
Abstract
Contactin-associated protein-like 2 (CNTNAP2) gene encodes for CASPR2, a presynaptic type 1 transmembrane protein, involved in cell–cell adhesion and synaptic interactions. Biallelic CNTNAP2 loss has been associated with “Pitt-Hopkins-like syndrome-1” (MIM#610042), while the pathogenic role of heterozygous variants remains controversial. We report 22 novel patients harboring mono- (n = 2) and bi-allelic (n = 20) CNTNAP2 variants and carried out a literature review to characterize the genotype–phenotype correlation. Patients (M:F 14:8) were aged between 3 and 19 years and affected by global developmental delay (GDD) (n = 21), moderate to profound intellectual disability (n = 17) and epilepsy (n = 21). Seizures mainly started in the first two years of life (median 22.5 months). Antiseizure medications were successful in controlling the seizures in about two-thirds of the patients. Autism spectrum disorder (ASD) and/or other neuropsychiatric comorbidities were present in nine patients (40.9%). Nonspecific midline brain anomalies were noted in most patients while focal signal abnormalities in the temporal lobes were noted in three subjects. Genotype–phenotype correlation was performed by also including 50 previously published patients (15 mono- and 35 bi-allelic variants). Overall, GDD (p < 0.0001), epilepsy (p < 0.0001), hyporeflexia (p = 0.012), ASD (p = 0.009), language impairment (p = 0.020) and severe cognitive impairment (p = 0.031) were significantly associated with the presence of biallelic versus monoallelic variants. We have defined the main features associated with biallelic CNTNAP2 variants, as severe cognitive impairment, epilepsy and behavioral abnormalities. We propose CASPR2-deficiency neurodevelopmental disorder as an exclusively recessive disease while the contribution of heterozygous variants is less likely to follow an autosomal dominant inheritance pattern.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Contactin-associated protein-like 2 (CNTNAP2) is one of the largest genes in the human genome located on chromosome 7q35-36.1 (Nakabayashi and Scherer 2001). It encodes for CASPR2, a member of the neurexin superfamily of cell adhesion proteins (Poliak et al. 1999). CASPR2 is a presynaptic type 1 transmembrane protein, with a large extracellular and smaller intracellular portion that participates in cell–cell adhesion and synaptic interactions. CNTNAP2 is expressed throughout the developing and adult central nervous system (CNS) (Peñagarikano 2011). Mouse studies have uncovered a role for CASPR2 in neuronal migration and postmitotic neuronal development (Canali et al. 2018; Fernandes et al. 2019). Experimental studies on knock-out mice and in human cell lines support the hypothesis that CASPR2 is involved in neuronal migration, myelination, and neuronal transmission with a reduction in both inhibitory GABAergic neuronal numbers and excitatory neurotransmission (Peñagarikano 2011).
A homozygous 1-bp deletion (c.3709delG) of CNTNAP2 was initially detected in an Old Order Amish kindred, whose nine affected children exhibited mild motor delay until the onset of intractable seizures during infancy, which were followed by deterioration in learning and language abilities, and social behavior (Strauss et al. 2006). Three subjects showed unilateral cortical dysplasia of the anterior temporal lobe, and neuronal migration defects from brain specimen biopsies. Altogether, this neurological disorder was named cortical dysplasia-focal epilepsy (CDFE) syndrome (Strauss et al. 2006). Subsequently, homozygous or compound heterozygous variants and/or intragenic deletions within CNTNAP2 were associated with Pitt-Hopkins like syndrome 1 (PTHSL1, MIM#610042), with variable features that included intellectual disability (ID), early seizure onset, regression of language ability, and hyper-breathing patterns (Strauss et al. 2006; Zweier et al. 2009; Smogavec et al. 2016). Given the lack of typical Pitt-Hopkins craniofacial features and hyper-breathing patterns in most patients, it has recently been proposed that biallelic loss of CNTNAP2 results in a disorder called “CASPR2-deficiency neurodevelopmental disorder (NDD)”, which includes severe ID, early infantile seizures, language regression, variable presence of autistic features, hyporeflexia and ataxia (Rodenas-Cuadrado et al. 2016).
A growing body of literature over the last two decades underscored a possible role of heterozygous chromosomal translocations and deletions, single nucleotide polymorphisms (SNPs), and rare heterozygous variants of CNTNAP2. These were found in a wide array of neuropsychiatric disorders, such as autism spectrum disorder (ASD), schizophrenia, obsessive–compulsive disorder, Gilles de la Tourette syndrome, attention deficit hyperactivity disorder (ADHD), dyslexia, specific language impairment and stuttering (Verkerk et al. 2003; Arking et al. 2008; Friedman et al. 2008; Mikhail et al. 2011; Newbury et al. 2011; Ji et al. 2013; Centanni et al. 2015). However, heterozygous CNTNAP2 variations are also present in the healthy population including healthy parents of children with either mono- or biallelic variants. Thus, the evidence for the role of heterozygous variants in CNTNAP2 in neuropsychiatric disorders has yet to be clarified (Toma et al. 2018). The identification and description of new patients with CNTNAP2 variants may further define the criteria of the syndrome and better characterize its genotype–phenotype correlation (Rodenas-Cuadrado et al. 2016). We report 22 patients harboring mono- or biallelic variants in CNTNAP2 and show genotype–phenotype correlations by including a further 50 previously reported patients.
Material and methods
Patient recruitment
We recruited 22 previously unreported patients from 17 unrelated families carrying mono or biallelic variants in CNTNAP2. Patients were followed up at 16 centers worldwide for developmental and epileptic encephalopathy (DEE) and/or neurodevelopmental disorders. Genetic analyses were performed either in a diagnostic or research setting. Subsequently, they were enrolled using the international platform GeneMatcher (Sobreira et al. 2015).
Firstly, the respective referring clinicians were asked to fill in a spreadsheet with all clinical and genetic information for each patient (Online Resource). Secondly, all available clinical and genetic data, electroencephalography (EEG) and neuroradiological images were reviewed by expert pediatric neurologists, neuroradiologist and geneticists. Written informed consent was obtained from parents or guardians.
Genetic testing
Most CNTNAP2 variants were detected by epilepsy Next Generation Sequencing (NGS) panel (n = 11) or autism/ID NGS panel (n = 1). Exome sequencing (ES) (singleton n = 3; trios n = 7) was performed in the respective collaborating centers using different analysis platforms according to the BWA/GATK’s based pipelines. Targeted Sanger sequencing using standard methods was also performed either for verification of identified variants or segregation analysis. Sequencing methods and additional genetic analyses performed per individual are summarized in Online Resource. All variants were classified according to the ACMG/AMP criteria (Richards et al. 2015). CNTNAP2 variants are listed according to the transcript NM_014141.6 and copy number variants (CNV) refer to the hg19/GRCh37 assembly.
Literature review
We performed a literature review on MEDLINE (accessed by PubMed, updated to December 2022) with the search term “CNTNAP2” and “CASPR2”, including articles with reported pathogenic or likely pathogenic variants or variants of uncertain significance (VUS) in CNTNAP2 that were suspected to contribute to the phenotype of patients. Patients with copy number variation (CNV) that encompassed other genes that were likely to contribute to the phenotype and/or reports without available clinical information were excluded. We also excluded reports of subjects with limited clinical information.
Statistical analysis
We used descriptive analysis to characterize our cohort and previously published CNTNAP2 patients. Based on both datasets, we compared the phenotypes of patients harboring a heterozygosity variant versus (vs.) patients with biallelic variants using chi-squared test or a Fisher’s exact test.
Results
Patients
We enrolled 22 patients (14 males) aged between 3 and 19 years (Table 1). Consanguinity was reported in six families (6/17, 35.3%); a family history of neurological diseases and/or disabilities was present in 11/17 subjects (64.7%).
Auxology and dysmorphology
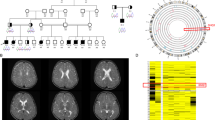
Five patients (5/22, 22.7%) presented with failure to thrive. Four subjects had microcephaly while one was found to be macrocephalic. A total of eight patients (8/22, 36.3%) exhibited non-specific facial dysmorphisms (Fig. 1a). In addition, café-au-lait stains were observed in two patients.

a Ind- 5, -6, -7, -8, -12, -14, -15, -16 and -17 iconography is shown (from left to right). Common facial dysmorphisms are shown including prominent ears (Ind-6 and Ind-7) and hypertelorism (Ind-6, Ind-12). Ind-7 shows mild ptosis of the left eyelid. Ind-14 presents with lips thickness, prognathism, and prominent philtrum. A lean, elongated face with mild lax skin is observed in Ind-15. Ind-17 has sparse hair. No noticeable dysmorphisms are appreciable in Ind-5, Ind-8, Ind-16 and Ind-17. b Brain MRI findings of the patients and a control; sagittal T1-weighted (first) and coronal and/or axial T2-weighted (middle and/or last) images. Inferior cerebellar vermis hypoplasia is noted in all the cases included in the figure (thin arrows) associated with mild superior cerebellar vermis atrophy in Ind-3, Ind-8, Ind-12, and Ind-17 (empty arrowheads). A thin corpus callosum is present in Ind-2, Ind-13 and Ind-16, while a thick posterior corpus callosum is noted in Ind-3 and Ind-8 (empty arrows). Mild white matter volume reduction with consequent ventricular enlargement is noted in Ind-3, Ind-13 and Ind-16 (asterisks). Cerebellar dentate nuclei T2 hyperintensity is visible in Ind-6 and Ind-12 (arrowheads). In Ind-2, Ind-8 and Ind-12 there are additional uni- or bilateral T2 hyperintensities at the level of the anterior temporal lobes (thick arrows) in keeping with focal cortical dysplasias
Neurodevelopment
Global developmental delay (GDD), of variable severity, is reported in almost all patients (21/22, 95.5%). Moreover, individual-1 (Ind-1) and Ind-3 had early normal development before the onset of epilepsy, leading to major irreversible regression, while Ind-17 experienced a partial recovery of her cognitive and motor skills after seizure control. Intellectual disability (ID) has been assessed as mild in 4 patients, moderate in 9, severe in 7 and profound in 1 subject, whereas Ind-22 had a borderline intelligence quotient.
Epilepsy
Epilepsy occurred in 21 patients (95.5%) with onset at median age of 22.5 months [17 25th percentile–29.2 75th percentile]. Major findings are summarized in Table 2. Seizures were mainly described as primary generalized tonic–clonic (GTC) seizures (11/21, 52.3%) or focal motor seizures with impaired awareness (FIA) (11/21, 52.3%) and focal to bilateral (7/21, 33.3%). Tonic seizures (5/21, 23.8%), absences (3/21, 14.3%) and atonic seizures (2/21, 9.5%) were also reported. In three patients fever represented a trigger (3/21, 14.3%). Status epilepticus has occurred in 2 individuals (2/21, 9.5%). Half of the cohort experienced daily seizures at onset (10/21, 47.6%). Median number of anti-seizure medications (ASMs), prescribed over the course of their history, was 3 [3 25th percentile–5 75th percentile]. Eight patients (8/21, 38%) achieved seizure freedom for more than one year, and the other 8 (8/21, 38%) benefited from ASMs by showing a considerable seizure frequency reduction greater than 50%. None of them discontinued ASMs nor did any of them undergo epilepsy surgery. EEG often showed epileptic discharges in the temporal or fronto-temporal regions (8/21, 38%) (Fig. 2).

EEG features. a. Ind-1, 10 years old. Sleep recording. High voltage bilateral anterior delta waves and focal spikes over the frontal regions of both hemispheres. b Ind-2, 2 years 9 months. Awake recording. Synchronous and asynchronous spikes on bilateral frontal–temporal regions. c Ind-10, 3 years old. Awake recording. Right central-temporal medium voltage sharp waves. d Ind-10, 3 years old. Sleep recording. Nearly sub-continuous trend of right central-temporal sharp waves in the N2 phase, with a tendency to spread
Neuropsychiatric features and other neurological and neurobehavioral findings
Expressive and/or receptive language was consistenly impaired in all patients. A formal diagnosis of ASD was reported in nine patients (9/22, 40.9%), variably associated with other neuropsychiatric comorbidities such as hyperactivity (4/21, 19%) and behavioral issues (4/21, 19%). More specifically, sudden episodes of aggressive and violent behavior were reported in Ind-3, Ind-6 and Ind-21, while psychomotor agitation occurred occasionally in Ind-8. Coprophagia was reported in two sisters (2/22, 9.1%) from family 2. No other psychiatric comorbidities have been identified in our population. Neurological examination revealed hypotonia of varying degrees in 12 cases (12/22, 55%) and hyporeflexia in 5 (5/22, 23%). Six patients exhibited an ataxic gait (6/22, 27%). Two patients presented with breathing disorders consisting of episodes of hyperpnea and apnea during the day (2/22, 9%). No sensorineural deficits or extrapyramidal disorders were noted.
Neuroimaging
Neuroimaging studies were performed in 21/22 subjects, including 18 brain magnetic resonance imaging (MRI) and 3 computed tomography (CT) studies (Ind-1, Ind-5 and Ind-7). Brain MRI revealed non-specific dysmorphisms in the majority of subjects (11/21, 52.4%) (Fig. 1b), including inferior cerebellar vermis hypoplasia (9/21, 42.9%), abnormalities of the corpus callosum (6/21, 28.5%; thick in two cases and thin in four other cases), superior cerebellar vermis atrophy (4/21, 19%), mild white matter volume reduction with ventricular enlargement (3/21, 14.3%), cerebellar dentate nuclei signal alterations (2/21, 9.5%), and mild cerebral atrophy (2/21, 9.5%). Signal abnormalities consistent with focal cortical dysplasia were noted at the level of the anterior temporal lobes in three subjects (Ind-2, Ind-8, and Ind-12). Neuroimaging was unremarkable in ten patients (10/21, 47.6%).
Other comorbidities
Extra-neurological comorbidities occurred in nine individuals (9/22, 40.9%), including recurrent respiratory infections, haematological disorders (pancytopenia, haemolytic anaemia) and rectal prolapse (2/22, 9%). Precocious puberty, asthma, hypogammaglobulinemia, gastroesophageal reflux and osteopenia were reported once (1/22, 4.5%). None of our patients presented with congenital abnormalities of any extra-CNS organ. Two patients in our cohort deceased: the first at the age of 13 (Ind-1) due to cachexia in the context of feeding difficulties and severe GDD, while the other (Ind-20) at 7 years for unknown reasons. No statistically significant differences were observed when comparing patients with a history of consanguinity and non-consanguinity.
Genetic results
A total of 18 distinct CNTNAP2 variants were identified, seven of which were novel (Online Resource). Except for two heterozygous variants, all other individuals were found to harbor biallelic variants; either homozygous (n = 16) or compound heterozygous (n = 4) variants. Variants included ten likely gene-disrupting (LGD) variants, four intragenic deletions (identified either by microarray or an epilepsy NSG gene panel) and three missense variants. Sanger sequencing confirmed variants segregation with the phenotype within these families. All variants were absent or extremely rare in human population variant databases (allele frequency ranging from 0 to 0.0001557 in the gnomAD database). None of the variants were reported in a homozygous state in healthy individuals. LGD variants were scattered throughout CNTNAP2 and included four different frameshift and four nonsense changes and two splice site variants. All frameshift and nonsense variants were predicted to result in premature termination codon and, therefore, likely be degraded through nonsense-mediated mRNA decay (NMD). As such these were classified as pathogenic/likely pathogenic. Of note, the frameshift variant c.1361_1362del p.(Asn454ArgfsTer24) was recurrent in eight subjects of families 1–4 of Croatian Roman ancestry and the nonsense variant c.3262C > Tp.(Arg1088Ter) was found in three subjects of two nonrelated Egyptian families suggesting these variants are likely to be founder mutations in these populations. Two individuals carried homozygous splicing variants as follows: the variant c.1777+2T > C (Ind-11) affects the consensus GT-splice donor site of intron 11 and was computationally predicted to cause a loss of a splice donor site disrupting the reading frame and resulting in NMD (Splice AI score 0.98). Thus, it was classified as likely pathogenic according to the ACMG criteria. The homozygous variant c.550+5G > T (Ind-15) predicts a loss of a splice donor site (Splice AI score 0.66), yet it remains a VUS according to the current ACMG guidelines. CGH-array revealed two intragenic CNTNAP2 deletions in Ind-17: a 31,949 bp deletion in 7q35(147,651,818–147,683,766) encompassing exon 15 and inherited by her mother and a paternally inherited deletion of 9317 bp in 7q36.1 (148,071,316–148,080,632), encompassing exon 22. These deletions were confirmed by multiplex ligation-dependent probe amplification. Epilepsy gene panel showed two compound heterozygous deletions in Ind-10, namely the c.98-?_402+? that encompasses exons 2–3 of CNTNAP2 and the heterozygous deletion c.98-?_1348+?, encompassing exons 2–8. Both deletions were confirmed by CGH-array. Individual-13 harbored the compound heterozygous missense variant c.400T > G p.(Trp134Gly) and c.2449G > A p.(Gly817Arg) that were classified as VUS. We included subjects harboring these biallelic VUS given supporting criteria of pathogenicity and consistent phenotype. The heterozygous missense variant c.3814A > T p.(Ile1272Phe) was found to be de novo in individual 21 while an ID/ASD panel identified in Ind-22 the frameshift variant c.1628del p.(Ser543Ilefs*13) that was maternally inherited. Both variants were classified as VUS.
Overall, we ascertained a diagnosis with biallelic CNTNAP2 pathogenic/likely pathogenic variants in 18 out of 22 subjects included in this study. No other pathogenic/likely pathogenic variants were identified in the currently known NDD-related genes in the ES data in these families. Additional VUS detected in our cohort either by ES or microarray are listed in Online Resource.
Previously published cases
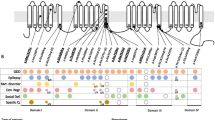
We identified 50 previously published patients from 17 articles (Strauss et al. 2006; Friedman et al. 2008; Jackman et al. 2009; Zweier et al. 2009; Gregor et al. 2011; Al Murrani et al. 2012; Watson et al. 2014; Pippucci et al. 2015; Smogavec et al. 2016; Rodenas-Cuadrado et al. 2016; Riccardi et al. 2019; Falsaperla et al. 2020; Freri et al. 2021; Lu et al. 2021; Mittal et al. 2021; Scala et al. 2021; Badshash et al. 2022) reporting the clinical phenotype of patients carrying pathogenic or likely pathogenic CNTNAP2 variants or VUS suspected to contribute to the phenotype (Online Resource). Figure 3 summarizes the main phenotypic features observed in our cohort and in the literature, distinguishing between heterozygous and homozygous variants, while variant positions are shown in Fig. 4.

Summary of the key clinical features of patients carrying mono- or bi-allelic pathogenic CNTNAP2 variants in our cohort and the literature. ID intellectual disability, NPsy neuropsychiatric findings. Statistical significance refers to patients with biallelic versus monoallelic variants

CNTNAP2 variants position in our cohort (in bold, # individual) and previously published patients. The arrow indicates a deletion, and the line a duplication
Genotype–phenotype correlation
Altogether, GDD and epilepsy were significantly more present in patients harboring homozygous variants than in heterozygous patients (p < 0.0001) (Online Resource). Similarly, ASD (p = 0.009), hyporeflexia (p = 0.012), language impairment (p = 0.020), as well as a moderate to severe degree of ID (p = 0.031) were more frequent in patients with biallelic variants.
Discussion
We reported a cohort of 22 new patients harboring either biallelic (20) or monoallelic variants (2) in CNTNAP2. To the best of our knowledge, this is the largest cohort of patients with CNTNAP2 variants reported together to date. Our study corroborates previous literature, confirming that CNTNAP2 deficiency due to biallelic variants leads to a distinct neurodevelopmental disorder typically characterized by developmental delay, seizure onset within the first 2 years followed by developmental regression, moderate to severe ID and variable occurrence of ASD and behavioral abnormalities. Similarly to previous reports, hypotonia and hyporeflexia are frequent, whereas only a few patients display ataxia. Likewise, occipital frontal circumference is normal in the majority of patients in contrast to the initial reports of relative macrocephaly. Furthermore, our patients harboring biallelic variants do not display the typical craniofacial features and abnormal breathing patterns reported for PTHS. Together, this supports previous literature suggesting that the name PTHS1 should be replaced by CASPR2-deficiency NDD (Rodenas-Cuadrado et al. 2016). In addition, the occurrence of epilepsy in virtually all patients within the first 2 years with consequent regression of development and cognitive impairment would suggest a DEE. Epilepsy is indeed a cardinal feature in patients with biallelic CNTNAP2 variants. The onset of seizures typically occurs in the first two to three years of life. Seizures initially are very frequent and difficult to treat. However, most patients achieve good seizure control within a few years after onset. Seizures are most frequently focal motor, at times with secondary generalization This is in line with previous descriptions in the literature (Strauss et al. 2006; Rodenas-Cuadrado et al. 2016; Smogavec et al. 2016). Cortical areas most typically involved seem to be the frontal and temporal regions (Strauss et al. 2006).
CASPR2 is found in the inhibitory presynaptic compartment and, to a lesser extent, in the excitatory postsynaptic compartment where it is involved in several pivotal processes, such as neurite development and synapse maturation, stability, and function (Horresh et al. 2008). It also localizes to juxtaparanodes of myelinated axons, where it is involved in neuron-glia interactions, and mediates the clustering of potassium channels via interaction with contactin 2 (also known as TAG-1) (Horresh et al. 2008). Similar to humans, Cntnap2−/−mice display epilepsy in addition to ASD features and cortical developmental abnormalities (Peñagarikano et al. 2011). RNAi-mediated knock-down of Caspr2 produced a cell-autonomous decrease in dendritic arborization and spine development in pyramidal neurons, decreasing the number of excitatory and inhibitory synapse numbers, and impairing synaptic transmission (Anderson et al. 2012). Together, these observations suggest that a perturbation of synaptic homeostasis and function due to CASPR2 deficiency leads to an imbalance of excitatory and inhibitory post-synaptic currents in neural networks that may contribute to epilepsy phenotypes (Anderson et al. 2012).
Strauss et al. (2006) described neuroimaging features of focal cortical dysplasia in three subjects that were consistent with findings of neuronal migration defects from brain biopsies. These results were in line with neuropathological and physiological studies in the Cntnap2−/−mice showing neuronal migration abnormalities, reduced number of interneurons and abnormal neuronal network activity (Peñagarikano et al. 2011). Subsequent to this, no further reports have described malformations of cortical development: however, cerebellar hypoplasia and nonspecific white matter abnormalities have been occasionally reported in subjects with biallelic CNTNAP2 variants (Zweier et al. 2009; Smogavec et al. 2016). Here, we describe the largest cohort of subjects for whom brain MRI was available, showing that three subjects had unilateral or bilateral anterior temporal lobe T2 hyperintensities consistent with focal cortical dysplasia, supporting the notion of malformation of cortical development due to CNTNAP2-deficiency. Interestingly, we also noted several nonspecific findings that have been described in subjects with PTHS, including callosal anomalies, white matter volume reduction, dentate nuclei signal alterations and other minor posterior fossa abnormalities.
All our patients suffered from severe speech impairment and one-third had ASD or other behavioral abnormalities including aggressive behavior and stereotypic movements. There is evidence that supports a role for CNTNAP2 in language development, including enriched expression during human brain development in frontotemporal-subcortical circuits known to be critical for human executive function (Alarcón et al. 2008). Despite some conflicting results (Sampath et al. 2013; Murdoch et al. 2015; Toma et al. 2018; Zhang et al. 2019), several studies have linked SNPs in CNTNAP2 variants with ASD and/or language-related disorders (Vernes et al. 2008; Li et al. 2010; Gregor et al. 2011; Uddin et al. 2021). Further, some SNPs (e.g. rs2710102 and rs7794745) have been associated with abnormal activation of the right inferior frontal gyrus (Broca’s area homologue) and right lateral temporal cortex in subject with ASD and reduced volume of specific grey matter areas (Whalley et al. 2011). Together this evidence supports an impact of CNTNAP2 variation on language related brain regions and phenotypes; however, it is not yet clear what role (if any) CASPR2 has in the development of language.
While the loss of function (LoF) mechanism due to biallelic CNTNAP2 variants is well understood, the impact of heterozygous CNTNAP2 variants is more controversial. It has been suggested that the phenotypic picture of each heterozygous variant may result from the combination of two mechanisms. On the one hand a dominant-negative effect on wild-type Caspr2 function might be due to endoplasmic reticulum (ER) retention mimicking the homozygous null phenotype (Canali and Goutebroze 2018). On the other hand a loss of function mechanism for adhesion-defective variant proteins, could enable the interaction with their extracellular partners (Canali and Goutebroze 2018). According to this model, the phenotype of our patient, Ind-21 (mild ID, epilepsy and behavioral abnormalities) harboring the de novo missense variant p.(Ile1272Phe) lying in the extracellular domains may be due to a LoF mechanism if the protein is secreted from the ER. However, the impact of the de novo frameshift variant in the patient (Ind-22) with isolated ASD remains controversial since it is predicted to undergo nonsense-mediated decay and thus it would unlikely exert a dominant negative effect. It is also noteworthy that CNTNAP2 is not constrained for missense and Lof variants in the gnomAD databse (Z score − 0.29, pLI score 0) indicating that heterozygous missense and Lof variants of CNTNAP2 are not subject to negative selection (Lek et al. 2016). This is in line with the fact that carrier parents of CNTNAP2 variants are healthy. Furthermore, large scale studies on gene enriched for de novo variants in NDD have failed to highlight this gene with any meaningful significance (Kaplanis et al. 2020; Satterstrom et al. 2020) and several other studies did not identify a significant burden for CNTNAP2 rare variants in patients with ASD or schizophrenia comparing to controls (Murdoch et al. 2015; Toma et al. 2018; Zhang et al. 2019), suggesting that CNTNAP2 is not a a primary risk gene for psychiatric disorders. Although it might be possible that CNTNAP2 heterozygous variants contribute to ASD and related neuropsychiatric phenotypes with a polygenic inheritance pattern, it seems unlikely based on the above observations that they solely result in a neuropsychiatric phenotype following a classical autosomal dominant Mendelian inheritance. Taken together, we propose CNTNAP2-related NDD as an exclusively recessive disorder while the dominant version is becoming weaker with the increase body of evidence in the literature and in human population variant databases.
In conclusion, we report the largest cohort of patients with CNTNAP2 variants to date and define the core phenotype associated with biallelic CNTNAP2 variants. These data suggest that patients with biallelic variants are likely to develop severe cognitive impairment, epilepsy and variable behavioral abnormalities.
In most cases, patients have an unremarkable perinatal history and a normal psychomotor development or slightly delayed during the first year of life. Concomitant with the epilepsy onset, occurring more often during the second year of life, developmental stagnation or regression is observed. Epilepsy can be difficult to control at the beginning, with a “stormy” phase, while during childhood seizures are usually well-controlled with ASMs. Response to ASMs may be associated with a slight cognitive improvement in some cases, although most patients still suffer from moderate to profound ID throughout their lives. In more severe cases, feeding difficulties, failure to thrive with increased potentially fatal comorbidities may be observed.
The role of heterozygous variants remains to be fully elucidated. Future studies should address the functional impact of heterozygous CNTNAP2 variants and the related pathomechanisms with ultimately important implications for patient management and counselling.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author. CNTNAP2 variants have been submitted to ClinVar.
References
Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM, Geschwind DH (2008) Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet 82(1):150–159. https://doi.org/10.1016/j.ajhg.2007.09.005
Al-Murrani A, Ashton F, Aftimos S, George AM, Love DR (2012) Amino-terminal microdeletion within the CNTNAP2 gene associated with variable expressivity of speech delay. Case Rep Genet 2012:172408. https://doi.org/10.1155/2012/172408
Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Südhof TC (2012) Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci USA 109(44):18120–18125. https://doi.org/10.1073/pnas.1216398109
Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, Chakravarti A (2008) A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 82(1):160–164. https://doi.org/10.1016/j.ajhg.2007.09.015
Badshah N, Mattison KA, Ahmad S, Chopra P, Johnston HR, Ahmad S, Khan SH, Sarwar MT, Cutler DJ, Taylor M, Vadlamani G, Zwick ME, Escayg A (2022) Novel missense CNTNAP2 variant identified in two consanguineous Pakistani families with developmental delay, epilepsy, intellectual disability, and aggressive behavior. Front Neurol 13:918022. https://doi.org/10.3389/fneur.2022.918022
Canali G, Goutebroze L (2018) CNTNAP2 heterozygous missense variants: risk factors for autism spectrum disorder and/or other pathologies? J Exp Neurosci 12:1179069518809666. https://doi.org/10.1177/1179069518809666
Canali G, Garcia M, Hivert B, Pinatel D, Goullancourt A, Oguievetskaia K, Saint-Martin M, Girault JA, Faivre-Sarrailh C, Goutebroze L (2018) Genetic variants in autism-related CNTNAP2 impair axonal growth of cortical neurons. Hum Mol Genet 27(11):1941–1954. https://doi.org/10.1093/hmg/ddy102
Centanni TM, Sanmann JN, Green JR, Iuzzini-Seigel J, Bartlett C, Sanger WG, Hogan TP (2015) The role of candidate-gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am J Med Genet B Neuropsychiatr Genet 168(7):536–543. https://doi.org/10.1002/ajmg.b.32325
Falsaperla R, Pappalardo XG, Romano C, Marino SD, Corsello G, Ruggieri M, Parano E, Pavone P (2020) Intronic variant in CNTNAP2 gene in a boy with remarkable conduct disorder minor facial features, mild intellectual disability, and seizures. Front Pediatr 8:550. https://doi.org/10.3389/fped.2020.00550
Fernandes D, Santos SD, Coutinho E, Whitt JL, Beltrão N, Rondão T, Leite MI, Buckley C, Lee HK, Carvalho AL (2019) Disrupted AMPA receptor function upon genetic- or antibody-mediated loss of autism-associated CASPR2. Cereb Cortex 29(12):4919–4931. https://doi.org/10.1093/cercor/bhz032
Freri E, Castellotti B, Canafoglia L, Ragona F, Solazzi R, Vannicola C, Magri S, Messina G, D’Arrigo S, Gellera C, DiFrancesco JC, Granata T (2021) Severe epilepsy in CNTNAP2-related Pitt-Hopkins-like syndrome successfully treated with stiripentol. Seizure 88:143–145. https://doi.org/10.1016/j.seizure.2021.04.012
Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BH, Knoers NV, Cahn W, Kahn RS, Edelmann L, Davis KL, Silverman JM, Brunner HG, van Kessel AG, Wijmenga C, Ophoff RA, Veltman JA (2008) CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry 13(3):261–266. https://doi.org/10.1038/sj.mp.4002049. (Erratum in: Mol Psychiatry. 2010 Nov;15(11):1121)
Gregor A, Albrecht B, Bader I, Bijlsma EK, Ekici AB, Engels H, Hackmann K, Horn D, Hoyer J, Klapecki J, Kohlhase J, Maystadt I, Nagl S, Prott E, Tinschert S, Ullmann R, Wohlleber E, Woods G, Reis A, Rauch A, Zweier C (2011) Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet 12:106. https://doi.org/10.1186/1471-2350-12-106
Horresh I, Poliak S, Grant S, Bredt D, Rasband MN, Peles E (2008) Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J Neurosci 28(52):14213–14222. https://doi.org/10.1523/JNEUROSCI.3398-08.2008
Jackman C, Horn ND, Molleston JP, Sokol DK (2009) Gene associated with seizures, autism, and hepatomegaly in an Amish girl. Pediatr Neurol 40(4):310–313. https://doi.org/10.1016/j.pediatrneurol.2008.10.013
Ji W, Li T, Pan Y, Tao H, Ju K, Wen Z, Fu Y, An Z, Zhao Q, Wang T, He L, Feng G, Yi Q, Shi Y (2013) CNTNAP2 is significantly associated with schizophrenia and major depression in the Han Chinese population. Psychiatry Res 207(3):225–228. https://doi.org/10.1016/j.psychres.2012.09.024
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, Gallone G, Lelieveld SH, Martin HC, McRae JF, Short PJ, Torene RI, de Boer E, Danecek P, Gardner EJ, Huang N, Lord J, Martincorena I, Pfundt R, Reijnders MRF, Yeung A, Yntema HG, Deciphering Developmental Disorders Study, Vissers LELM, Juusola J, Wright CF, Brunner HG, Firth HV, FitzPatrick DR, Barrett JC, Hurles ME, Gilissen C, Retterer K (2020) Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586(7831):757–762. https://doi.org/10.1038/s41586-020-2832-5
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. https://doi.org/10.1038/nature19057
Li X, Hu Z, He Y, Xiong Z, Long Z, Peng Y, Bu F, Ling J, Xun G, Mo X, Pan Q, Zhao J, Xia K (2010) Association analysis of CNTNAP2 polymorphisms with autism in the Chinese Han population. Psychiatr Genet 20(3):113–117. https://doi.org/10.1097/YPG.0b013e32833a216f
Lu P, Wang F, Zhou S, Huang X, Sun H, Zhang YW, Yao Y, Zheng H (2021) A Novel CNTNAP2 mutation results in abnormal neuronal E/I balance. Front Neurol 12:712773. https://doi.org/10.3389/fneur.2021.712773
Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, Carroll AJ (2011) Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A 155A(10):2386–2396. https://doi.org/10.1002/ajmg.a.34177
Mittal R, Kumar A, Ladda R, Mainali G, Aliu E (2021) Pitt hopkins-like syndrome 1 with Novel CNTNAP2 mutation in siblings. Child Neurol Open. 8:2329048X211055330. https://doi.org/10.1177/2329048X211055330
Murdoch JD, Gupta AR, Sanders SJ, Walker MF, Keaney J, Fernandez TV, Murtha MT, Anyanwu S, Ober GT, Raubeson MJ, DiLullo NM, Villa N, Waqar Z, Sullivan C, Gonzalez L, Willsey AJ, Choe SY, Neale BM, Daly MJ, State MW (2015) No evidence for association of autism with rare heterozygous point mutations in contactin-associated protein-like 2 (CNTNAP2), or in other contactin-associated proteins or contactins. PLoS Genet 11(1):e1004852. https://doi.org/10.1371/journal.pgen.1004852
Nakabayashi K, Scherer SW (2001) The human contactin-associated protein-like 2 gene (CNTNAP2) spans over 2 Mb of DNA at chromosome 7q35. Genomics 73(1):108–112. https://doi.org/10.1006/geno.2001.6517
Newbury DF, Paracchini S, Scerri TS, Winchester L, Addis L, Richardson AJ, Walter J, Stein JF, Talcott JB, Monaco AP (2011) Investigation of dyslexia and SLI risk variants in reading- and language-impaired subjects. Behav Genet 41(1):90–104. https://doi.org/10.1007/s10519-010-9424-3
Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH (2011) Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147(1):235–246. https://doi.org/10.1016/j.cell.2011.08.040
Pippucci T, Licchetta L, Baldassari S, Palombo F, Menghi V, D’Aurizio R, Leta C, Stipa C, Boero G, d’Orsi G, Magi A, Scheffer I, Seri M, Tinuper P, Bisulli F (2015) Epilepsy with auditory features: a heterogeneous clinico-molecular disease. Neurol Genet. 1(1):e5. https://doi.org/10.1212/NXG.0000000000000005
Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E (1999) Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24(4):1037–1047. https://doi.org/10.1016/s0896-6273(00)81049-1
Riccardi F, Urquhart J, McCullagh G, Lawrence P, Douzgou S (2019) A patient with a novel CNTNAP2 homozygous variant: further delineation of the CASPR2 deficiency syndrome and review of the literature. Clin Dysmorphol 28(2):66–70. https://doi.org/10.1097/MCD.0000000000000259
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Rodenas-Cuadrado P, Pietrafusa N, Francavilla T, La Neve A, Striano P, Vernes SC (2016) Characterisation of CASPR2 deficiency disorder–a syndrome involving autism, epilepsy and language impairment. BMC Med Genet 17:8. https://doi.org/10.1186/s12881-016-0272-8
Sampath S, Bhat S, Gupta S, O’Connor A, West AB, Arking DE, Chakravarti A (2013) Defining the contribution of CNTNAP2 to autism susceptibility. PLoS One 8(10):e77906. https://doi.org/10.1371/journal.pone.0077906
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, Stevens C, Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, Brand H, Schwartz G, Nguyen R, Guerrero EE, Dias C, Autism Sequencing Consortium; iPSYCH-Broad Consortium, Betancur C, Cook EH, Gallagher L, Gill M, Sutcliffe JS, Thurm A, Zwick ME, Børglum AD, State MW, Cicek AE, Talkowski ME, Cutler DJ, Devlin B, Sanders SJ, Roeder K, Daly MJ, Buxbaum JD (2020) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180(3):568-584.e23. https://doi.org/10.1016/j.cell.2019.12.036
Scala M, Anijs M, Battini R, Madia F, Capra V, Scudieri P, Verrotti A, Zara F, Minetti C, Vernes SC, Striano P (2021) Hyperkinetic stereotyped movements in a boy with biallelic CNTNAP2 variants. Ital J Pediatr 47(1):208. https://doi.org/10.1186/s13052-021-01162-w
Smogavec M, Cleall A, Hoyer J, Lederer D, Nassogne MC, Palmer EE, Deprez M, Benoit V, Maystadt I, Noakes C, Leal A, Shaw M, Gecz J, Raymond L, Reis A, Shears D, Brockmann K, Zweier C (2016) Eight further individuals with intellectual disability and epilepsy carrying bi-allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J Med Genet 53(12):820–827. https://doi.org/10.1136/jmedgenet-2016-103880
Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36(10):928–930. https://doi.org/10.1002/humu.22844
Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH (2006) Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med 354(13):1370–1377. https://doi.org/10.1056/NEJMoa052773
Toma C, Pierce KD, Shaw AD, Heath A, Mitchell PB, Schofield PR, Fullerton JM (2018) Comprehensive cross-disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet 14(12):e1007535. https://doi.org/10.1371/journal.pgen.1007535
Uddin MS, Azima A, Aziz MA, Aka TD, Jafrin S, Millat MS, Siddiqui SA, Uddin MG, Hussain MS, Islam MS (2021) CNTNAP2 gene polymorphisms in autism spectrum disorder and language impairment among Bangladeshi children: a case-control study combined with a meta-analysis. Hum Cell 34(5):1410–1423. https://doi.org/10.1007/s13577-021-00546-8
Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA, Tourette Syndrome Association International Consortium for Genetics (2003) CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 82(1):1–9. https://doi.org/10.1016/s0888-7543(03)00097-1
Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcón M, Oliver PL, Davies KE, Geschwind DH, Monaco AP, Fisher SE (2008) A functional genetic link between distinct developmental language disorders. N Engl J Med 359(22):2337–2345. https://doi.org/10.1056/NEJMoa0802828
Watson CM, Crinnion LA, Tzika A, Mills A, Coates A, Pendlebury M, Hewitt S, Harrison SM, Daly C, Roberts P, Carr IM, Sheridan EG, Bonthron DT (2014) Diagnostic whole genome sequencing and split-read mapping for nucleotide resolution breakpoint identification in CNTNAP2 deficiency syndrome. Am J Med Genet A 164A(10):2649–2655. https://doi.org/10.1002/ajmg.a.36679
Whalley HC, O’Connell G, Sussmann JE, Peel A, Stanfield AC, Hayiou-Thomas ME, Johnstone EC, Lawrie SM, McIntosh AM, Hall J (2011) Genetic variation in CNTNAP2 alters brain function during linguistic processing in healthy individuals. Am J Med Genet B Neuropsychiatr Genet 156B(8):941–948. https://doi.org/10.1002/ajmg.b.31241
Zhang T, Zhang J, Wang Z, Jia M, Lu T, Wang H, Yue W, Zhang D, Li J, Wang L (2019) Association between CNTNAP2 polymorphisms and autism: A family-based study in the chinese han population and a meta-analysis combined with GWAS data of psychiatric genomics consortium. Autism Res 12(4):553–561. https://doi.org/10.1002/aur.2078
Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A, Schenck A, Rauch A (2009) CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 85(5):655–666. https://doi.org/10.1016/j.ajhg.2009.10.004. (Epub 2009 Nov 5)
Acknowledgements
We gratefully acknowledge patients and their families for their participation in this study and Raúl Jiménez Heredia, St. Anna Children’s Cancer Research Institute (CCRI), Vienna, Austria for his technical support.
Funding
Open access funding provided by Università degli Studi di Genova within the CRUI-CARE Agreement. SS receives funding from the National Institutes of Health National Institute of Neurological Disorders and Stroke (K23NS119666). VN is supported by the Ludwig Boltzmann Gesellschaft core funding, the Austrian Science Fund (FWF): P 32924 and TAI 202 1000 Ideas Project.
Author information
Authors and Affiliations
Contributions
AA, TK, SCV, VS, CM, FZ, PS and VN: contributed to the study conception and design. GD’O made the literature review and drafted the manuscript together with VN. HC, AB, KGJ, SM, TZ, KG, NB, VD, AB, DPR, IB, FB, WF, PH, CM, RV, VG, AB, CR, IAA, NM, MMUR, CAA, GN, MM, TG, SS, JRA, HT, SM, MI, AR, MS, FM, JBR, MYS, SE, RM, HH: were involved in the clinical care of the patients and/or in collecting clinical and genetic data. MSS reviewed neuroimaging images. AP: performed the statistical analysis. All authors have critically revised the manuscript and approved the final one as submitted.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki and procedures were in accordance with the ethical standards and approvals of the Medical Ethics Committees at the various medical centers where the patients were treated or sequenced.
Consent to participate
Informed consent was obtained from the parents of all individual participants included in the study.
Consent to publish
The authors affirm that human research participants provided informed consent for publication of the images in Figs. 1 and 2.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Onofrio, G., Accogli, A., Severino, M. et al. Genotype–phenotype correlation in contactin-associated protein-like 2 (CNTNAP-2) developmental disorder. Hum Genet 142, 909–925 (2023). https://doi.org/10.1007/s00439-023-02552-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-023-02552-2