares depict males, filled circles and squares depict affected individuals, and unfilled circles and squares depict controls. DLD, developmental language disorders; ADHD, attention-deficit hyperactivity disorder; ASD, autism spectrum disorder. * Individuals with a heterozygous deletion including APBA1 exons 12 and 13
The pathogenicity of DIP2C, TXLNA, and AQR was supported by curating large datasets of genetic information of NDD and ASD individuals published by Zhou and colleagues (Zhou et al. 2022). Two loss-of-function DIP2C variants, NM_014974.3:c.2548del (p.Ser850ProfsTer10) and NM_014974.3:c.1969dup (p.Ala657GlyfsTer12), were reported in two ASD individuals out of 16,877 ASD individuals and 5,764 neurotypical controls (Zhou et al. 2022). Both variants were absent from the gnomAD v4.0 database. Similarly, one TXLNA frameshift variant, NM_175852.4:c.164del (p.Pro55ArgfsTer21), and one AQR nonsense variant, NM_014691.3:c.3136C>T (p.Arg1046Ter) (rs749141492), were each reported in one participant out of 31,058 with ASD (Zhou et al. 2022). NM_175852.4(TXLNA):c.164del (p.Pro55ArgfsTer21) was absent from gnomAD v4.0. On the other hand, rs749141492 was reported heterozygous in gnomAD v4.0 database with an allele frequency of 0.000002 in the European population.
Differences in cognitive, speech, and language-related measures based on the genetic groups
We wanted to test if there were any differences in quantitative IQ and language-related measures between those with a putative genetic diagnosis (n = 16, also including the candidate genes) and those without (n = 43). We detected a statistically significant difference between these groups for NWR_PCC (t = 2.71, df = 59, two-tailed p value = 0.009). No statistically significant differences were detected in the other measures (Supplementary Table S1).
Association with polygenic risk scores and the phenotypes
Next, we calculated PRS for two diagnoses (ASD and ADHD) and eight traits using earlier published source GWAS data (Demontis et al. 2023; Eising et al. 2022; Grove et al. 2019; Lee et al. 2018; Okbay et al. 2022). First, we tested if any significant differences could be seen in the distribution of the PRS values in the families by grouping the individuals, including probands, for neurodevelopmental difficulties (Supplementary Figs. S4–S13 for distribution of the PRS). Using GEE logistic regression models adjusted for the family structure, sex, and PC1–10, we did not detect any statistically significant association between neurodevelopmental difficulties and polygenic scores of ASD, ADHD, educational attainment, and cognitive performance (Supplementary Fig. S15).
Next, we used linear regression models to test available quantitative measures in the probands and the corresponding PRS values (Supplementary Table S2). No statistically significant associations were detected. Also, no significant differences were seen when we tested parents-to-probands PRS transmission disequilibrium for all the calculated PRS values (Supplementary Fig. S14).
Discussion
Here, we studied the contribution of rare sequence-level variants to the genetic landscape of severe DLD in 53 individuals, followed by segregation analysis. Before, we have reported an aggregation of neurodevelopmental difficulties in the families of those probands (Kalnak et al. 2012, 2014). In a subsequent study, we reported, in the same cohort, an enrichment of rare CNVs in the probands but not in their family members (Kalnak et al. 2014). Herein, in our quest to explain the aggregation of these difficulties within those families, we investigated the association between the neurodevelopmental difficulties in the families and multiple neurodevelopmental traits/phenotypes PRS. We also investigated the association between multiple quantitative tests in the probands and their cognate PRS, and the transmission disequilibrium of neurodevelopmental traits/phenotypes PRS to the probands.
Our study adds to the growing evidence that individuals with DLD have similar rates of rare pathogenic genetic variants as in other NDD, ASD for instance (Srivastava et al. 2019). We also showed that individuals with monogenic/genomic DLD perform worse than those without monogenic/genomic DLD on a subtype of nonword repetition test, the percentage of correct consonants subtype. Nonword repetition has been identified as a clinical marker for Swedish-speaking school-aged children diagnosed with DLD (Kalnak et al. 2014) and in several other languages around the world (Schwob et al. 2021). A similar trend of scoring lower in many tests was seen but not significant. Therefore, further studies should investigate if indeed there are differences in these quantitative language measures within DLD based on the genetic subgrouping in larger samples.
We reached a molecular diagnostic yield of 7.5% (4/53) using WES. When we added on our earlier study for rare CNV contribution (Kalnak et al. 2018), the combined diagnostic yield in our cohort was 13.7% (8/58). A previous study reported a diagnostic yield of 26.8% on investigating a larger cohort of 127 DLD cases using a combination of karyotyping analysis, repeat expansion testing, targeted gene panels, SNP array genotyping, and WES (Plug et al. 2021). Our lower diagnostic yield was probably due to several reasons, including our smaller sample size, lesser range of genetic investigations used, and more conservative gene and variant pathogenicity cutoffs applied. In addition, we identified six variants in six different genes with less/no association with DLD/NDD in humans (Table 2; Fig. 2). The relevance of those candidate genes to DLD is discussed below.
DIP2C encodes a highly expressed protein in the brain and a member of the highly conserved DIP2 (Disco-interacting protein 2) family implicated in neuronal morphology and development and lipids’ metabolism (Ma et al. 2019; Mondal et al. 2022; Mukhopadhyay et al. 2002; Nitta et al. 2017; Noblett et al. 2019; Oo et al. 2020). A previous study reported 19 patients with submicroscopic deletion of chromosome 10p15.3 that included DIP2C and/or ZMYND11 in all the cases. All the examined patients had developmental difficulties (n = 11) or language disorder (n = 10) (Descipio et al. 2012). A recent study reported a case of a 17-month-old female with focal infantile epilepsy, dysmorphic features, and developmental delays in motor developmental coordination and in receptive and expressive language (Yang et al. 2022). WES identified the de novo variant NM_014974.3 (DIP2C): c.1057 + 2T>G that was shown by minigene transfection assay to lead to DIP2C alternative splicing and an 80 bp deletion in Exon 8 (Yang et al. 2022). Furthermore, we detected two loss-of-function DIP2C variants in two ASD individuals upon mining published genetic data from 16,877 ASD individuals (Zhou et al. 2022).
PARD3 is essential for cell polarity and nervous system development (Chen et al. 2017a, b; Hirose et al. 2022). Two CNVs that involved only PARD3 were reported before in association with ASD and neural tube defects. The first was a 139 kb deletion in a case presented with craniorachischisis, cleft lip and palate, and bilateral adrenal hypoplasia (Chen et al. 2013). The second was a de novo 209 kb duplication in a Turkish individual with ASD (Özaslan et al. 2021). Regarding rare sequence-level variants, a heterozygous frameshift variant, NM_019619.4(PARD3): c.1012dupG (p.Glu338GlyfsTer26), was identified in six patients from a Chinese family manifesting nonsyndromic isolated cleft palate. Ethmoid plate patterning defects observed in zebrafish supported the gene’s candidacy (Cui et al. 2022).
ZBTB38 is a ubiquitously expressed ZNF transcription factor that belongs to the POZ/BTB family (Costoya 2007; Sasai et al. 2005). In mice, heterozygous loss of ZBTB38 decreased the expression of Nanog and Sox2 and led to embryonic developmental failure and early embryonic lethality (Nishio et al. 2022). Heterozygous SOX2 loss-of-function leads to anophthalmia/microphthalmia syndrome (OMIM 206900) that almost always includes NDD, sometimes without ocular abnormalities (Amlie-Wolf et al. 2022; Fantes et al. 2003). Multiple members of the POZ/BTB family are also associated with monogenic NDD, e.g., ZBTB7A (OMIM 619769) (Ohishi et al. 2020), ZBTB11 (OMIM 618383) (Fattahi et al. 2018), ZBTB18 (OMIM 612337) (de Munnik et al. 2014), and ZBTB20 (OMIM 259050) (Cordeddu et al. 2014).
AQR encodes a retinoic acid-responsive DNA/RNA helicase that serves in homologous recombination repair and resolving R-loops (Sakasai et al. 2017; Sam et al. 1998; Sollier et al. 2014). Multiple proteins are involved in R-loop regulation, including DNA/RNA helicases and RNA degrading enzymes. Mutations in R-loops regulatory proteins are associated with multiple cancers and neurological disorders (Khan and Danckwardt 2022). One prominent example is SETX that encodes a DNA/RNA helicase and when mutated causes a recessive form of spinocerebellar ataxia (OMIM 606002) and a dominant form of juvenile amyotrophic lateral sclerosis (OMIM 602433) (Chen et al. 2004; Moreira et al. 2004). Interestingly, we detected one nonsense AQR variant in an NDD individual upon curating published genetic data from 31,058 NDD patients (Zhou et al. 2022).
APBA1 encodes a member in a putative presynaptic organizer complex (CASK/APBA1/LIN-7) essential for synaptogenesis and synaptic transmission (Brouwer et al. 2019; Butz et al. 1998; Leonoudakis et al. 2004; Motodate et al. 2019). Mutations in CASK cause variable NDD phenotypes (OMIM 300422 and 300749), while mutations in LIN7B were proposed to cause ASD (Becker et al. 2020; Mizuno et al. 2015; Najm et al. 2008; Piluso et al. 2009). We predicted that the nonsense variant NM_001163.4:c.2497C>T (p.Gln833Ter) in the two siblings from the family F54 would not abolish the APBA1 protein expression by nonsense-mediated decay, given its predicted termination merely five amino acids prior to its canonical endpoint. Yet, these five amino acids reside within the highly conserved C-terminal tail of the protein; thus, their absence may perturb the tail-mediated regulatory mechanisms.
TXLNA encodes α-taxilin, a constituent of the centriolar subdistal appendage essential for centrosomal microtubules’ anchorage (Ma et al. 2022). Mutations in genes encoding components of the distal and subdistal centriolar appendages are implicated in ciliopathies and developmental defects, including brain defects (Ma et al. 2023). For example, homozygous mutations in NIN and KIF2A, encoding two components of the subdistal centriolar appendage, are linked to Sickler syndrome type 7 (OMIM 614851) and a syndrome of complex brain malformations (OMIM 615411), respectively (Dauber et al. 2012; Poirier et al. 2013). Knockout of TXLNA in human retinal pigment epithelial-1 and HeLa cell lines impairs microtubules dynamics and its knockdown alters the centrosomal localization of CEP170, another component of the subdistal centriolar appendage (Ma et al. 2022). Interestingly, we detected one carrier with frameshift TXLNA variant upon curating published genetic data from 31,058 ASD participants (Zhou et al. 2022).
More complex inheritance patterns and scenarios constitute a major challenge in NDD, including DLD and other genetic conditions. These scenarios include dual molecular diagnosis, oligogenic inheritance, and polygenic inheritance (Centanni et al. 2015; Dale et al. 2020; Posey et al. 2017). Dual molecular diagnosis is possible in family F60, in which we detected a high-impact variant in PARD3 (Fig. 2E) and a variant of uncertain significance in CHD3 (Supplementary Fig. S3). Pathogenic variants in CHD3 show variable expressivity in Snijders Blok–Campeau syndrome (OMIM 618205), while PARD3 was associated before with neural tube defects, cleft lip, cleft palate, ASD, and bilateral adrenal hypoplasia (Chen et al. 2013; Cui et al. 2022; Özaslan et al. 2021; van der Spek et al. 2022). The variant in CHD3 was a predicted splice-site variant, and we did not functionally validate its effect. Thus, we could not conclude whether the mild phenotype in family F60 was a case of variable expressivity, an unknown disease-modifying genetic interaction between CHD3 and PARD3, or other modifying factors. Another challenge that we want to highlight is the shared symptomatology and pathways between DLD and other NDD. However, this challenge is rather an opportunity to readdress the NDD nosology and phenotype-based classifications and to consider more gene- or pathway-oriented classifications. The emerging concept of NDD continuum is a major leap forward to reformulating NDD nosology (Morris-Rosendahl and Crocq 2020). Our results were in line with the NDD continuum concept as despite excluding individuals with ASD and/or ID, we still identified pathogenic/likely pathogenic variants in genes that can cause ASD and/or ID.
Regarding our PRS analyses, we did not detect any association between probands PRS and the assessed quantitative clinical measures. Additionally, PRS for ASD, ADHD, educational attainment, and cognitive performance were not associated with neurodevelopmental difficulties in the families. A negative association between educational attainment PRS and language delay has been reported before (Dale et al. 2020). The lack of association between ASD, ADHD, educational attainment, and cognitive performance polygenic scores and neurodevelopmental difficulties in the participant families could be due to one or more causes. These causes include a true lack of association, the small size of our cohort, the impact of environmental factors complicating investigating the association between those traits and PRS, the heterogeneity in our cohort as while all the probands had DLD their family members could have other forms of neurodevelopmental difficulties, and, except for educational attainment and cognitive performance, the relatively small size of the base GWAS. The small size of our cohort is not ideal for polygenic associations, and larger studies should be conducted to investigate better associations between PRS and phenotypes in DLD probands. Similarly, there is a pressing need for larger GWAS for the language diagnoses and even other NDD.
Study limitations
Our sample size was small for investigating the polygenic factors in DLD. Another limitation was the lack of functional validation of both variant and gene pathogenicity. This limitation is not unique to our study and constitutes a challenge for studying DLD and other genetic conditions. Mitigating strategies that support and encourage functional studies include sharing novel candidate genes and variants to platforms such as GeneMatcher (Sobreira et al. 2015) and ClinVar and initiatives such as the DECIPHER project (Firth et al. 2009). One example is the NM_001377142.1(PLCB4):c.1898G>A (p.Arg633His) that has been functionally validated after repeated detection in auriculocondylar syndrome 2 (Kanai et al. 2022). Another example is when we used the datasets published by Zhou and colleagues (Zhou et al. 2022) to support the pathogenicity of DIP2C, TXLNA, and AQR. Also, we identified several matches in GeneMatcher for future studies. Functional studies also come with limitations in that the results cannot always be extrapolated to humans (Damianidou et al. 2022).
Another limitation was that we did not perform a physical clinical examination for participants and their families which could have added a broader phenotypic description and could have resolved possible report bias. Also, we performed WES only in the probands which reduced our chances of identifying families with more than one genetic cause and discouraged us from looking for small CNVs in WES data.
Conclusion
Achieving a diagnostic success rate of 7.5%, we provide clear evidence of the usefulness of WES in searching for molecular diagnoses in children with DLD. We also provide preliminary results, indicating that individuals without monogenic/genomic DLD perform better than those with monogenic/genomic DLD on the percentage of correct consonants nonword repetition test, a finding to be verified in larger cohorts. We did not detect significant associations between language quantitative measures and language-related polygenic scores. Overall, more genetic studies focusing on DLD and related speech and language phenotypes should be prioritized, so that sample size would be comparable to the other more studied NDD, such as ASD and ADHD.
Data availability
The data supporting the findings of this study (not including participants’ personal information) are available from the corresponding author upon reasonable request and subject to necessary clearances. Clinically relevant variants have been submitted to ClinVar database (submission SUB13751313).
References
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM (2018) Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 39(11):1517–1524. https://doi.org/10.1002/humu.23626
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
American Psychiatric Association (2022) Diagnostic and statistical manual of mental disorders: DSM-5-TR. American Psychiatric Association, United States
Amlie-Wolf L, Bardakjian T, Kopinsky SM, Reis LM, Semina EV, Schneider A (2022) Review of 37 patients with SOX2 pathogenic variants collected by the Anophthalmia/Microphthalmia Clinical Registry and DNA research study. Am J Med Genet A 188(1):187–198. https://doi.org/10.1002/AJMG.A.62518
Antonarakis SE, Holoubek A, Rapti M, Rademaker J, Meylan J, Iwaszkiewicz J, Zoete V, Wilson C, Taylor J, Ansar M, Borel C, Menzel O, Kuželová K, Santoni FA (2021) Dominant monoallelic variant in the PAK2 gene causes Knobloch syndrome type 2. Hum Mol Genet 31(1):1–9. https://doi.org/10.1093/HMG/DDAB026
Becker M, Mastropasqua F, Reising JP, Maier S, Ho ML, Rabkina I, Li D, Neufeld J, Ballenberger L, Myers L, Moritz V, Kele M, Wincent J, Willfors C, Sitnikov R, Herlenius E, Anderlid BM, Falk A, Bölte S, Tammimies K (2020) Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl Psychiatry 10(1):312. https://doi.org/10.1038/s41398-020-00994-0
Bishop DVM, Snowling MJ, Thompson PA, Greenhalgh T, Adams C, Archibald L, Baird G, Bauer A, Bellair J, Boyle C, Brownlie E, Carter G, Clark B, Clegg J, Cohen N, Conti-Ramsden G, Dockrell J, Dunn J, Ebbels S et al (2016) CATALISE: a multinational and multidisciplinary delphi consensus study Identifying language impairments in children. PLoS ONE 11(7):e0158753. https://doi.org/10.1371/JOURNAL.PONE.0158753
Brouwer M, Farzana F, Koopmans F, Chen N, Brunner JW, Oldani S, Li KW, van Weering JR, Smit AB, Toonen RF, Verhage M (2019) SALM1 controls synapse development by promoting F-actin/PIP2-dependent Neurexin clustering. EMBO J 38(17):e101289. https://doi.org/10.15252/EMBJ.2018101289
Butz S, Okamoto M, Südhof TC (1998) A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94(6):773–782. https://doi.org/10.1016/S0092-8674(00)81736-5
Calder SD, Brennan-Jones CG, Robinson M, Whitehouse A, Hill E (2022) The prevalence of and potential risk factors for Developmental Language Disorder at 10 years in the Raine Study. J Paediatr Child Health 58(11):2044–2050. https://doi.org/10.1111/jpc.16149
Centanni TM, Green JR, Iuzzini-Seigel J, Bartlett CW, Hogan TP (2015) Evidence for the multiple hits genetic theory for inherited language impairment: A case study. Front Genet 6:272. https://doi.org/10.3389/FGENE.2015.00272
Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4(1):7. https://doi.org/10.1186/S13742-015-0047-8/2707533
Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, de Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74(6):1128–1135. https://doi.org/10.1086/421054
Chen X, Shen Y, Gao Y, Zhao H, Sheng X, Zou J, Lip V, Xie H, Guo J, Shao H, Bao Y, Shen J, Niu B, Gusella JF, Wu BL, Zhang T (2013) Detection of copy number variants reveals association of cilia genes with neural tube defects. PLoS ONE 8(1):e54492. https://doi.org/10.1371/JOURNAL.PONE.0054492
Chen X, An Y, Gao Y, Guo L, Rui L, Xie H, Sun M, Lam Hung S, Sheng X, Zou J, Bao Y, Guan H, Niu B, Li Z, Finnell RH, Gusella JF, Wu BL, Zhang T (2017a) Rare deleterious PARD3 variants in the aPKC-binding region are implicated in the pathogenesis of human cranial neural tube defects via disrupting apical tight junction formation. Hum Mutat 38(4):378–389. https://doi.org/10.1002/HUMU.23153
Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, Newbury DF, Fisher SE (2017b) Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci Rep 7:46105. https://doi.org/10.1038/SREP46105
Cordeddu V, Redeker B, Stellacci E, Jongejan A, Fragale A, Bradley TEJ, Anselmi M, Ciolfi A, Cecchetti S, Muto V, Bernardini L, Azage M, Carvalho DR, Espay AJ, Male A, Molin AM, Posmyk R, Battisti C, Casertano A et al (2014) Mutations in ZBTB20 cause Primrose syndrome. Nat Genet 46(8):815–817. https://doi.org/10.1038/NG.3035
Costoya JA (2007) Functional analysis of the role of POK transcriptional repressors. Brief Funct Genomic Proteomic 6(1):8–18. https://doi.org/10.1093/BFGP/ELM002
Cui R, Chen D, Li N, Cai M, Wan T, Zhang X, Zhang M, Du S, Ou H, Jiao J, Jiang N, Zhao S, Song H, Song X, Ma D, Zhang J, Li S (2022) PARD3 gene variation as candidate cause of nonsyndromic cleft palate only. J Cell Mol Med 26(15):4292–4304. https://doi.org/10.1111/JCMM.17452
Dale PS, von Stumm S, Selzam S, Hayiou-Thomas ME (2020) Does the inclusion of a genome-wide polygenic score improve early risk prediction for later language and literacy delay? J Speech Lang Hear Res 63(5):1467–1478. https://doi.org/10.1044/2020_JSLHR-19-00161
Damianidou E, Mouratidou L, Kyrousi C (2022) Research models of neurodevelopmental disorders: the right model in the right place. Front Neurosci 16:1846. https://doi.org/10.3389/FNINS.2022.1031075/BIBTEX
Dauber A, LaFranchi SH, Maliga Z, Lui JC, Moon JE, McDeed C, Henke K, Zonana J, Kingman GA, Pers TH, Baron J, Rosenfeld RG, Hirschhorn JN, Harris MP, Hwa V (2012) Novel microcephalic primordial dwarfism disorder associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab 97(11):E2140–E2151. https://doi.org/10.1210/JC.2012-2150
de Munnik SA, García-Miñaúr S, Hoischen A, van Bon BW, Boycott KM, Schoots J, Hoefsloot LH, Knoers NVAM, Bongers EMHF, Brunner HG (2014) A de novo non-sense mutation in ZBTB18 in a patient with features of the 1q43q44 microdeletion syndrome. EJHG 22(6):844–846. https://doi.org/10.1038/EJHG.2013.249
Delaneau O, Zagury JF, Robinson MR, Marchini JL, Dermitzakis ET (2019) Accurate, scalable and integrative haplotype estimation. Nat Commun 10(1):5436. https://doi.org/10.1038/s41467-019-13225-y
Demontis D, Walters GB, Athanasiadis G, Walters R, Therrien K, Nielsen TT, Farajzadeh L, Voloudakis G, Bendl J, Zeng B, Zhang W, Grove J, Als TD, Duan J, Satterstrom FK, Bybjerg-Grauholm J, Bækved-Hansen M, Gudmundsson OO, Magnusson SH et al (2023) Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. Nat Genet 55(2):198–208. https://doi.org/10.1038/s41588-022-01285-8
Descipio C, Conlin L, Rosenfeld J, Tepperberg J, Pasion R, Patel A, McDonald MT, Aradhya S, Ho D, Goldstein J, McGuire M, Mulchandani S, Medne L, Rupps R, Serrano AH, Thorland EC, Tsai ACH, Hilhorst-Hofstee Y, Ruivenkamp CAL et al (2012) Subtelomeric deletion of chromosome 10p15.3: clinical findings and molecular cytogenetic characterization. Am J Med Genet A 158A(9):2152–2161. https://doi.org/10.1002/AJMG.A.35574
Desvignes J-P, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, Salgado D (2018) VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res 46(W1):W545–W553. https://doi.org/10.1093/nar/gky471
Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, Hildebrand MS, Webster R, Ma A, Mazoyer B, Francks C, Bahlo M, Scheffer IE, Morgan AT, Shriberg LD, Fisher SE (2019) A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry 24(7):1065–1078. https://doi.org/10.1038/S41380-018-0020-X
Eising E, Mirza-Schreiber N, de Zeeuw EL, Wang CA, Truong DT, Allegrini AG, Shapland CY, Zhu G, Wigg KG, Gerritse ML, Molz B, Alagoz G, Gialluisi A, Abbondanza F, Rimfeld K, van Donkelaar M, Liao Z, Jansen PR, Andlauer TFM et al (2022) Genome-wide analyses of individual differences in quantitatively assessed reading- and language-related skills in up to 34,000 people. Proc Natl Acad Sci USA 119(35):e2202764119. https://doi.org/10.1073/PNAS.2202764119/SUPPL_FILE/PNAS.2202764119.SD15.XLSX
Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JRO, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR (2003) Mutations in SOX2 cause anophthalmia. Nat Genet 33(4):461–463. https://doi.org/10.1038/NG1120
Fattahi Z, Sheikh TI, Musante L, Rasheed M, Taskiran II, Harripaul R, Hu H, Kazeminasab S, Alam MR, Hosseini M, Larti F, Ghaderi Z, Celik A, Ayub M, Ansar M, Haddadi M, Wienker TF, Ropers HH, Kahrizi K et al (2018) Biallelic missense variants in ZBTB11 can cause intellectual disability in humans. Hum Mol Genet 27(18):3177–3188. https://doi.org/10.1093/HMG/DDY220
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Human Genet 84(4):524–533. https://doi.org/10.1016/J.AJHG.2009.03.010
Ge T, Chen CY, Ni Y, Feng YCA, Smoller JW (2019) Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10(1):1776. https://doi.org/10.1038/s41467-019-09718-5
Gordon CT, Vuillot A, Marlin S, Gerkes E, Henderson A, AlKindy A, Holder-Espinasse M, Park SS, Omarjee A, Sanchis-Borja M, Ben Bdira E, Oufadem M, Sikkema-Raddatz B, Stewart A, Palmer R, McGowan R, Petit F, Delobel B, Speicher MR et al (2013) Heterogeneity of mutational mechanisms and modes of inheritance in auriculocondylar syndrome. J Med Genet 50(3):174–186. https://doi.org/10.1136/JMEDGENET-2012-101331
Granadillo JL, Stegmann APA, Guo H, Xia K, Angle B, Bontempo K, Ranells JD, Newkirk P, Costin C, Viront J, Stumpel CT, Sinnema M, Panis B, Pfundt R, Krapels IPC, Klaassens M, Nicolai J, Li J, Jiang Y et al (2020) Pathogenic variants in TNRC6B cause a genetic disorder characterised by developmental delay/intellectual disability and a spectrum of neurobehavioural phenotypes including autism and ADHD. J Med Genet 57(10):717–724. https://doi.org/10.1136/JMEDGENET-2019-106470
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, Awashti S, Belliveau R, Bettella F, Buxbaum JD, Bybjerg-Grauholm J, Bækvad-Hansen M, Cerrato F, Chambert K, Christensen JH et al (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51(3):431. https://doi.org/10.1038/S41588-019-0344-8
Halekoh U, Højsgaard S, Yan J (2006) The R package geepack for generalized estimating equations. J Stat Softw 15(2):1–11. https://doi.org/10.18637/JSS.V015.I02
Hirose T, Sugitani Y, Kurihara H, Kazama H, Kusaka C, Noda T, Takahashi H, Ohno S (2022) PAR3 restricts the expansion of neural precursor cells by regulating hedgehog signaling. Development (cambridge, England) 149(21):dev199931. https://doi.org/10.1242/DEV.199931
Kalnak N, Sahlén B (2022) Description and prediction of reading decoding skills in Swedish children with Developmental Language Disorder. Logoped Phoniatr Vocol 47(2):84–91. https://doi.org/10.1080/14015439.2020.1839964
Kalnak N, Peyrard-Janvid M, Sahlén B, Forssberg H (2012) Family history interview of a broad phenotype in specific language impairment and matched controls. Genes Brain Behav 11(8):921–927. https://doi.org/10.1111/J.1601-183X.2012.00841.X
Kalnak N, Peyrard-Janvid M, Forssberg H, Sahlén B (2014) Nonword repetition—a clinical marker for specific language impairment in Swedish associated with parents’ language-related problems. PLoS ONE 9(2):e89544. https://doi.org/10.1371/JOURNAL.PONE.0089544
Kalnak N, Stamouli S, Peyrard-Janvid M, Rabkina I, Becker M, Klingberg T, Kere J, Forssberg H, Tammimies K (2018) Enrichment of rare copy number variation in children with developmental language disorder. Clin Genet 94(3–4):313–320. https://doi.org/10.1111/CGE.13389
Kanai SM, Heffner C, Cox TC, Cunningham ML, Perez FA, Bauer A, Reigan P, Carter C, Murray SA, Clouthier DE (2022) Auriculocondylar Syndrome 2 results from dominant negative action of PLCB4 variants. DMM Dis Models Mech. https://doi.org/10.1242/DMM.049320/VIDEO-2
Khan ES, Danckwardt S (2022) Pathophysiological role and diagnostic potential of R-loops in cancer and beyond. Genes 13(12):2181. https://doi.org/10.3390/GENES13122181
Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen-Viet TA, Bowers P, Sidorenko J, Karlsson Linnér R, Fontana MA, Kundu T, Lee C, Li H, Li R, Royer R, Timshel PN, Walters RK, Willoughby EA et al (2018) Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50(8):1112–1121. https://doi.org/10.1038/S41588-018-0147-3
Leonoudakis D, Conti LR, Radeke CM, McGuire LMM, Vandenberg CA (2004) A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J Biol Chem 279(18):19051–19063. https://doi.org/10.1074/JBC.M400284200
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987. https://doi.org/10.1093/BIOINFORMATICS/BTR509
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li D, Choque Olsson N, Becker M, Arora A, Jiao H, Norgren N, Jonsson U, Bölte S, Tammimies K (2022) Rare variants in the outcome of social skills group training for autism. Autism Res 15(3):434–446. https://doi.org/10.1002/AUR.2666
Li Q, Jiang Z, Zhang L, Cai S, Cai Z (2023) Auriculocondylar syndrome: pathogenesis, clinical manifestations and surgical therapies. J Formos Med Assoc 122(9):822–842. https://doi.org/10.1016/J.JFMA.2023.04.024
Ma J, Chen L, He XX, Wang YJ, Yu HL, He ZX, Zhang LQ, Zheng YW, Zhu XJ (2019) Functional prediction and characterization of Dip2 gene in mice. Cell Biol Int 43(4):421–428. https://doi.org/10.1002/CBIN.11106
Ma D, Wang F, Wang R, Hu Y, Chen Z, Huang N, Tian Y, Xia Y, Teng J, Chen J (2022) α-/γ-Taxilin are required for centriolar subdistal appendage assembly and microtubule organization. Elife 11:e73252. https://doi.org/10.7554/ELIFE.73252
Ma D, Wang F, Teng J, Huang N, Chen J (2023) Structure and function of distal and subdistal appendages of the mother centriole. J Cell Sci 136(3):jcs60560. https://doi.org/10.1242/JCS.260560
Marees AT, de Kluiver H, Stringer S, Vorspan F, Curis E, Marie-Claire C, Derks EM (2018) A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int J Methods Psychiatr Res 27(2):e1608. https://doi.org/10.1002/MPR.1608
McGregor KK (2020) How we fail children with developmental language disorder. Lang Speech Hear Serv Sch 51(4):981–992. https://doi.org/10.1044/2020_LSHSS-20-00003
Mizuno M, Matsumoto A, Hamada N, Ito H, Miyauchi A, Jimbo EF, Momoi MY, Tabata H, Yamagata T, Nagata KI (2015) Role of an adaptor protein Lin-7B in brain development: possible involvement in autism spectrum disorders. J Neurochem 132(1):61–69. https://doi.org/10.1111/JNC.12943
Mondal S, Kinatukara P, Singh S, Shambhavi S, Patil GS, Dubey N, Singh SH, Pal B, Shekar PC, Kamat SS, Sankaranarayanan R (2022) DIP2 is a unique regulator of diacylglycerol lipid homeostasis in eukaryotes. Elife 11:e77665. https://doi.org/10.7554/ELIFE.77665
Moreira MC, Klur S, Watanabe M, Németh AH, le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schöls L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE et al (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 36(3):225–227. https://doi.org/10.1038/NG1303
Morris-Rosendahl DJ, Crocq MA (2020) Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci 22(1):65–72. https://doi.org/10.31887/DCNS.2020.22.1/MACROCQ
Motodate R, Saito H, Sobu Y, Hata S, Saito Y, Nakaya T, Suzuki T (2019) X11 and X11-like proteins regulate the level of extrasynaptic glutamate receptors. J Neurochem 148(4):480–498. https://doi.org/10.1111/JNC.14623
Mountford HS, Braden R, Newbury DF, Morgan AT (2022) The genetic and molecular basis of developmental language disorder: a review. Children (basel, Switzerland) 9(5):586. https://doi.org/10.3390/CHILDREN9050586
Mukhopadhyay M, Pelka P, DeSousa D, Kablar B, Schindler A, Rudnicki MA, Campos AR (2002) Cloning, genomic organization and expression pattern of a novel Drosophila gene, the disco-interacting protein 2 (dip2), and its murine homolog. Gene 293(1–2):59–65. https://doi.org/10.1016/S0378-1119(02)00694-7
Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, Christian SL, Ullmann R, Kuechler A, Haas CA, Flubacher A, Charnas LR, Uyanik G, Frank U, Klopocki E, Dobyns WB, Kutsche K (2008) Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet 40(9):1065–1067. https://doi.org/10.1038/NG.194
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814. https://doi.org/10.1093/nar/gkg509
Nishio M, Matsuura T, Hibi S, Ohta S, Oka C, Sasai N, Ishida Y, Matsuda E (2022) Heterozygous loss of Zbtb38 leads to early embryonic lethality via the suppression of Nanog and Sox2 expression. Cell Prolif 55(4):e13215. https://doi.org/10.1111/CPR.13215
Nitin R, Shaw DM, Rocha DB, Walters CE, Chabris CF, Camarata SM, Gordon RL, Below JE (2022) Association of developmental language disorder with comorbid developmental conditions using algorithmic phenotyping. JAMA Netw Open 5(12):e2248060. https://doi.org/10.1001/JAMANETWORKOPEN.2022.48060
Nitta Y, Yamazaki D, Sugie A, Hiroi M, Tabata T (2017) DISCO Interacting Protein 2 regulates axonal bifurcation and guidance of Drosophila mushroom body neurons. Dev Biol 421(2):233–244. https://doi.org/10.1016/J.YDBIO.2016.11.015
Noblett N, Wu Z, Ding ZH, Park S, Roenspies T, Flibotte S, Chisholm AD, Jin Y, Colavita A (2019) DIP-2 suppresses ectopic neurite sprouting and axonal regeneration in mature neurons. J Cell Biol 218(1):125–133. https://doi.org/10.1083/JCB.201804207
Ohishi A, Masunaga Y, Iijima S, Yamoto K, Kato F, Fukami M, Saitsu H, Ogata T (2020) De novo ZBTB7A variant in a patient with macrocephaly, intellectual disability, and sleep apnea: implications for the phenotypic development in 19p13.3 microdeletions. J Hum Genet 65(2):181–186. https://doi.org/10.1038/S10038-019-0690-5
Okbay A, Wu Y, Wang N, Jayashankar H, Bennett M, Nehzati SM, Sidorenko J, Kweon H, Goldman G, Gjorgjieva T, Jiang Y, Hicks B, Tian C, Hinds DA, Ahlskog R, Magnusson PKE, Oskarsson S, Hayward C, Campbell A et al (2022) Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat Genet 54(4):437–449. https://doi.org/10.1038/s41588-022-01016-z
Oo ZM, Adlat S, Sah RK, Myint MZZ, Hayel F, Chen Y, Htoo H, Bah FB, Bahadar N, Chan MK, Zhang L, Feng X, Zheng Y (2020) Brain transcriptome study through CRISPR/Cas9 mediated mouse Dip2c gene knock-out. Gene 758:144975. https://doi.org/10.1016/J.GENE.2020.144975
Özaslan A, Kayhan G, İşeri E, Ergün MA, Güney E, Perçin FE (2021) Identification of copy number variants in children and adolescents with autism spectrum disorder: a study from Turkey. Mol Biol Rep 48(11):7371–7378. https://doi.org/10.1007/S11033-021-06745-8
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell-Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE, Tayoun AA, Berg JS, Cutting GR, Ellard S, Kang P, Karbassi I et al (2022) Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet 109(12):2163–2177. https://doi.org/10.1016/j.ajhg.2022.10.013
Piluso G, D’Amico F, Saccone V, Bismuto E, Rotundo IL, di Domenico M, Aurino S, Schwartz CE, Neri G, Nigro V (2009) A missense mutation in CASK causes FG syndrome in an Italian family. Am J Hum Genet 84(2):162–177. https://doi.org/10.1016/J.AJHG.2008.12.018
Plug MB, van Wijngaarden V, de Wilde H, van Binsbergen E, Stegeman I, van den Boogaard MJH, Smit AL (2021) Clinical Characteristics and Genetic Etiology of Children With Developmental Language Disorder. Front Pediatr 9:651995. https://doi.org/10.3389/FPED.2021.651995
Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, Lacombe D, Geneviève D, Fontana E, Darra F, Cances C, Barth M, Bonneau D, Bernadina BD, N’Guyen S et al (2013) Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45(6):639–647. https://doi.org/10.1038/NG.2613
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, Xia F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE et al (2017) Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 376(1):21–31. https://doi.org/10.1056/NEJMOA1516767/SUPPL_FILE/NEJMOA1516767_DISCLOSURES.PDF
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Rieder MJ, Green GE, Park SS, Stamper BD, Gordon CT, Johnson JM, Cunniff CM, Smith JD, Emery SB, Lyonnet S, Amiel J, Holder M, Heggie AA, Bamshad MJ, Nickerson DA, Cox TC, Hing AV, Horst JA, Cunningham ML (2012) A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am J Hum Genet 90(5):907–914. https://doi.org/10.1016/J.AJHG.2012.04.002
Rubinacci S, Delaneau O, Marchini J (2020) Genotype imputation using the Positional Burrows Wheeler Transform. PLoS Genet 16(11):e1009049. https://doi.org/10.1371/JOURNAL.PGEN.1009049
Sakasai R, Isono M, Wakasugi M, Hashimoto M, Sunatani Y, Matsui T, Shibata A, Matsunaga T, Iwabuchi K (2017) Aquarius is required for proper CtIP expression and homologous recombination repair. Sci Rep 7(1):1–11. https://doi.org/10.1038/s41598-017-13695-4
Sam M, Wurst W, Klüppel M, Jin O, Heng H, Bernstein A (1998) Aquarius, a novel gene isolated by gene trapping with an RNA-dependent RNA polymerase motif. Dev Dyn 212(2):304–317. https://doi.org/10.1002/(sici)1097-0177(199806)212:2%3c304::aid-aja15%3e3.0.co;2-3
Sasai N, Matsuda E, Sarashina E, Ishida Y, Kawaichi M (2005) Identification of a novel BTB-zinc finger transcriptional repressor, CIBZ, that interacts with CtBP corepressor. Genes Cells 10(9):871–885. https://doi.org/10.1111/J.1365-2443.2005.00885.X
Schwob S, Eddé L, Jacquin L, Leboulanger M, Picard M, Oliveira PR, Skoruppa K (2021) Using nonword repetition to identify developmental language disorder in monolingual and bilingual children: a systematic review and meta-analysis. J Speech Lang Hear Res 64(9):3578–3593. https://doi.org/10.1044/2021_JSLHR-20-00552
Snijders Blok L, Hiatt SM, Bowling KM, Prokop JW, Engel KL, Cochran JN, Bebin EM, Bijlsma EK, Ruivenkamp CAL, Terhal P, Simon MEH, Smith R, Hurst JA, McLaughlin H, Person R, Crunk A, Wangler MF, Streff H, Symonds JD et al (2018) De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum Genet 137(5):375–388. https://doi.org/10.1007/S00439-018-1887-Y
Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36(10):928–930. https://doi.org/10.1002/humu.22844
Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA (2014) Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56(6):777–785. https://doi.org/10.1016/J.MOLCEL.2014.10.020
Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, Firth HV, Frazier T, Hansen RL, Prock L, Brunner H, Hoang N, Scherer SW, Sahin M, Miller DT (2019) Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med 21(11):2413–2421. https://doi.org/10.1038/S41436-019-0554-6
Tomblin JB, Records NL, Buckwalter P, Zhang X, Smith E, O’Brien M (1997) Prevalence of specific language impairment in kindergarten children. JSLHR 40(6):1245–1260. https://doi.org/10.1044/JSLHR.4006.1245
Trivisano M, de Dominicis A, Micalizzi A, Ferretti A, Dentici ML, Terracciano A, Calabrese C, Vigevano F, Novelli G, Novelli A, Specchio N (2022) MED13 mutation: a novel cause of developmental and epileptic encephalopathy with infantile spasms. Seizure 101:211–217. https://doi.org/10.1016/J.SEIZURE.2022.09.002
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35(suppl 2):W71–W74. https://doi.org/10.1093/nar/gkm306
van der Spek J, den Hoed J, Snijders Blok L, Dingemans AJM, Schijven D, Nellaker C, Venselaar H, Astuti GDN, Barakat TS, Bebin EM, Beck-Wödl S, Beunders G, Brown NJ, Brunet T, Brunner HG, Campeau PM, Čuturilo G, Gilissen C, Haack TB et al (2022) Inherited variants in CHD3 show variable expressivity in Snijders Blok-Campeau syndrome. Genet Med 24(6):1283–1296. https://doi.org/10.1016/J.GIM.2022.02.014
Vegas N, Demir Z, Gordon CT, Breton S, Romanelli Tavares VL, Moisset H, Zechi-Ceide R, Kokitsu-Nakata NM, Kido Y, Marlin S, Gherbi Halem S, Meerschaut I, Callewaert B, Chung B, Revencu N, Lehalle D, Petit F, Propst EJ, Papsin BC et al (2022) Further delineation of auriculocondylar syndrome based on 14 novel cases and reassessment of 25 published cases. Hum Mutat 43(5):582–594. https://doi.org/10.1002/HUMU.24349
Villanueva P, Nudel R, Hoischen A, Fernández MA, Simpson NH, Gilissen C, Reader RH, Jara L, Echeverry MM, Francks C, Baird G, Conti-Ramsden G, O’Hare A, Bolton PF, Hennessy ER, Palomino H, Carvajal-Carmona L, Veltman JA, Cazier JB et al (2015) Exome sequencing in an admixed isolated population indicates NFXL1 variants confer a risk for specific language impairment. PLoS Genet 11(3):e1004925. https://doi.org/10.1371/JOURNAL.PGEN.1004925
Wang Y, Zeng C, Li J, Zhou Z, Ju X, Xia S, Li Y, Liu A, Teng H, Zhang K, Shi L, Bi C, Xie W, He X, Jia Z, Jiang Y, Cai T, Wu J, Xia K, Sun ZS (2018) PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell Rep 24(8):2029–2041. https://doi.org/10.1016/J.CELREP.2018.07.061
Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, Werge T, Hougaard DM, Taylor J, Skuse D, Devlin B, Anney R, Sanders SJ, Bishop S, Mortensen PB et al (2017) Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 49(7):978. https://doi.org/10.1038/NG.3863
Xiang J, Peng J, Baxter S, Peng Z (2020) AutoPVS1: an automatic classification tool for PVS1 interpretation of null variants. Hum Mutat 41(9):1488–1498. https://doi.org/10.1002/humu.24051
Yang L, Zhao S, Ma N, Liu L, Li D, Li X, Wang Z, Song X, Wang Y, Wang D (2022) Novel DIP2C gene splicing variant in an individual with focal infantile epilepsy. Am J Med Genet A 188(1):210–215. https://doi.org/10.1002/AJMG.A.62524
Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, Obiajulu JU, Wright JR, Murali SC, Xu SX, Brueggeman L, Thomas TR, Marchenko O, Fleisch C, Barns SD, Snyder LG, Han B, Chang TS, Turner TN et al (2022) Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet 54(9):1305–1319. https://doi.org/10.1038/s41588-022-01148-2
Acknowledgements
The authors are grateful to the study participants, their families, and the schools for their cooperation. The authors thank Ingegerd Fransson for technical help with DNA extraction and the core facility for Bioinformatics and Expression Analysis at Karolinska Institutet for genotyping. The authors thank Maria Argyroudi for technical assistance with variant validation. Anna Gellerbring, Emma Sernstad, Emilia Ottosson, and Valtteri Wirta from Clinical Genomics Stockholm core facility, Karolinska Institutet, and Science for Life Laboratory for conducting exome sequencing experiments and variant calling. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at Uppsala University partially funded by the Swedish Research Council through Grant Agreements Nos. 2022-06725 and 2018-05973.
Funding
Open access funding provided by Karolinska Institute. The Swedish Brain Foundation, Stiftelsen Promobilia; Stiftelsen Sunnerdahls Handikappfond; Stiftelsen Sven Jerrings Fond; Aina Börjesons Foundation, Marcus Borgströms Foundation, the Strategic Neuroscience Program at Karolinska Institutet, The Foundation for Strategic Research.
Author information
Authors and Affiliations
Contributions
AY, NK, and KT designed the study. NK collected phenotype information. AY and DL analyzed the WES data. AY performed PRS and statistical calculations. SR provided technical assistance. AY and SL conducted Sanger sequencing experiments. AY and KT drafted the manuscript. KT obtained funds for implementing the study procedures. All authors critically revised and approved the final version of the manuscript and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Consent to participate
Written informed consent was obtained from the families.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. The study was approved by the local ethics committee in Stockholm (Dnr. 2008/543-31/3, 2008/1052-32, and 2010/1746-32).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yahia, A., Li, D., Lejerkrans, S. et al. Whole exome sequencing and polygenic assessment of a Swedish cohort with severe developmental language disorder. Hum. Genet. 143, 169–183 (2024). https://doi.org/10.1007/s00439-023-02636-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-023-02636-z