
Abstract
Sensorineural hearing loss (SNHL) is a major cause of functional disability in both the developed and developing world. While hearing aids and cochlear implants provide significant benefit to many with SNHL, neither targets the cellular and molecular dysfunction that ultimately underlies SNHL. The successful development of more targeted approaches, such as growth factor, stem cell, and gene therapies, will require a yet deeper understanding of the underlying molecular mechanisms of human hearing and deafness. Unfortunately, the human inner ear cannot be biopsied without causing significant, irreversible damage to the hearing or balance organ. Thus, much of our current understanding of the cellular and molecular biology of human deafness, and of the human auditory system more broadly, has been inferred from observational and experimental studies in animal models, each of which has its own advantages and limitations. In 2013, researchers described a protocol for the generation of inner ear organoids from pluripotent stem cells (PSCs), which could serve as scalable, high-fidelity alternatives to animal models. Here, we discuss the advantages and limitations of conventional models of the human auditory system, describe the generation and characteristics of PSC-derived inner ear organoids, and discuss several strategies and recent attempts to model hereditary deafness in vitro. Finally, we suggest and discuss several focus areas for the further, intensive characterization of inner ear organoids and discuss the translational applications of these novel models of the human inner ear.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An estimated 430 million people worldwide (13 million in the United States) have moderate-to-profound hearing loss (GBD Hearing Loss Collaborators 2021; Goman and Lin 2016). Hearing loss is not only a quality-of-life issue, with hearing impaired persons reporting feelings of isolation, frustration, and anxiety (Khan et al. 2020; Lindburg et al. 2021), but also a significant contributor to the global disability burden (GBD Hearing Loss Collaborators 2021). Reduced hearing is associated with language and other developmental delay (Figueras et al. 2008; Tomblin et al. 2015), cognitive decline (Lin et al. 2013), and depression (Li et al. 2014), with working-age adults demonstrating lower average wages and reduced labor force participation (Jung and Bhattacharyya 2012) and elderly persons reporting increased difficulty in completing activities of daily living (Dalton et al. 2003; Gopinath et al. 2012). Most permanent hearing loss is of the sensorineural type (SNHL). Causes of SNHL include aging (Agrawal et al. 2008; Yamoah et al. 2020), infection (Bedford et al. 2001; Brown et al. 2009; Goderis et al. 2014), noise exposure (Lie et al. 2016), ototoxic drugs (Farzal et al. 2016; Frisina et al. 2016), traumatic disruption of the otic capsule (Honeybrook et al. 2017), and a long—and growing—list of single-gene mutations (Shearer et al. 1993; Toriello and Smith 2013). Regardless of the specific etiology, all SNHL ultimately results from the loss, dysfunction, or malformation of cochlear hair cells, spiral ganglion neurons, and/or the synapses in between. Though the causes of SNHL are generally well established, the underlying pathophysiologic mechanisms of even the most common causes remain poorly elucidated at the cellular and molecular level.
The successful development of new therapeutic approaches, such as growth factor, stem cell, and gene therapies, will require a yet deeper understanding of the biology of hearing and deafness, as well as high-fidelity models for pre-clinical testing. Since the human inner ear cannot be biopsied without causing significant, irreversible damage to the hearing or balance organ, the biological study of human inner ear cells has traditionally been limited to scarce fetal or cadaveric tissues. Thus, much of our current understanding of the cellular and molecular biology of human deafness, and of the human auditory system more broadly, has been inferred from observational and experimental studies in animal models [e.g., mouse (Mus musculus), chicken (Gallus gallus), zebrafish (Danio rerio), and African clawed frog (Xenopus laevis)]. Scientists have developed over 50 mouse models of hereditary deafness (Friedman et al. 2007), as well as rodent models for cochlear toxicity (Fernandez et al. 2019), noise-induced hearing loss (Escabi et al. 2019; Holt et al. 2019), infection (Yun et al. 2015), age-related hearing loss (presbycusis) (Cai et al. 2018; Hunter and Willott 1987), and cochlear ischemia, which is implicated in sudden SNHL (Gyo 2013). The anatomical and histological similarity of the human and rodent inner ears has also made rodents useful for modeling the many technical and biological hurdles to stem cell and gene therapy in the inner ear (Al-Moyed et al. 2019; Chen et al. 2012; Gyorgy et al. 2019; Pandit et al. 2011).
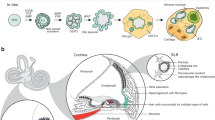
While each animal model has its own advantages and limitations (Fig. 1), all exhibit important differences from the human auditory system (Fig. 1). The hair cells of zebrafish and other non-mammalian vertebrates display a robust spontaneous regenerative response not seen in the mammalian organ of Corti (Corwin and Cotanche 1988; Harris et al. 2003; Roberson and Rubel 1994; Ryals and Rubel 1988; Schuck and Smith 2009), while those of the rodent cochlea do not acquire an adult-like morphology until the early postnatal period (Lenoir et al. 1987), in contrast to the appearance of adult-like hair cells in the third trimester of human gestation (Lavigne-Rebillard and Pujol 1986). Indeed, the stages of mouse and human inner ear development are hardly equivalent (Yamoah et al. 2020). In addition, while more than 99% of genes in the mouse genome have a human homologue (with approximately 80% having a one-to-one orthologue) (Mouse Genome Sequencing Consortium 2002), the targeted introduction of human deafness-related mutations into the mouse genome does—in some cases—fail to produce a deafness phenotype (Lu et al. 2014; Tona et al. 2020), suggesting that the sequence homology of a gene does not necessarily translate to functional identicality of its end-product. This was demonstrated more systematically by Liao and Zhang (2008), who found that more than 20% of a sample of one-to-one mouse orthologues of human essential genes (i.e., those genes required for survival to reproductive age or reproduction itself) were non-essential.

Chart comparing the characteristics of non-mammalian vertebrate (left), rodent (left-middle), 2D cell culture (right-middle), and organoid (right) models of the human auditory system
In vitro generation of inner ear tissue
An alternative and complementary approach to generating animal models is the in vitro derivation of inner ear tissues from mouse or human pluripotent stem cells (PSCs) or tissue-specific progenitor cells. Specialized tissues are generated from PSCs through a process termed directed differentiation, which involves the precisely timed addition of growth factors and small molecules to recapitulate the signaling events of in vivo development. Culture systems are more scalable than animal models, and cells in vitro can be easily accessed for electrophysiological, molecular, and imaging studies (Fig. 1). PSCs and tissue-specific progenitor cells can be distinguished by their respective capacities for cell fate specification (virtually unlimited versus lineage-restricted), and in theory, self-renewal (infinite versus limited). However, PSCs may spontaneously differentiate in culture, and researchers have demonstrated using multiple cytogenetic methods that, over long-term culture, PSCs have a tendency to accumulate chromosomal aberrations (Dekel-Naftali et al. 2012; Martins-Taylor 2011; Merkle et al. 2017; Narva et al. 2010; International Stem Cell Initiative 2011; Yang et al. 2010), which, in turn, have been shown to affect their differentiation capacity (Fazeli et al. 2011; Markouli et al. 2019; Yang et al. 2010). The combination of spontaneous differentiation and accumulated chromosomal aberrations likely imposes a de facto passaging limit on PSCs in culture.
From a culturing standpoint, there are at least two important differences between mouse and human PSCs. First, in vitro tissues arise more slowly from human PSCs (hPSCs) than from mouse PSCs (mPSCs) (Compare Koehler et al. 2013, 2017), reflecting the longer gestational period in humans (280 days versus 20 days). Second, mPSCs and hPSCs have distinct states of pluripotency—the ground (naïve) and primed states, respectively (Nichols and Smith 2009). The ground state is embodied by the inner cell mass of the pre-implantation mouse blastocyst, while the primed state is embodied by the epiblast of the post-implantation mouse embryo. Mouse induced PSCs (miPSCs), like mESCs, exhibit the properties of naïve pluripotency. Conversely, hPSCs, even when derived as hESCs from the pre-implantation inner cell mass, more closely resemble post-implantation mouse epiblast cells (Brons et al. 2007; Tesar et al. 2007) and are, therefore, primed PSCs. This difference is important, as primed cells demonstrate poorer survival in single-cell suspension and are poised to differentiate along certain lineages. Together, these differences generally lead to the slower, less efficient, and more inconsistent in vitro derivation of inner ear tissues from hPSCs than from mESCs.
Despite the advantages of using mPSCs, in vitro models of the mouse inner ear obviously do not provide the ability to research human tissues. Conversely, hPSC-based culture systems allow researchers to study auditory system development, structure, physiology, and regeneration in living tissue that is genetically identical to the human inner ear. However, the transcriptional similarity of these models is highly dependent on the fidelity of the culture system. Conventional 2D culture systems, in which cells are grown on a glass or plastic substrate, have generally failed to yield transcriptionally, morphologically, and physiologically mature hair cell-like cells in appreciably high numbers (Chen et al. 2012; Ealy et al. 2016; Oshima et al. 2010; Ouji et al. 2012; Ronaghi et al. 2014). Indeed, it is likely that the 2D microenvironment is unable to accurately recapitulate the cell–cell and cell–ECM (extracellular matrix) interactions seen in vivo, which provide important cues for differentiation and gene expression. The use of utricular feeder cells or stromal conditioned medium can overcome this limitation to some degree (Oshima et al. 2010; Ouji et al. 2012), but these practices can lead to highly variable cultures. There is also a second, inherent limitation to modeling SNHL in 2D culture. That is, 2D culture systems are—by definition—incapable of modeling the complex, 3D process of mammalian inner ear morphogenesis or the precise, 3D spatial organization of the adult inner ear and its embryologic forerunners.
It is perhaps unsurprising, then, that 3D culture systems can overcome many of the limitations of 2D culture, resulting in higher fidelity models (Fig. 1). The term “organoid,” meaning “resembling an organ,” refers to culture systems in which cells self-organize into 3D tissues that recapitulate—to at least some degree—the cellular diversity, 3D spatial organization, and functional properties of native organs. Researchers have developed protocols for generating intestinal (Spence et al. 2011), cerebral (Lancaster et al. 2013), retinal (Eiraku et al. 2011; Nakano et al. 2012), kidney (Freedman et al. 2015; Morizane et al. 2015; Takasato et al. 2015), lung (Dye et al. 2015), and—of course—inner ear (Koehler et al. 2013, 2017) organoids from PSCs. LGR5+ tissue-specific progenitor cells have also been expanded into 3D in vitro tissues resembling pyloric epithelium (Barker et al. 2010), small intestinal crypt-villus units (Sato et al. 2009), colonic crypts (Sato et al. 2011), and hepatocytes (Huch et al. 2013). A population of LGR5+ support cells that displays some characteristics of tissue-specific progenitors is also present in the mammalian cochlea (Shi et al. 2012). McLean et al. (2017) recently described a protocol for expanding these LGR5+ support cells in vitro into 3D vesicles lined by hair cell- and support cell-like cells. However, these LGR5+ support cell-derived vesicles fail to recapitulate the full cellular diversity of the mammalian cochlea.
PSC-derived inner ear organoids, in contrast, comprise not only hair cell- and support cell-like cells, but also neuron-like cells and distinct regions of PAX8+ non-sensory otic-like epithelium and TFAP2A+/SLUG+ periotic-like mesenchyme (Bouchard et al. 2010; Koehler et al. 2013, 2017). To date, all publications describing the successful generation of PSC-derived inner ear organoids have built upon the foundations of the stepwise induction protocol described by Koehler et al. (2013) (see DeJonge et al. 2016; Hartman et al. 2018; Koehler et al. 2017; Liu et al. 2016; Perny et al. 2017; Schaefer et al. 2018; Tang et al. 2019). PSC-derived aggregates are treated with BMP-4 and the TGF-beta inhibitor SB-431542, which promote the specification of non-neural ectoderm, while simultaneously inhibiting the formation of mesendoderm. Next, FGF-2 and LDN-193189, a BMP-4 inhibitor, are used to drive non-neural ectoderm toward a pre-placodal, rather than an epidermal fate. Under the influence of endogenous Wnt signaling (DeJonge et al. 2016; Koehler et al. 2013), the pre-placodal ectoderm-like tissues adopt an otic placodal fate, and mimicking the sequential genesis of the otic pit and vesicle, invaginate to form distinct vesicles (Koehler et al. 2013, 2017). DeJonge et al. (2016) later discovered that otic placodal differentiation could be enhanced by exogenous Wnt signaling through addition of the GSK3 inhibitor CHIR-99021. After a period of self-directed maturation, the vesicles contain luminal patches of MYO7A+ hair cell-like cells, which abut a dense, basal layer of SOX2+ support cell-like cells and form putative synapses with TUJ1+ neuron-like cells (Koehler et al. 2013, 2017).
The further, intensive characterization of inner ear organoids by Koehler et al. (2013, 2017) and others has revealed an impressive similarity to native inner ear tissues. Besides Myo7a, PSC-derived hair cell-like cells were found to express a number of other hair cell-specific genes, including Pou4f3 (Koehler et al. 2013, 2017; Xiang et al. 1998, 2003), Otof (Tang et al. 2019; Yasunaga et al. 1999), and Tmc1 (Kurima et al. 2002; Tang et al. 2019). The majority of the hair cell-like cells also labeled for a handful of proteins expressed in the type II vestibular hair cells—but not in the inner, outer, or type I hair cells—of adult mice, namely CALB2 (Desai et al. 2005; Koehler et al. 2013, 2017), SOX2 (Koehler et al. 2013, 2017; Oesterle et al. 2008), and ANXA4 (Koehler et al. 2017; Liu et al. 2016; McInturff et al. 2018). Consistent with this, most hair cells in mouse inner ear organoids also adopted the morphology of type II vestibular cells, as well as voltage response, fast inward rectifier, and large outward delayed rectifier currents that resembled those of the type II vestibular hair cells of the postnatal day 4 (P4) mouse utricle (Liu et al. 2016). Amazingly, these currents underwent a maturation process similar to that in the native mouse utricle. That is, the prevalence of hyperpolarization-activated cation channels increased after culture day 22, while that of voltage-dependent Na+ currents declined (Liu et al. 2016).
In addition, the hair cell-like cells in inner ear organoids seem to have the ‘necessary parts’ for mechanotransduction. PSC-derived hair cell-like cells display F-actin+/ESPN+ stereocilia bundles, PCDH15+/CDH23+ tip link-like structures, and a single acetylated-alpha-tubulin+ kinocilium on their luminal surfaces, reminiscent of the mechanotransduction apparatus of native vestibular hair cells (Koehler et al. 2013; Tang et al. 2019). The hair bundle-like structures of these cells often demonstrated a pattern of local alignment that was reminiscent of the organization seen in the mouse utricle (Liu et al. 2016), and by day 24, the length of stereocilia on mouse PSC-derived hair cells fell within the normal range for adult mouse utricular hair cells. FM4-64 and FM1-43 uptake assays suggested the presence of functional mechanosensitive channels (Koehler et al. 2013; Liu et al. 2016), which was confirmed by the measurement of mechanotransduction currents by day 25 (Liu et al. 2016). Furthermore, CTBP2+ punctae were observed at the base of hair cell-like cells in close proximity to the neurite-like extensions of neuron-like cells, which, in turn, expressed multiple postsynaptic markers (Koehler et al. 2013, 2017). Hair cell-like cells also exhibited the depolarization-activated Ca2+ currents necessary for neurotransmitter release in vivo (Liu et al. 2016).
Modeling hereditary deafness in 2D and 3D culture
An estimated 80% of prelingual deafness in the developed world is thought to be attributable to genetic causes (Shearer et al. 1993). Modeling hereditary deafness, therefore, represents an incredibly valuable application of PSC-derived inner ear organoids. There are two general approaches to modeling hereditary deafness in vitro (Fig. 2). The first approach involves the targeted introduction of deafness-associated mutations into wild-type ESC lines via CRISPR-Cas9 (Cong et al. 2013; Mali et al. 2013), prime editing (Anzalone et al. 2019), or another precision genome-editing technique. The second is to harvest somatic cells—often dermal fibroblasts or peripheral blood mononuclear cells—from patients with hereditary deafness and convert them into induced PSCs (iPSCs) (Takahashi et al. 2007; Takahashi and Yamanaka 2006). Stepwise induction protocols can then be used for the directed differentiation of inner ear-like tissues from iPSCs or CRISPR-Cas9-edited ESCs or iPSCs.

Graphical representation of the generation and analysis of PSC-derived inner ear organoid models of hereditary deafness. a Patient-specific iPSCs with mutation in deafness-related gene (X) are generated from somatic cells through induced expression of pluripotency genes. b Precision genome editing is used to create targeted, deafness-related mutations in ESCs. c The coding sequence of a fluorophore (e.g., tdTomato) is inserted downstream of the promoter for a gene-of-interest (e.g., deafness-related gene, cell type-specific gene, or regionally expressed transcription factor) to generate a fluorescent reporter PSC line. d Fluorescent cells within organoids can be selectively harvested, either manually or automatically (e.g., by FACS), for further analyses. ESCs embryonic stem cells, FACS fluorescence-activated cell sorting, iPSC induced pluripotent stem cell
iPSCs certainly hold more therapeutic potential than ESCs, as iPSC-derived donor cells can be used for autologous cell-based inner ear therapy without concern for rejection. However, several studies have revealed the presence of significant genetic background variation among iPSCs, which, in turn, leads to significant variability in the directed differentiation process (Burrows et al. 2016; Kyttala et al. 2016; Rouhani et al. 2014). Indeed, DeJonge et al. (2016) reported that the optimal timing of FGF-2/LDN-193189 treatment in the stepwise induction of inner ear organoids varied among four miPSC lines, while Koehler et al. (2017) found that exogenous BMP-4 was necessary for the induction of non-neural ectoderm in hiPSC-, but not hESC-, derived aggregates. In contrast, in the absence of off-target mutations, genetically engineered ESCs have a homogenous genetic background, allowing for the more consistent derivation of inner ear organoids, as well as the ability to control for genetic background noise when comparing wild-type and mutant PSC-derived cells or tissues. Another limitation of iPSCs is the rarity of specific single-gene mutations, which are often only reported in individual families. The routine use of iPSC-based culture systems in modeling hereditary deafness will, therefore, likely require a significant collaborative effort with the establishment of an iPSC biorepository.
Nonetheless, researchers have used hiPSC-based, 2D culture systems to model two forms of autosomal recessive non-syndromic deafness, DFNB2 (Tang et al. 2016) and DFNB3 (Chen et al. 2016a), as well as Pendred syndrome (Hosoya et al. 2017, 2019), the inherited peripheral neuropathy Charcot–Marie–Tooth disease type 1A (CMT1A) (Shi et al. 2018), and myoclonic epilepsy with ragged-red fibers (MERRF) (Chen et al. 2018), a mitochondrial disorder characterized by SNHL, myopathy, ataxia, and—as the name implies—epilepsy. Shi et al. (2018) generated iPSCs from CMT1A and healthy patients, and then differentiated these cells into neural crest stem cell-like cells in 2D culture. Under the appropriate conditions, CMT1A and wild-type neural crest stem cell-like cells were able to differentiate into osteoblast-, adipocyte-, chondrocyte-, smooth muscle cell-, and neuron-like cells with similar efficiency. However, under conditions in which wild-type cells gave rise to GFAP+/S100B+ Schwann cell-like cells, CMT1A hiPSC-derived neural crest stem cell-like cells instead produced CD34+ endoneurial fibroblast-like cells, suggesting that the pathogenesis of CMT1A may be related to the aberrant differentiation of Schwann cell progenitors. Hosoya et al. (2017) went a step further, not only modeling Pendred syndrome in an iPSC-based 2D culture, but also using this model to test the therapeutic effects of low-dose rapamycin and metformin. This iPSC-based Pendred syndrome model has since been used to determine the minimum effective dose of rapamycin in preventing otic-like cell death in vitro (Hosoya et al. 2019).
Several other studies have used CRISPR-Cas9 genome editing to generate in vitro models of genetic deafness. Barhl1 is a mammalian homologue of the Drosophila homeobox gene BarH1 (Bulfone et al. 2000). Barhl1 encodes a homeodomain transcription factor expressed in the inner ear and central nervous system. While there is no human deafness phenotype currently associated with BARHL1 mutation, Barhl1-null mice exhibit progressive SNHL (Li et al. 2002). Zhejiang University scientists have used CRISPR-Cas9 technology to generate frameshift mutations in the coding region (Zhong et al. 2018) and 3’ enhancer (Hou et al. 2019) of the Barhl1 gene in mESCs. To investigate the underlying mechanism of SNHL in Barhl1 mutants, both wild-type and Barhl1-mutant mESCs were subjected to a stepwise induction protocol for deriving hair cell-like cells in 2D culture. The authors used several analytical methods to investigate the effects of Barhl1 mutation on hair cell differentiation, revealing a significant downregulation of hair cell-specific genes in both mutant cell lines (Hou et al. 2019; Zhong et al. 2018). Downregulated hair cell-specific genes were then cross-referenced with potential BARHL1 targets (Zhong et al. 2018). The results of this analysis suggested that the effects of Barhl1 mutation on hair cell differentiation may be mediated by the downregulation of Clic5 and Ush1g.
Similarly, Tang et al. (2019) used CRISPR-Cas9 to generate Tmprss3-KO mESCs. Tmprss3 encodes a type II transmembrane serine protease. Mutations in TMPRSS3 are the cause of autosomal recessive non-syndromic deafness DFNB8/10, which is characterized by prelingual SNHL (Scott et al. 2001). Mice homozygous for the nonsense mutation Tmprss3Y260X exhibit rapid hair cell degeneration at P12 following a period of normal hair cell development (Fasquelle et al. 2011). Despite the presence of this mouse model, the exact functional role of TMPRSS3 in the inner ear remains poorly understood, and the precise pathophysiologic mechanism underlying DFNB8/10 has yet to be revealed. Thus, Tmprss3-KO and wild-type mESCs were subjected to directed differentiation into inner ear organoids (Tang et al. 2019). The researchers found that, while Tmprss3-KO PSCs initially gave rise to hair cell-like cells with normal hair bundles and FM1-43 uptake, by day 38 (equivalent to P12-P14 in vivo), Tmprss3-KO hair cell-like cells exhibited significantly higher levels of the apoptosis protein caspase-3. The organoid format permitted easy access to inner ear-like tissues for single-cell RNA sequencing (scRNA-seq), which revealed potential roles for calcium ion homeostasis and extracellular matrix maintenance in TMPRSS3-related deafness.
Unsettled questions in inner ear organoid research
Despite the promise of inner ear organoids in modeling human deafness, we must exercise caution when applying findings in inner ear organoids to the human auditory system. A recent scRNA-seq analysis revealed that the specification of distinct cellular subtypes was not achieved in cortical organoid culture and suggested that this was due, at least in part, to high levels of endoplasmic reticulum (ER) stress (Bhaduri et al. 2020). Only time will tell if this ER stress-induced inhibition of cell type specification is an inherent feature of organoid culture or simply a matter of optimizing culture conditions, or—for that matter—whether these results are even generalizable to organoids on the whole. It is certainly auspicious for the future of inner ear organoid research that many deafness-related genes are expressed in inner ear organoids with a similar spatiotemporal pattern to the native inner ear (Table 1), while many others have been detected in putative otic-like sensory epithelial cells by scRNA-seq (Tang et al. 2019). Nonetheless, the process of characterizing inner ear organoids is still in its infancy, and establishing the inner ear organoid as a valid model of the human auditory system in health, disease, and development will require the continued dedication and collaboration of stem cell biologists, cellular electrophysiologists, bioinformaticians, and others. We will use this opportunity to suggest and discuss areas for the further, intensive characterization of inner ear organoids.
Perhaps the single greatest open question in inner ear organoid research—which is also a significant limitation of inner ear organoids—relates to the conspicuous absence of hair cell-like cells with a cochlear phenotype. Indeed, until cochlear-type inner ear organoids are derived, these 3D culture systems will never be able to fully recapitulate the human auditory system. There are several possible explanations for the notable absence of cochlear hair cell-like cells in organoid culture. First, the non-physiologic culture environment may be causing ER stress that, in turn, inhibits cell type specification, as observed in cortical organoids (Bhaduri et al. 2020). It is not exactly clear why ER stress would specifically inhibit the differentiation of cochlear hair cell-like cells. However, it is worth noting that vestibular hair cells express many hair cell-specific genes at earlier developmental timepoints than cochlear hair cells (Table 1) and share several features with immature hair cells that are eventually lost in cochlear hair cells, including the presence of a kinocilium and—in type II hair cells—expression of SOX2. This suggests that the differentiation of vestibular hair cells may require less molecular specification of hair cell progenitors than cochlear hair cells. Another possibility is that the innervation by PSC-derived neuron-like cells in inner ear organoids is too limited to provide adequate trophic support for the survival of early cochlear hair cell-like cells. Indeed, Kersigo and Fritzsch (2015) elegantly demonstrated that denervation results in the progressive loss of hair cells in mice, but that vestibular hair cells are significantly more resilient. It is, therefore, possible that nascent cochlear hair cell-like cells are originally present in inner ear organoids but that inadequate innervation by neuron-like cells eventually leads to their preferential loss over vestibular hair cell-like cells.
Alternatively, it may be that current protocols produce a signaling environment favoring the differentiation of vestibular hair cell-like cells from hair cell progenitor-like cells. A series of elegant studies has demonstrated that development of the cochlea and vestibule are regulated by the opposing effects of ventral and dorsal signals. Sonic hedgehog (SHH) induces expression of ventral transcription factors such as Otx2, Pax2, and Ngn1, while inhibiting expression of the dorsal marker Dlx5 (Riccomagno et al. 2002). Conversely, Wnt and BMP both induce the expression of Dlx5, and BMP inhibits Otx2 (Ohta et al. 2016; Riccomagno et al. 2005). These signals are so critical to the specification of the cochlea and vestibule that their loss or ectopic expression has been shown to produce dramatic malformations. For example, the loss of SHH results in complete absence of the cochlear duct in mice (Riccomagno et al. 2002). Thus, the preferential differentiation of vestibular hair cell-like cells could be explained by the relative overactivity of dorsal signals, and therefore, corrected by the precisely timed addition of a ventralizing molecule, such as the SHH agonist purmorphamine, or an inhibitor of dorsal signals, such as LDN-193189. Jeong et al. (2018) recently reported the generation of inner ear organoids with hair cell-like cells that expressed some markers of cochlear hair cells. Notably, this was achieved with only slight modifications to the protocol described by Koehler et al. (2013), such as the maintenance of PSCs on feeder cells. However, the purported cochlear hair cell-like cells were incompletely characterized, and the result has yet to be replicated. If these results are replicated, it will be important to investigate the mechanism(s) by which the modified protocol produced cochlear-type hair cell-like cells.
Cell–cell junctions, including gap junctions, tight junctions, and tricellular junctions, are broadly present in the membranous labyrinth of the mammalian inner ear (Forge et al. 1999; Kitajiri et al. 2004) and are essential in maintaining the functional barrier between the perilymphatic and endolymphatic compartments, as well as facilitating neuronal growth and promoting cellular organization (Reviewed in Jagger and Forge 2015; Kitajiri and Katsuno 2016). Indeed, the cell–cell junctions of the inner ear are essential for normal hearing, evidenced by the fact that deficiencies in several junctional proteins are associated with hereditary deafness (Shearer et al. 1993; Toriello and Smith 2013), including GJB2 (connexin 26) deficiency (DFNB1A), the most common cause of autosomal recessive non-syndromic deafness in humans. Phalloidin staining has revealed the presence of an apical F-actin network in inner ear organoids, reminiscent of the network of cell–cell tight junctions between support cells of the mouse inner ear (Koehler et al. 2013; Schaefer et al. 2018), and scRNA-seq data have revealed the expression of Cldn9, which encodes the tight junction protein claudin-9, in putative mouse hair cell-like cells (Tang et al. 2019). However, the subcellular co-localization of F-actin, claudins, and other tight junction proteins, such as occludins and junctional adhesion molecules, at cell–cell interfaces has yet to be demonstrated in inner ear organoids. The extent to which inner ear organoids recapitulate the many, highly specialized, non-neurosensory cell types of the native inner ear is another open question in inner ear organoid research. However, this subject was recently discussed in great depth by van der Valk et al. (2021), and we will direct the interested reader to this excellent review.
Should inner ear organoids prove to accurately recapitulate the structural, functional, and molecular features of the native inner ear, then it is reasonable to assume that the same extrinsic insults that lead to SNHL in humans would be similarly deleterious to inner ear organoids, allowing researchers to study the pathophysiologic mechanisms by which they act. However, it is not obvious how certain causes of SNHL (e.g., noise-induced hearing loss) could be simulated in the culture environment, while limits on culture duration will likely preclude any meaningful study of non-genetic, age-related hearing loss. Nonetheless, a wide range of chemical and infectious insults remain potentially amenable for study in inner ear organoids. To date, few studies have focused on the ability of inner ear organoids to model these insults. The ototoxic aminoglycoside dihydrostreptomycin has been shown to reversibly block stimulus-evoked currents in the hair cell-like cells of mouse organoids (Liu et al. 2016), while another aminoglycoside, gentamicin, failed to produce any obvious loss of hair bundle-like structures (Schaefer et al. 2018). Further studies are needed to better characterize inner ear organoids’ susceptibility to aminoglycoside antibiotics and other extrinsic insults known to cause SNHL.
Genome editing and single-cell omics in inner ear organoids
Genome editing encompasses a number of technologies that allow researchers to alter the genetic code of a cell or organism. Genome-editing technologies have become increasingly targeted over the years. For example, fluorescent reporter PSC lines are generated by the targeted insertion of a fluorescent protein coding sequence downstream of the promoter of a gene-of-interest, which—in the case of inner ear organoids—could be a deafness-related gene, cell type-specific gene, or regionally expressed transcription factor. PSC reporter lines for PAX2, FBXO2, and ATOH1, have all been generated and differentiated into inner ear organoids (DeJonge et al. 2016; Hartman et al. 2018; Koehler et al. 2017; Liu et al. 2016; Schaefer et al. 2018), with numerous applications. For example, Liu et al. (2016) used the eGFP signal emitted by the hair cell-like cells of Atoh1-eGFP PSC-derived aggregates to selectively harvest hair cell-like cell bearing vesicles for electrophysiological recording. FACS sorting of organoid cells derived from reporter lines could be similarly used for the selective isolation of otic-like cell types for single-cell analyses. Fluorescent reporters can also been used for expression monitoring, allowing researchers to observe the expression patterns of developmental transcription factors or cell type-specific marker genes over the course of organoid development. For example, Hartman et al. (2018) used a Venus reporter for Fbxo2, an otic lineage-specific gene, to demonstrate surprising differences in spatiotemporal expression patterns between the native mouse vestibular epithelium and mouse inner ear organoids. Another application of fluorescent reporters is lineage tracing. Chimeric inner ear organoids could be generated from a mixture of mutant and wild-type cells to study the role of paracrine signaling in different types of hereditary deafness or to control for batch-to-batch variability in comparisons of wild-type and mutant cells. Combining this technology with fluorescent reporter lines would allow for the easy identification of genotype in chimeric organoids via fluorescent microscopy or single-cell transcriptomics.
scRNA-seq has a number of applications in both auditory research (Reviewed in Pyle and Hoa 2020) and PSC-derived organoids (Reviewed in Camp and Treutlein 2017; Qin and Tape 2020), and will undoubtedly facilitate the further characterization of organoids and their application in disease modeling. Several unsettled questions in inner ear organoid research could be answered by the generation of a single-cell transcriptional atlas (Kolla et al. 2020; Korrapati et al. 2019; Petitpre et al. 2018; Shrestha et al. 2018; Sun et al. 2018) for inner ear organoids or by scRNA-seq analyses that focus on the expression of signaling molecules, stress response genes, or regionally expressed transcription factors. scRNA-seq has already been used to compare the transcriptional profiles of mutant and wild-type inner ear organoids in the hopes of elucidating the underlying molecular mechanisms of hereditary deafness Tang et al. (2019). Meanwhile, single-cell RT-qPCR, has been used to compare the gene expression between PSC-derived otic-like tissues and the native inner ear to investigate the transcriptional fidelity of a 2D culture system and optimize the protocol (Ealy et al. 2016). In the future, comparative analyses could be performed to investigate transcriptional heterogeneity among organoids derived from different cell lines or according to different protocols, as has been demonstrated in kidney organoids (Wu et al. 2018). Other single-cell technologies, such as scATAC-seq (single-cell assay for transposase-accessible chromatin with high-throughput sequencing), could be similarly applied to compare, for example, the epigenetic landscape in inner ear organoids and the native inner ear. Multiple single-cell technologies can also be simultaneously applied (single-cell multi-omics), and the resulting data integrated for analysis with computational tools such as Seurat v3 (Stuart et al. 2019) or LIGER (Welch et al. 2019). Protocols have even been developed which would allow for simultaneous characterization of the transcriptome, electrophysiology, and/or morphology of single PSC-derived hair cell- and neuron-like cells (Bardy et al. 2016; Cadwell et al. 2016; Chen et al. 2016b; Foldy et al. 2016; Fuzik et al. 2016; Ranum et al. 2019), permitting the remarkably detailed comparison of native and PSC-derived cells, and possibly, the identification of cellular subtypes in inner ear organoids.
Translational applications of inner ear organoids
The potential translational applications of inner ear organoids in disease modeling are incredibly numerous. Should inner ear organoids prove to be sensitive to the ototoxic compounds, they would be incredibly useful in the scalable, high-throughput testing of drug toxicity in pre-clinical trials. Conversely, when combined with developmental or regeneration studies, this same scalability could be applied toward the large-scale, high-throughput, pre-clinical screening of otoprotective or oto-regenerative compounds. Inner ear organoids could also serve as a platform for testing the effects of CRISPR-Cas9 genetic correction of single-gene mutations. This could even one day be employed in a patient-specific manner, with the generation of inner ear organoids from CRISPR-Cas9-corrected patient-specific iPSCs being a routine quality check before cell-based therapy is performed for SNHL. Lastly, inner ear organoid models of inner ear disease could be used for the testing of emerging therapies such as cell-based and gene therapy. Successfully employing inner ear organoids for these applications will almost certainly require that researchers find a way to overcome the previously reported high variability and low efficiency of organoid generation (Schaefer et al. 2018; Koehler et al. 2017), as well as the overgrowth of periotic-like mesenchyme in late-stage culture, which hinders analytic methods such as expression monitoring, whole-mount imaging, and single-cell isolation. Fortunately, progress toward these goals is already underway. For example, Chang et al. (2020) recently reported increased efficiency of inner ear organoid generation with the use of photobiomodulation and hanging droplet techniques, while Hocevar et al. (2021) described a method for dissecting organoids away from their aggregates and reported that, when cultured in media with Matrigel, organoids display the same autonomy seen in vivo (Swanson et al. 1990).
Summary
Since the human inner ear cannot be biopsied without causing significant, irreversible damage to the hearing or balance organ, the biological study of human inner ear cells has traditionally been limited to scarce fetal and cadaveric tissues. Researchers recently described a protocol for generating inner ear organoids, which could serve as a scalable, high-fidelity alternative to animal models. However, many questions and challenges remain, including how cochlear-type hair cell-like cells can be derived, whether cell–cell junctions are present, whether non-neurosensory inner cell types are represented, and whether inner ear organoids are susceptible to the same extrinsic insults that cause deafness in humans and other mammals. With the continued dedication of stem cell biologists, cellular electrophysiologists, and bioinformaticians, and the utilization of fluorescent reporter lines and single-cell omics, it is likely that these questions and others will be answered in the coming years. If, through these efforts, high-fidelity, human cochlear inner ear organoids are successfully generated, then a wide array of translational applications await inner ear organoids, including high-throughput drug and toxicity screens and pre-clinical testing and patient-specific quality checks for stem cell and gene therapies.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C et al (1997) A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 15(2):157–164. https://doi.org/10.1038/ng0297-157
Agrawal Y, Platz EA, Niparko JK (2008) Prevalence of hearing loss and differences by demographic characteristics among US adults: data from the National Health and Nutrition Examination Survey, 1999–2004. Arch Intern Med 168(14):1522–1530. https://doi.org/10.1001/archinte.168.14.1522
Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ et al (2001) Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 69(1):25–34. https://doi.org/10.1086/321277
Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM et al (2003a) Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet 72(5):1315–1322. https://doi.org/10.1086/375122
Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF et al (2003b) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12(24):3215–3223. https://doi.org/10.1093/hmg/ddg358
Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M et al (2006) The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci 26(26):7022–7034. https://doi.org/10.1523/JNEUROSCI.1163-06.2006
Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR et al (2001) Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 10(16):1709–1718. https://doi.org/10.1093/hmg/10.16.1709
Al-Moyed H, Cepeda AP, Jung S, Moser T, Kugler S, Reisinger E (2019) A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. https://doi.org/10.15252/emmm.201809396
Andrade LR (2015) Evidence for changes in beta- and gamma-actin proportions during inner ear hair cell life. Cytoskeleton (hoboken) 72(6):282–291. https://doi.org/10.1002/cm.21227
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM et al (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576(7785):149–157. https://doi.org/10.1038/s41586-019-1711-4
Bardy C, van den Hurk M, Kakaradov B, Erwin JA, Jaeger BN, Hernandez RV et al (2016) Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol Psychiatry 21(11):1573–1588. https://doi.org/10.1038/mp.2016.158
Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH et al (2010) Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6(1):25–36. https://doi.org/10.1016/j.stem.2009.11.013
Bedford H, de Louvois J, Halket S, Peckham C, Hurley R, Harvey D (2001) Meningitis in infancy in England and Wales: follow up at age 5 years. BMJ 323(7312):533–536. https://doi.org/10.1136/bmj.323.7312.533
Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D et al (2020) Cell stress in cortical organoids impairs molecular subtype specification. Nature 578(7793):142–148. https://doi.org/10.1038/s41586-020-1962-0
Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG et al (2001) Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet 27(1):108–112. https://doi.org/10.1038/83667
Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S et al (2007) Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet 81(6):1169–1185. https://doi.org/10.1086/522090
Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL et al (2001) Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68(1):26–37. https://doi.org/10.1086/316954
Bouchard M, de Caprona D, Busslinger M, Xu P, Fritzsch B (2010) Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev Biol 10:89. https://doi.org/10.1186/1471-213X-10-89
Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM et al (2007) Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448(7150):191–195. https://doi.org/10.1038/nature05950
Brown ED, Chau JK, Atashband S, Westerberg BD, Kozak FK (2009) A systematic review of neonatal toxoplasmosis exposure and sensorineural hearing loss. Int J Pediatr Otorhinolaryngol 73(5):707–711. https://doi.org/10.1016/j.ijporl.2009.01.012
Bulfone A, Menguzzato E, Broccoli V, Marchitiello A, Gattuso C, Mariani M et al (2000) Barhl1, a gene belonging to a new subfamily of mammalian homeobox genes, is expressed in migrating neurons of the CNS. Hum Mol Genet 9(9):1443–1452. https://doi.org/10.1093/hmg/9.9.1443
Burrows CK, Banovich NE, Pavlovic BJ, Patterson K, Gallego Romero I, Pritchard JK, Gilad Y (2016) Genetic variation, not cell type of origin, underlies the majority of identifiable regulatory differences in iPSCs. PLoS Genet 12(1):e1005793. https://doi.org/10.1371/journal.pgen.1005793
Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M et al (2016) Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol 34(2):199–203. https://doi.org/10.1038/nbt.3445
Cai R, Montgomery SC, Graves KA, Caspary DM, Cox BC (2018) The FBN rat model of aging: investigation of ABR waveforms and ribbon synapse changes. Neurobiol Aging 62:53–63. https://doi.org/10.1016/j.neurobiolaging.2017.09.034
Camp JG, Treutlein B (2017) Human organomics: a fresh approach to understanding human development using single-cell transcriptomics. Development 144(9):1584–1587. https://doi.org/10.1242/dev.150458
Chang SY, Carpena NT, Mun S, Jung JY, Chung PS, Shim H et al (2020) Enhanced inner-ear organoid formation from mouse embryonic stem cells by photobiomodulation. Mol Ther Methods Clin Dev 17:556–567. https://doi.org/10.1016/j.omtm.2020.03.010
Chen W, Jongkamonwiwat N, Abbas L, Eshtan SJ, Johnson SL, Kuhn S et al (2012) Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 490(7419):278–282. https://doi.org/10.1038/nature11415
Chen JR, Tang ZH, Zheng J, Shi HS, Ding J, Qian XD et al (2016a) Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ 23(8):1347–1357. https://doi.org/10.1038/cdd.2016.16
Chen X, Zhang K, Zhou L, Gao X, Wang J, Yao Y et al (2016b) Coupled electrophysiological recording and single cell transcriptome analyses revealed molecular mechanisms underlying neuronal maturation. Protein Cell 7(3):175–186. https://doi.org/10.1007/s13238-016-0247-8
Chen YC, Tsai CL, Wei YH, Wu YT, Hsu WT, Lin HC, Hsu YC (2018) ATOH1/RFX1/RFX3 transcription factors facilitate the differentiation and characterisation of inner ear hair cell-like cells from patient-specific induced pluripotent stem cells harbouring A8344G mutation of mitochondrial DNA. Cell Death Dis 9(4):437. https://doi.org/10.1038/s41419-018-0488-y
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. https://doi.org/10.1126/science.1231143
Corwin JT, Cotanche DA (1988) Regeneration of sensory hair cells after acoustic trauma. Science 240(4860):1772–1774. https://doi.org/10.1126/science.3381100
Dalton DS, Cruickshanks KJ, Klein BE, Klein R, Wiley TL, Nondahl DM (2003) The impact of hearing loss on quality of life in older adults. Gerontologist 43(5):661–668. https://doi.org/10.1093/geront/43.5.661
DeJonge RE, Liu XP, Deig CR, Heller S, Koehler KR, Hashino E (2016) Modulation of Wnt signaling enhances inner ear organoid development in 3D culture. PLoS ONE 11(9):e0162508. https://doi.org/10.1371/journal.pone.0162508
Dekel-Naftali M, Aviram-Goldring A, Litmanovitch T, Shamash J, Reznik-Wolf H, Laevsky I et al (2012) Screening of human pluripotent stem cells using CGH and FISH reveals low-grade mosaic aneuploidy and a recurrent amplification of chromosome 1q. Eur J Hum Genet 20(12):1248–1255. https://doi.org/10.1038/ejhg.2012.128
Desai SS, Zeh C, Lysakowski A (2005) Comparative morphology of rodent vestibular periphery. I. Saccular and utricular maculae. J Neurophysiol 93(1):251–266. https://doi.org/10.1152/jn.00746.2003
Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R et al (2015) In vitro generation of human pluripotent stem cell derived lung organoids. Elife. https://doi.org/10.7554/eLife.05098
Ealy M, Ellwanger DC, Kosaric N, Stapper AP, Heller S (2016) Single-cell analysis delineates a trajectory toward the human early otic lineage. Proc Natl Acad Sci U S A 113(30):8508–8513. https://doi.org/10.1073/pnas.1605537113
Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S et al (2011) Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472(7341):51–56. https://doi.org/10.1038/nature09941
Escabi CD, Frye MD, Trevino M, Lobarinas E (2019) The rat animal model for noise-induced hearing loss. J Acoust Soc Am 146(5):3692. https://doi.org/10.1121/1.5132553
Farzal Z, Kou YF, St John R, Shah GB, Mitchell RB (2016) The role of routine hearing screening in children with cystic fibrosis on aminoglycosides: a systematic review. Laryngoscope 126(1):228–235. https://doi.org/10.1002/lary.25409
Fasquelle L, Scott HS, Lenoir M, Wang J, Rebillard G, Gaboyard S et al (2011) Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem 286(19):17383–17397. https://doi.org/10.1074/jbc.M110.190652
Fazeli A, Liew CG, Matin MM, Elliott S, Jeanmeure LF, Wright PC et al (2011) Altered patterns of differentiation in karyotypically abnormal human embryonic stem cells. Int J Dev Biol 55(2):175–180. https://doi.org/10.1387/ijdb.103177af
Fernandez K, Wafa T, Fitzgerald TS, Cunningham LL (2019) An optimized, clinically relevant mouse model of cisplatin-induced ototoxicity. Hear Res 375:66–74. https://doi.org/10.1016/j.heares.2019.02.006
Figueras B, Edwards L, Langdon D (2008) Executive function and language in deaf children. J Deaf Stud Deaf Educ 13(3):362–377. https://doi.org/10.1093/deafed/enm067
Foldy C, Darmanis S, Aoto J, Malenka RC, Quake SR, Sudhof TC (2016) Single-cell RNAseq reveals cell adhesion molecule profiles in electrophysiologically defined neurons. Proc Natl Acad Sci U S A 113(35):E5222-5231. https://doi.org/10.1073/pnas.1610155113
Forge A, Becker D, Casalotti S, Edwards J, Evans WH, Lench N, Souter M (1999) Gap junctions and connexin expression in the inner ear. Novartis Found Symp 219:134–150. https://doi.org/10.1002/9780470515587.ch9 (discussion 151-136)
Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V et al (2015) Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 6:8715. https://doi.org/10.1038/ncomms9715
Friedman LM, Dror AA, Avraham KB (2007) Mouse models to study inner ear development and hereditary hearing loss. Int J Dev Biol 51(6–7):609–631. https://doi.org/10.1387/ijdb.072365lf
Frisina RD, Wheeler HE, Fossa SD, Kerns SL, Fung C, Sesso HD et al (2016) Comprehensive audiometric analysis of hearing impairment and tinnitus after cisplatin-based chemotherapy in survivors of adult-onset cancer. J Clin Oncol 34(23):2712–2720. https://doi.org/10.1200/JCO.2016.66.8822
Fuzik J, Zeisel A, Mate Z, Calvigioni D, Yanagawa Y, Szabo G et al (2016) Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat Biotechnol 34(2):175–183. https://doi.org/10.1038/nbt.3443
GBD Hearing Loss Collaborators (2021) Hearing loss prevalence and years lived with disability, 1990–2019: findings from the Global Burden of Disease Study 2019. Lancet 397(10278):996–1009. https://doi.org/10.1016/S0140-6736(21)00516-X
Goderis J, De Leenheer E, Smets K, Van Hoecke H, Keymeulen A, Dhooge I (2014) Hearing loss and congenital CMV infection: a systematic review. Pediatrics 134(5):972–982. https://doi.org/10.1542/peds.2014-1173
Goman AM, Lin FR (2016) Prevalence of hearing loss by severity in the United States. Am J Public Health 106(10):1820–1822. https://doi.org/10.2105/AJPH.2016.303299
Gopinath B, Schneider J, McMahon CM, Teber E, Leeder SR, Mitchell P (2012) Severity of age-related hearing loss is associated with impaired activities of daily living. Age Ageing 41(2):195–200. https://doi.org/10.1093/ageing/afr155
Gyo K (2013) Experimental study of transient cochlear ischemia as a cause of sudden deafness. World J Otorhinolaryngol 3(1):1–15. https://doi.org/10.5319/wjo.v3.i1.1
Gyorgy B, Nist-Lund C, Pan B, Asai Y, Karavitaki KD, Kleinstiver BP et al (2019) Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat Med 25(7):1123–1130. https://doi.org/10.1038/s41591-019-0500-9
Harris JA, Cheng AG, Cunningham LL, MacDonald G, Raible DW, Rubel EW (2003) Neomycin-induced hair cell death and rapid regeneration in the lateral line of zebrafish (Danio rerio). J Assoc Res Otolaryngol 4(2):219–234. https://doi.org/10.1007/s10162-002-3022-x
Hartman BH, Bscke R, Ellwanger DC, Keymeulen S, Scheibinger M, Heller S (2018) Fbxo2(VHC) mouse and embryonic stem cell reporter lines delineate in vitro-generated inner ear sensory epithelia cells and enable otic lineage selection and Cre-recombination. Dev Biol 443(1):64–77. https://doi.org/10.1016/j.ydbio.2018.08.013
Hocevar SE, Liu L, Duncan RK (2021) Matrigel is required for efficient differentiation of isolated, stem cell-derived otic vesicles into inner ear organoids. Stem Cell Res 53:102295. https://doi.org/10.1016/j.scr.2021.102295
Holt AG, Kuhl A, Braun RD, Altschuler R (2019) The rat as a model for studying noise injury and otoprotection. J Acoust Soc Am 146(5):3681. https://doi.org/10.1121/1.5131344
Honeybrook A, Patki A, Chapurin N, Woodard C (2017) Hearing and mortality outcomes following temporal bone fractures. Craniomaxillofac Trauma Reconstr 10(4):281–285. https://doi.org/10.1055/s-0037-1601885
Hosoya M, Fujioka M, Sone T, Okamoto S, Akamatsu W, Ukai H et al (2017) Cochlear cell modeling using disease-specific iPSCs unveils a degenerative phenotype and suggests treatments for congenital progressive hearing loss. Cell Rep 18(1):68–81. https://doi.org/10.1016/j.celrep.2016.12.020
Hosoya M, Saeki T, Saegusa C, Matsunaga T, Okano H, Fujioka M, Ogawa K (2019) Estimating the concentration of therapeutic range using disease-specific iPS cells: low-dose rapamycin therapy for Pendred syndrome. Regen Ther 10:54–63. https://doi.org/10.1016/j.reth.2018.11.001
Hou K, Jiang H, Karim MR, Zhong C, Xu Z, Liu L et al (2019) A critical E-box in Barhl1 3’ enhancer is essential for auditory hair cell differentiation. Cells. https://doi.org/10.3390/cells8050458
Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M et al (2013) In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 494(7436):247–250. https://doi.org/10.1038/nature11826
Hunter KP, Willott JF (1987) Aging and the auditory brainstem response in mice with severe or minimal presbycusis. Hear Res 30(2–3):207–218. https://doi.org/10.1016/0378-5955(87)90137-7
International Stem Cell Initiative, Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S et al (2011) Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol 29(12):1132–1144. https://doi.org/10.1038/nbt.2051
Jagger DJ, Forge A (2015) Connexins and gap junctions in the inner ear–it’s not just about K(+) recycling. Cell Tissue Res 360(3):633–644. https://doi.org/10.1007/s00441-014-2029-z
Jeong M, O’Reilly M, Kirkwood NK, Al-Aama J, Lako M, Kros CJ, Armstrong L (2018) Generating inner ear organoids containing putative cochlear hair cells from human pluripotent stem cells. Cell Death Dis 9(9):922. https://doi.org/10.1038/s41419-018-0967-1
Jung D, Bhattacharyya N (2012) Association of hearing loss with decreased employment and income among adults in the United States. Ann Otol Rhinol Laryngol 121(12):771–775. https://doi.org/10.1177/000348941212101201
Kalatzis V, Sahly I, El-Amraoui A, Petit C (1998) Eya1 expression in the developing ear and kidney: towards the understanding of the pathogenesis of Branchio-Oto-Renal (BOR) syndrome. Dev Dyn 213(4):486–499. https://doi.org/10.1002/(SICI)1097-0177(199812)213:4%3c486::AID-AJA13%3e3.0.CO;2-L
Kersigo J, Fritzsch B (2015) Inner ear hair cells deteriorate in mice engineered to have no or diminished innervation. Front Aging Neurosci 7:33. https://doi.org/10.3389/fnagi.2015.00033
Khan A, Mubdi N, Budnick A, Feldman DR, Williams SW, Patel S, Tonorezos ES (2020) The experience of hearing loss in adult survivors of childhood and young adult cancer: a qualitative study. Cancer 126(8):1776–1783. https://doi.org/10.1002/cncr.32698
Kitajiri S, Katsuno T (2016) Tricellular tight junctions in the inner ear. Biomed Res Int 2016:6137541. https://doi.org/10.1155/2016/6137541
Kitajiri SI, Furuse M, Morita K, Saishin-Kiuchi Y, Kido H, Ito J, Tsukita S (2004) Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear Res 187(1–2):25–34. https://doi.org/10.1016/s0378-5955(03)00338-1
Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500(7461):217–221. https://doi.org/10.1038/nature12298
Koehler KR, Nie J, Longworth-Mills E, Liu XP, Lee J, Holt JR, Hashino E (2017) Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat Biotechnol 35(6):583–589. https://doi.org/10.1038/nbt.3840
Kolla L, Kelly MC, Mann ZF, Anaya-Rocha A, Ellis K, Lemons A et al (2020) Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat Commun 11(1):2389. https://doi.org/10.1038/s41467-020-16113-y
Korrapati S, Taukulis I, Olszewski R, Pyle M, Gu S, Singh R et al (2019) Single cell and single nucleus RNA-Seq reveal cellular heterogeneity and homeostatic regulatory networks in adult mouse stria vascularis. Front Mol Neurosci 12:316. https://doi.org/10.3389/fnmol.2019.00316
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S et al (2002) Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 30(3):277–284. https://doi.org/10.1038/ng842
Kyttala A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK et al (2016) Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep 6(2):200–212. https://doi.org/10.1016/j.stemcr.2015.12.009
Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB (2005) Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol 280(2):295–306. https://doi.org/10.1016/j.ydbio.2005.01.015
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME et al (2013) Cerebral organoids model human brain development and microcephaly. Nature 501(7467):373–379. https://doi.org/10.1038/nature12517
Lavigne-Rebillard M, Pujol R (1986) Development of the auditory hair cell surface in human fetuses. A scanning electron microscopy study. Anat Embryol (berl) 174(3):369–377. https://doi.org/10.1007/BF00698787
Lenoir M, Puel JL, Pujol R (1987) Stereocilia and tectorial membrane development in the rat cochlea. A SEM Study. Anat Embryol (berl) 175(4):477–487. https://doi.org/10.1007/BF00309683
Li S, Price SM, Cahill H, Ryugo DK, Shen MM, Xiang M (2002) Hearing loss caused by progressive degeneration of cochlear hair cells in mice deficient for the Barhl1 homeobox gene. Development 129(14):3523–3532. https://www.ncbi.nlm.nih.gov/pubmed/12091321
Li CM, Zhang X, Hoffman HJ, Cotch MF, Themann CL, Wilson MR (2014) Hearing impairment associated with depression in US adults, National Health and Nutrition Examination Survey 2005–2010. JAMA Otolaryngol Head Neck Surg 140(4):293–302. https://doi.org/10.1001/jamaoto.2014.42
Liao BY, Zhang J (2008) Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A 105(19):6987–6992. https://doi.org/10.1073/pnas.0800387105
Lie A, Skogstad M, Johannessen HA, Tynes T, Mehlum IS, Nordby KC et al (2016) Occupational noise exposure and hearing: a systematic review. Int Arch Occup Environ Health 89(3):351–372. https://doi.org/10.1007/s00420-015-1083-5
Lin FR, Yaffe K, Xia J, Xue QL, Harris TB, Purchase-Helzner E, Health ABC Study Group et al (2013) Hearing loss and cognitive decline in older adults. JAMA Intern Med 173(4):293–299. https://doi.org/10.1001/jamainternmed.2013.1868
Lindburg M, Ead B, Jeffe DB, Lieu JEC (2021) Hearing loss-related issues affecting quality of life in preschool children. Otolaryngol Head Neck Surg 164(6):1322–1329. https://doi.org/10.1177/0194599820962475
Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD (1997a) Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet 16(2):188–190. https://doi.org/10.1038/ng0697-188
Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD (1997b) Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet 17(3):268–269. https://doi.org/10.1038/ng1197-268
Liu XP, Koehler KR, Mikosz AM, Hashino E, Holt JR (2016) Functional development of mechanosensitive hair cells in stem cell-derived organoids parallels native vestibular hair cells. Nat Commun 7:11508. https://doi.org/10.1038/ncomms11508
Lu YC, Wu CC, Yang TH, Lin YH, Yu IS, Lin SW et al (2014) Differences in the pathogenicity of the p.H723R mutation of the common deafness-associated SLC26A4 gene in humans and mice. PLoS ONE 8(6):e64906. https://doi.org/10.1371/journal.pone.0064906
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826. https://doi.org/10.1126/science.1232033
Markouli C, Couvreu De Deckersberg E, Regin M, Nguyen HT, Zambelli F, Keller A et al (2019) Gain of 20q11.21 in human pluripotent stem cells impairs TGF-beta-dependent neuroectodermal commitment. Stem Cell Reports 13(1):163–176. https://doi.org/10.1016/j.stemcr.2019.05.005
Martins-Taylor K, Nisler BS, Taapken SM, Compton T, Crandall L, Montgomery KD et al (2011) Recurrent copy number variations in human induced pluripotent stem cells. Nat Biotechnol 29(6):488–491. https://doi.org/10.1038/nbt.1890
McInturff S, Burns JC, Kelley MW (2018) Characterization of spatial and temporal development of Type I and Type II hair cells in the mouse utricle using new cell-type-specific markers. Biol Open. https://doi.org/10.1242/bio.038083
McLean WJ, Yin X, Lu L, Lenz DR, McLean D, Langer R et al (2017) Clonal expansion of Lgr5-positive cells from mammalian cochlea and high-purity generation of sensory hair cells. Cell Rep 18(8):1917–1929. https://doi.org/10.1016/j.celrep.2017.01.066
Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R et al (2001) MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 69(3):635–640. https://doi.org/10.1086/323156
Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C et al (2017) Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545(7653):229–233. https://doi.org/10.1038/nature22312
Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV (2015) Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 33(11):1193–1200. https://doi.org/10.1038/nbt.3392
Mosrati MA, Hammami B, Rebeh IB, Ayadi L, Dhouib L, Ben Mahfoudh K et al (2011) A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. Eur J Med Genet 54(5):e484-488. https://doi.org/10.1016/j.ejmg.2011.06.001
Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420(6915):520–562. https://doi.org/10.1038/nature01262
Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K et al (2012) Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10(6):771–785. https://doi.org/10.1016/j.stem.2012.05.009
Narva E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D et al (2010) High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol 28(4):371–377. https://doi.org/10.1038/nbt.1615
Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr, Khan SN et al (2004) Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet 41(8):591–595. https://doi.org/10.1136/jmg.2004.018523
Nichols J, Smith A (2009) Naive and primed pluripotent states. Cell Stem Cell 4(6):487–492. https://doi.org/10.1016/j.stem.2009.05.015
Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR (2008) Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol 9(1):65–89. https://doi.org/10.1007/s10162-007-0106-7
Ohta S, Wang B, Mansour SL, Schoenwolf GC (2016) BMP regulates regional gene expression in the dorsal otocyst through canonical and non-canonical intracellular pathways. Development 143(12):2228–2237. https://doi.org/10.1242/dev.137133
Oshima K, Shin K, Diensthuber M, Peng AW, Ricci AJ, Heller S (2010) Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 141(4):704–716. https://doi.org/10.1016/j.cell.2010.03.035
Ouji Y, Ishizaka S, Nakamura-Uchiyama F, Yoshikawa M (2012) In vitro differentiation of mouse embryonic stem cells into inner ear hair cell-like cells using stromal cell conditioned medium. Cell Death Dis 3:e314. https://doi.org/10.1038/cddis.2012.56
Pandit SR, Sullivan JM, Egger V, Borecki AA, Oleskevich S (2011) Functional effects of adult human olfactory stem cells on early-onset sensorineural hearing loss. Stem Cells 29(4):670–677. https://doi.org/10.1002/stem.609
Perny M, Ting CC, Kleinlogel S, Senn P, Roccio M (2017) Generation of otic sensory neurons from mouse embryonic stem cells in 3D culture. Front Cell Neurosci 11:409. https://doi.org/10.3389/fncel.2017.00409
Petitpre C, Wu H, Sharma A, Tokarska A, Fontanet P, Wang Y et al (2018) Neuronal heterogeneity and stereotyped connectivity in the auditory afferent system. Nat Commun 9(1):3691. https://doi.org/10.1038/s41467-018-06033-3
Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A et al (1998) SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet 18(2):171–173. https://doi.org/10.1038/ng0298-171
Pyle MP, Hoa M (2020) Applications of single-cell sequencing for the field of otolaryngology: a contemporary review. Laryngoscope Investig Otolaryngol 5(3):404–431. https://doi.org/10.1002/lio2.388
Qin X, Tape CJ (2020) Deciphering organoids: high-dimensional analysis of biomimetic cultures. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2020.10.013
Ranum PT, Goodwin AT, Yoshimura H, Kolbe DL, Walls WD, Koh JY et al (2019) Insights into the biology of hearing and deafness revealed by single-cell RNA sequencing. Cell Rep 26(11):3160-3171 e3163. https://doi.org/10.1016/j.celrep.2019.02.053
Riccomagno MM, Martinu L, Mulheisen M, Wu DK, Epstein DJ (2002) Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes Dev 16(18):2365–2378. https://doi.org/10.1101/gad.1013302
Riccomagno MM, Takada S, Epstein DJ (2005) Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev 19(13):1612–1623. https://doi.org/10.1101/gad.1303905
Roberson DW, Rubel EW (1994) Cell division in the gerbil cochlea after acoustic trauma. Am J Otol 15(1):28–34. https://www.ncbi.nlm.nih.gov/pubmed/8109626
Ronaghi M, Nasr M, Ealy M, Durruthy-Durruthy R, Waldhaus J, Diaz GH et al (2014) Inner ear hair cell-like cells from human embryonic stem cells. Stem Cells Dev 23(11):1275–1284. https://doi.org/10.1089/scd.2014.0033
Rouhani F, Kumasaka N, de Brito MC, Bradley A, Vallier L, Gaffney D (2014) Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet 10(6):e1004432. https://doi.org/10.1371/journal.pgen.1004432
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT et al (2004) SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A 101(21):8090–8095. https://doi.org/10.1073/pnas.0308475101
Ryals BM, Rubel EW (1988) Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240(4860):1774–1776. https://doi.org/10.1126/science.3381101
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE et al (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459(7244):262–265. https://doi.org/10.1038/nature07935
Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S et al (2011) Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141(5):1762–1772. https://doi.org/10.1053/j.gastro.2011.07.050
Schaefer SA, Higashi AY, Loomis B, Schrepfer T, Wan G, Corfas G et al (2018) From otic induction to hair cell production: Pax2(EGFP) cell line illuminates key stages of development in mouse inner ear organoid model. Stem Cells Dev 27(4):237–251. https://doi.org/10.1089/scd.2017.0142
Schuck JB, Smith ME (2009) Cell proliferation follows acoustically-induced hair cell bundle loss in the zebrafish saccule. Hear Res 253(1–2):67–76. https://doi.org/10.1016/j.heares.2009.03.008
Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R et al (2001) Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 27(1):59–63. https://doi.org/10.1038/83768
Sekerkova G, Zheng L, Mugnaini E, Bartles JR (2006) Differential expression of espin isoforms during epithelial morphogenesis, stereociliogenesis and postnatal maturation in the developing inner ear. Dev Biol 291(1):83–95. https://doi.org/10.1016/j.ydbio.2005.12.021
Shearer AE, Hildebrand MS, Smith RJH (1993) Hereditary hearing loss and deafness overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A (eds) GeneReviews((R)). Seattle (WA)
Shi F, Kempfle JS, Edge AS (2012) Wnt-responsive Lgr5-expressing stem cells are hair cell progenitors in the cochlea. J Neurosci 32(28):9639–9648. https://doi.org/10.1523/JNEUROSCI.1064-12.2012
Shi L, Huang L, He R, Huang W, Wang H, Lai X et al (2018) Modeling the pathogenesis of charcot-marie-tooth disease type 1A using patient-specific iPSCs. Stem Cell Rep 10(1):120–133. https://doi.org/10.1016/j.stemcr.2017.11.013
Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV (2018) Sensory neuron diversity in the inner ear is shaped by activity. Cell 174(5):1229-1246 e1217. https://doi.org/10.1016/j.cell.2018.07.007
Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K et al (2011) Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470(7332):105–109. https://doi.org/10.1038/nature09691
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd et al (2019) Comprehensive integration of single-cell data. Cell 177(7):1888-1902 e1821. https://doi.org/10.1016/j.cell.2019.05.031
Sun S, Babola T, Pregernig G, So KS, Nguyen M, Su SM et al (2018) Hair cell mechanotransduction regulates spontaneous activity and spiral ganglion subtype specification in the auditory system. Cell 174(5):1247-1263 e1215. https://doi.org/10.1016/j.cell.2018.07.008
Swanson GJ, Howard M, Lewis J (1990) Epithelial autonomy in the development of the inner ear of a bird embryo. Dev Biol 137(2):243–257. https://doi.org/10.1016/0012-1606(90)90251-d
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. https://doi.org/10.1016/j.cell.2006.07.024
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872. https://doi.org/10.1016/j.cell.2007.11.019
Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C et al (2015) Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526(7574):564–568. https://doi.org/10.1038/nature15695
Tang ZH, Chen JR, Zheng J, Shi HS, Ding J, Qian XD et al (2016) Genetic correction of induced pluripotent stem cells from a deaf patient with MYO7A mutation results in morphologic and functional recovery of the derived hair cell-like cells. Stem Cells Transl Med 5(5):561–571. https://doi.org/10.5966/sctm.2015-0252
Tang PC, Alex AL, Nie J, Lee J, Roth AA, Booth KT et al (2019) Defective Tmprss3-associated hair cell degeneration in inner ear organoids. Stem Cell Rep 13(1):147–162. https://doi.org/10.1016/j.stemcr.2019.05.014
Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL et al (2007) New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448(7150):196–199. https://doi.org/10.1038/nature05972
Tomblin JB, Harrison M, Ambrose SE, Walker EA, Oleson JJ, Moeller MP (2015) Language outcomes in young children with mild to severe hearing loss. Ear Hear 36(Suppl 1):76S-91S. https://doi.org/10.1097/AUD.0000000000000219
Tona R, Lopez IA, Fenollar-Ferrer C, Faridi R, Anselmi C, Khan AA et al (2020) Mouse models of human pathogenic variants of TBC1D24 associated with non-syndromic deafness DFNB86 and DFNA65 and syndromes involving deafness. Genes (basel). https://doi.org/10.3390/genes11101122
Toriello HV, Smith SD (eds) (2013) Hereditary hearing loss and its syndromes, 3rd edn. Oxford University Press, New York
Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N et al (1998) Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279(5358):1950–1954. https://doi.org/10.1126/science.279.5358.1950
van der Valk WH, Steinhart MR, Zhang J, Koehler KR (2021) Building inner ears: recent advances and future challenges for in vitro organoid systems. Cell Death Differ 28(1):24–34. https://doi.org/10.1038/s41418-020-00678-
van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW et al (2003) A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet 40(12):879–884. https://doi.org/10.1136/jmg.40.12.879
Vincent C, Kalatzis V, Abdelhak S, Chaib H, Compain S, Helias J et al (1997). BOR and BO syndromes are allelic defects of EYA1. Eur J Hum Genet 5(4):242–246. https://www.ncbi.nlm.nih.gov/pubmed/9359046
Wakaoka T, Motohashi T, Hayashi H, Kuze B, Aoki M, Mizuta K et al (2013) Tracing Sox10-expressing cells elucidates the dynamic development of the mouse inner ear. Hear Res 302:17–25. https://doi.org/10.1016/j.heares.2013.05.003
Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J et al (1995) Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374(6517):60–61. https://doi.org/10.1038/374060a0
Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M et al (1997) The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 16(2):191–193. https://doi.org/10.1038/ng0697-191
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ (2019) Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177(7):1873-1887 e1817. https://doi.org/10.1016/j.cell.2019.05.006
Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD (2018) Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23(6):869-881 e868. https://doi.org/10.1016/j.stem.2018.10.010
Xiang M, Gao WQ, Hasson T, Shin JJ (1998) Requirement for Brn-3c in maturation and survival, but not in fate determination of inner ear hair cells. Development 125(20):3935–3946. https://www.ncbi.nlm.nih.gov/pubmed/9735355
Xiang M, Maklad A, Pirvola U, Fritzsch B (2003) Brn3c null mutant mice show long-term, incomplete retention of some afferent inner ear innervation. BMC Neurosci 4:2. https://doi.org/10.1186/1471-2202-4-2
Xu J, Li J, Zhang T, Jiang H, Ramakrishnan A, Fritzsch B et al (2021) Chromatin remodelers and lineage-specific factors interact to target enhancers to establish proneurosensory fate within otic ectoderm. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.2025196118
Yamoah EN, Li M, Shah A, Elliott KL, Cheah K, Xu PX et al (2020) Using Sox2 to alleviate the hallmarks of age-related hearing loss. Ageing Res Rev 59:101042. https://doi.org/10.1016/j.arr.2020.101042
Yang S, Lin G, Tan YQ, Deng LY, Yuan D, Lu GX (2010) Differences between karyotypically normal and abnormal human embryonic stem cells. Cell Prolif 43(3):195–206. https://doi.org/10.1111/j.1365-2184.2010.00669.x
Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N et al (1999) A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 21(4):363–369. https://doi.org/10.1038/7693
Yun NE, Ronca S, Tamura A, Koma T, Seregin AV, Dineley KT et al (2015) Animal model of sensorineural hearing loss associated with lassa virus infection. J Virol 90(6):2920–2927. https://doi.org/10.1128/JVI.02948-15
Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX (2003) The role of Six1 in mammalian auditory system development. Development 130(17):3989–4000. https://doi.org/10.1242/dev.00628
Zhong C, Chen Z, Luo X, Wang C, Jiang H, Shao J et al (2018) Barhl1 is required for the differentiation of inner ear hair cell-like cells from mouse embryonic stem cells. Int J Biochem Cell Biol 96:79–89. https://doi.org/10.1016/j.biocel.2018.01.013
Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL et al (2003) Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 73(5):1082–1091. https://doi.org/10.1086/379286
Funding
This work was supported by the National Institutes of Health (K08-DC016034 to R.F.N.) and the Triological Society and American College of Surgeons (Clinician Scientist Development Award to R.F.N.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Romano, D.R., Hashino, E. & Nelson, R.F. Deafness-in-a-dish: modeling hereditary deafness with inner ear organoids. Hum Genet 141, 347–362 (2022). https://doi.org/10.1007/s00439-021-02325-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02325-9