
Abstract
Located in the critical 1p36 microdeletion region, the chromodomain helicase DNA-binding protein 5 (CHD5) gene encodes a subunit of the nucleosome remodeling and deacetylation (NuRD) complex required for neuronal development. Pathogenic variants in six of nine chromodomain (CHD) genes cause autosomal dominant neurodevelopmental disorders, while CHD5-related disorders are still unknown. Thanks to GeneMatcher and international collaborations, we assembled a cohort of 16 unrelated individuals harboring heterozygous CHD5 variants, all identified by exome sequencing. Twelve patients had de novo CHD5 variants, including ten missense and two splice site variants. Three familial cases had nonsense or missense variants segregating with speech delay, learning disabilities, and/or craniosynostosis. One patient carried a frameshift variant of unknown inheritance due to unavailability of the father. The most common clinical features included language deficits (81%), behavioral symptoms (69%), intellectual disability (64%), epilepsy (62%), and motor delay (56%). Epilepsy types were variable, with West syndrome observed in three patients, generalized tonic–clonic seizures in two, and other subtypes observed in one individual each. Our findings suggest that, in line with other CHD-related disorders, heterozygous CHD5 variants are associated with a variable neurodevelopmental syndrome that includes intellectual disability with speech delay, epilepsy, and behavioral problems as main features.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The chromodomain-helicase-DNA-binding protein 5 gene (CHD5) belongs to a highly conserved family of genes encoding ATP-dependent chromatin remodeling complex subunits comprising nine members, named CHD1–CHD9 (Delmas et al. 1993; Woodage et al. 1997). CHD proteins carry out multiple functions essential for cell survival and embryonic development, including chromatin remodeling, transcriptional regulation, and DNA repair (Tyagi et al. 2016). They are composed of two N-terminal chromodomains important for histone tail binding, a central and conserved SNF2-like helicase motif that uses ATP-hydrolysis for chromatin remodeling, and a less-defined C-terminal DNA-binding domain (Delmas et al. 1993; Woodage et al. 1997). The CHD protein family is further divided into three subfamilies based on the presence or absence of additional domains (Tyagi et al. 2016). Subfamily I (CHD1 and CHD2) features a C-terminal DNA-binding domain that preferentially binds to AT-rich DNA motifs (Tyagi et al. 2016). Subfamily III (CHD6 to CHD9) is characterized by the presence of additional C-terminal functional domains (BRK motif or SANT domain) that define their binding properties (Tyagi et al. 2016). CHD3, CHD4, and CHD5 are part of subfamily II, and, unlike other CHD members, they possess two N-terminal Plant-Homeo Domains (PHD) with histone-binding activity. These three proteins represent mutually exclusive subunits of a large protein complex known as Nucleosome Remodeling and Deacetylase (NuRD) complex (Tyagi et al. 2016).
Subfamily II members are characterized by different expression profiles, with CHD3 and CHD4 being ubiquitously expressed, whereas CHD5 is mainly expressed in brain and testis (Marfella and Imbalzano 2007; Zhuang et al. 2014). Furthermore, the three encoded proteins have distinct, non-redundant properties and functions within the NuRD complex and neuronal defects induced by the specific knockdown of one subunit cannot be rescued by overexpression of another CHD protein (Nitarska et al. 2016). A coordinated sequential switch of these subunits is crucial for mouse cortical development (Nitarska et al. 2016). CHD3 ensures proper layer specification, CHD4 induces early proliferation of the basal progenitors, while CHD5 mediates neuronal differentiation, radial migration, and neuronal cell identity (Nitarska et al. 2016). CHD5 is required both for activation of genes promoting neuronal differentiation programs and for repression of non-neuronal Polycomb target genes (Egan et al. 2013). Moreover, CHD5 directly interacts with the repressive H3K27me3 histone mark via its PHD and chromodomains (Egan et al. 2013).
All CHD genes are evolutionary constrained in human populations, with significantly fewer truncating and missense variants than expected by chance (Karczewski et al. 2020), but only six of the nine CHD members have been associated with human disorders so far (Zentner et al. 2010; Merner et al. 2016; Weiss et al. 2016, 2020; Pilarowski et al. 2018; Blok et al. 2018; Chen et al. 2020). Together with CHD6 and CHD9, CHD5 has not yet been associated with a human disease. However, CHD5 is located on chromosome 1p36.31, a region commonly deleted in monosomy 1p36, and CHD5 haploinsufficiency was hypothesized to contribute to the clinical features of this syndrome, which include neurodevelopmental deficits (intellectual disability with limited language ability), delayed growth, hypotonia, seizures, craniofacial and skeletal features, hearing and vision impairment, as well as cardiac anomalies (Shimada et al. 2015). In this study, we assembled a cohort of 16 unrelated patients with de novo or inherited heterozygous variants in CHD5. Comparison of the clinical features of these affected subjects showed that genetic alterations of CHD5 are associated with a variable neurodevelopmental disorder frequently characterized by intellectual disability (ID), speech delay, motor delay, behavioral problems, and epilepsy.
Materials and methods
Following the identification by exome sequencing of a de novo missense variant in CHD5 in a patient with ID, autism spectrum disorder (ASD), and epilepsy, we collected data from additional patients with CHD5 variants through GeneMatcher (Sobreira et al. 2015). We systematically included all patients with de novo variants as well as patients with either truncating or predicted damaging missense variants inherited from affected parents. Only patients without a detailed clinical history and/or inheritance information were excluded from the study. Exome sequencing was performed at the respective institutions. Referring physicians provided detailed developmental, neurological, and behavioral history of the patients. Patient information was anonymized before data sharing. Variants were described on the CHD5 NM_015557.3 RefSeq transcript using HGVS recommendations (den Dunnen et al. 2016) and classified according to ACMG guidelines (Richards et al. 2015). All variants have been submitted to the ClinVar Database and have been assigned the following accession numbers: SCV001477999–SCV001478015. Multiple algorithms were used to assess the pathogenicity of CHD5 variants, including Mutation Taster, Polyphen-2, and SIFT (Ng 2003; Adzhubei et al. 2010; Schwarz et al. 2014). Combined annotation-dependent depletion (CADD) scores (Rentzsch et al. 2019) were calculated for each variant using the GRCh37-v1.6 version (Online Resource Table 1). Prediction of the consequences of the two splicing variants were carried out with Alamut® Visual, a mutation analysis software which includes a splicing module integrating a number of prediction algorithms and splicing prediction data. Nucleotide conservation across 100 vertebrate species was calculated for each variant using the PhastCons score obtained with the phastCons100way.UCSC.hg19 R package (Siepel 2005) and represents the probability that a given nucleotide is conserved (range 0–1). Codon conservation scores were calculated as the mean nucleotide conservation of each triplet. Known CHD5 NM_015557.3 variants were retrieved from gnomAD v2.1.1 (Karczewski et al. 2020), restricting to loss-of-function, missense, and synonymous single nucleotide variants.
Results
CHD5 variant spectrum
We report 16 different genetic alterations in CHD5, including eleven missense variants [c.577C > T, p.(Arg193Trp); c.578G > A, p.(Arg193Gln), c.1279G > A, p.(Glu427Lys); c.2735C > T, p.(Ser912Phe); c.3250G > A, p.(Asp1084Asn); c.3371C > T, p.(Pro1124Leu); c.3407G > A, p.(Arg1136His); c.3419A > T, p.(Asn1140Ile); c.4257C > G, p.(Ile1419Met); c.4463A > T, p.(Asp1488Val) and c.5141A > G, p.(Glu1714Gly)], one duplication of a single base leading to a frameshift [c.612dup, p.(Ser205Leufs*88)], two nonsense substitutions [c.940G > T, p.(Glu314*) and c.1786C > T, p.(Arg596*)], and two splice site variants (c.4079-3C > G and c.4171 + 1G > C). All variants were either absent from gnomAD or present with an allele frequency below 0.0001% (Online Resource Table 1). All missense variants affect highly conserved amino acids of CHD5 (up to zebrafish, Online Resource Fig. 1), had CADD scores above 22, and were predicted to be damaging by at least two algorithms among Polyphen-2, SIFT, and Mutation Taster (Online Resource Table 1). The conservation score of nucleotides and corresponding codons calculated based on the alignment of 100 species additionally indicated that all the affected nucleotides, with the exception of c.4257C > G [resulting in p.(Ile1419Met)], were subject to a great level of conservation during evolution (score 1 in a 0 to 1 scale) (Online Resource Table 1).
Ten of the eleven missense substitutions and the two splice site variants occurred de novo in patients without family history, while one missense and the two nonsense variants segregated with neurodevelopmental phenotypes in three families (Fig. 1). The frameshift variant identified in Patient 3 was absent from her mother but inheritance could not be assessed further, since her father was not available for genetic analysis. Notably, one of the missense variants segregating in a larger family [p.(Arg193Trp)] occurred at the same highly conserved residue as one of the de novo missense variants [p.(Arg193Gln)]. Two de novo missense variants [p.(Asn1140Ile) and p.(Ile1419Met)] were mosaic in patients 11 and 14. Both mosaic variants, identified by WES, were present in less than 25% of the total reads on blood DNA and were confirmed by Sanger sequencing.

Family trees of the inherited mutations. In family 1, Individual III-3 corresponds to Patient 1. In family 2, Individual III-1 corresponds to Patient 4. In family 3, Individual III-1 corresponds to Patient 6. The variants in CHD5 identified in these three families are associated with incomplete penetrance and variable expressivity
In addition to these sixteen predicted damaging variants, a de novo variant [c.815C > T, p.(Ala272Val)] absent from gnomAD was identified in a male patient (VUS 1, Online Resource Tables 1 and 2). This variant alters a poorly conserved amino acid located outside of any known domain and is not predicted to alter splicing, but affects a highly conserved nucleotide (score 1) (Online Resource Fig. 1). Because of consistent benign predictions by all algorithms and a CADD score below 20, this variant was considered as of unknown significance despite its de novo occurrence.
Missense variants in CHD5 tend to cluster in functional domains
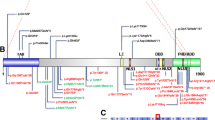
CHD5 comprises nine protein domains: an N-terminal domain of chromo domain-associated helicases (CHDNT), two PHD domains (PHD1 and PHD2) and two chromodomains (Chd1 and Chd2) important for histone binding, one bipartite Helicase domain with ATPase catalytic activity, two conserved Domains with Unknown Function (DUF1087 and DUF1086), and a C-terminal domain B of chromo domain-associated CHD-like helicases (CHDCT2) mediating the interaction with GATA2D (Pierson et al. 2019) (Fig. 2a). The helicase, PHD, and C-terminal regions are the most conserved and constrained domains (Samocha et al. 2017; Havrilla et al. 2019). Strikingly, missense variants with CADD scores above or equal to 22 reported in gnomAD alter fewer residues in the helicase C-terminal domain (17%) than in other regions (31–42%), contrary to gnomAD synonymous and missense variants with CADD scores below 22, which appeared in 30% of the residues of this part of the helicase domain (other regions 25–60%) (Fig. 2b).

Distribution of the CHD5 variants based on position and conservation of the affected amino acids. a Schematic representation of the CHD5 protein and its domains, with position of the identified mutations relative to exon and domain distribution. The CHDNT domain is indicated in yellow, the PHD domains in red, the Chd domains in green, the helicase domains in light blue, the DUFs domains in purple, and the CHDCT2 domain in lilac. Inherited variants are indicated in blue and de novo variants in black. Putative loss-of-function variants are indicated with a triangle, likely pathogenic missense substitutions with a filled circle and the VUS with an empty circle. b Comparison of the distribution of the variants identified in our cohort with the synonymous and missense variants reported in gnomAD, with relative position of each affected CHD5 residue across the protein domains. c Comparison of a portion of the highly conserved C-terminal Helicase domain among yeast SNF2 (black) and human CHD3 (red), CHD4 (green), and CHD5 (blue). Pathogenic missense substitutions altering residues in this domain are indicated with the color corresponding to the CHD protein where the variant was identified. The amino acids altered by the substitutions are indicated with a square whose color corresponds to the CHD protein where the variant was identified
The 11 predicted damaging missense substitutions alter 10 different amino acids and all but two affect a functional domain of CHD5: one variant affects the PHD2 domain, two variants each affect the CHDNT and the DUF1086 domains, and four variants alter the C-terminal part of the helicase domain depleted in missense variants in gnomAD (Fig. 2a, b, Online Resource Fig. 1). Two missense substitutions in the helicase domain match positions altered by two previously published pathogenic variants in CHD3 (Fig. 2c): CHD5-Asp1084 (Patient 8) corresponds to CHD3-Asp1120, whereas CHD5-Arg1136 (Patient 10) corresponds to CHD3-Arg1172 (Blok et al. 2018). Additionally, Pro1124 (Patient 9) is positioned within a stretch of amino acids that were found to be altered by missense substitutions of CHD3 (Trp1158, Asn1159, and His1161) or CHD4 (Asp1147, Trp1148) (Blok et al. 2018; Weiss et al. 2020). Importantly, CHD3 residues Arg1172, Trp1158, and Asn1159 were proven essential for either the ATPase activity of or the ability to carry out chromatin remodeling (Blok et al. 2018).
The nonsense and frameshift variants identified in the present cohort are located in exons 5, 7, and 11, and are therefore predicted to result in transcripts that are subject to nonsense-mediated mRNA decay or to generate a truncated protein, if expressed. The two splice site variants are predicted to, respectively, abolish the acceptor and donor splice sites of exon 27 with high probability (Online Resource Fig. 2a, b). Since variants altering canonical splice sites frequently lead to skipping of the corresponding exon, these two variants possibly induce the same in-frame deletion of the 93 nucleotides of exon 27. Given the preferential expression of CHD5 in brain and testis, we postulated that the effects of these two variants on CHD5 splicing could not be assessed. Surprisingly, we were able to amplify CHD5 transcript from RNA extracted from blood and fibroblasts. A blood sample of Patient 13 could be subsequently obtained, and the resulting analysis showed the in-frame exclusion of exon 27 in the mutant allele, predicted to cause a deletion of 31 amino acids at the protein level [p.(Glu1360_Ser1391delinsGly)] (Online Resource Fig. 2c). CHD5 splicing in Patient 12 could not be determined due to the impossibility to obtain additional material.
CHD5 variants are associated with developmental delay, intellectual disability, behavioral disturbances, epilepsy, and craniosynostosis
With the exclusion of Patient VUS1, the present cohort comprises seven females and nine males aged from 3 to 24 years (median age 9 years 6 months). Detailed phenotypical information for each patient is provided in Online Resource Table 2. For most patients, pregnancy was unremarkable, birth parameters were normal, and the neonatal period was uneventful. Measurements at the last evaluation were also mainly on average, with only two and four patients presenting with more than two standard deviations above the mean of growth standards for weight and height, respectively. The most frequent clinical features observed in this cohort are summarized in Table 1, and comprise speech delay (n = 13/16), behavioral disturbances (n = 11/16), epilepsy (n = 10/16), subtle facial dysmorphism (n = 11/16), motor delay (n = 9/16), intellectual disability (n = 9/14), hypotonia (n = 7/14), and craniosynostosis (n = 3/7). The level of intellectual disability could be assessed for six of the nine patients and was moderate in two patients and severe in four. Four patients presented with normal IQ, with a full-scale IQ ranged between 85 and 105, and one patient was reported to have an IQ above average. Developmental milestones were delayed in the majority of the patients, with language acquisition being more affected than motor development. Sitting and walking independently were achieved at a median age of 13 and 28 months, respectively. The first words were pronounced at a median age of 24 months. Three patients were still non-verbal at 3, 9, and 24 years of age. Four patients with an age range between 11 and 22 years could only speak a few words. Dysphasia, stuttering, and echolalia were also reported in single patients. Autism spectrum disorder and obsessive–compulsive tendencies were the most frequently observed behavioral problems in this cohort. Self-injurious behavior, poor eye contact, outbursts of anger, and aggressive behavior were also noted. Seizures occurred in more than half of the patients (n = 10) with a median age of onset of 10 months. The earliest onset was at day one and the latest at 16 years of age. Patients could be divided into three groups based on the severity of the seizures, although a significant intra-group variability was also observed: (1) three patients experienced one to five seizures only and were not under antiepileptic therapy; (2) three others had a generalized epilepsy and were still receiving antiepileptic drugs at the time of description; (3) four patients had a diagnosis of developmental and epileptic encephalopathy, and their EEG showed a suppression-burst pattern or hypsarrhythmia. Seizure types included generalized tonic–clonic febrile and afebrile seizures, infantile spasms, generalized staring spells, and myoclonus. Most of the patients were seizure-free at the time of the study with or without specific therapy. Hypotonia was the most frequent finding upon neurological examination (n = 7/14), while dysmetria and ataxia were each reported in single patients. Brain Magnetic Resonance Imaging (MRI) were mainly normal (n = 8/12) or showed nonspecific abnormalities. Dysmorphic facial features (Fig. 3) were rather nonspecific and did not suggest 1p36 deletion syndrome. They comprised ear anomalies (n = 4/16), including low set and posteriorly rotated ears, mildly cupped or smaller ears, prominent nasal bridge and tip (n = 3/16), short philtrum (n = 3/16), thin upper lip (n = 3/16), upslanting palpebral fissures (n = 3/16), synophrys (n = 2/16), epicanthic folds (n = 2/16), frontal bossing (n = 2/16), and micrognathia (n = 2/16). However, no distinctive facial gestalt emerges from the series. Eye anomalies/visual impairment were noted in some patients but appeared inconsistent within the cohort. Notably, three patients displayed craniosynostosis (Fig. 3). Specifically, Patient 1 displayed sagittal craniosynostosis, Patient 4 had metopic craniosynostosis, and Patient 5 was diagnosed with trigonocephaly. Craniosynostosis of Patients 1 (and of her cousin Individual 1-III-2, see Fig. 1) and of Patient 4 was diagnosed via computed tomography scans by craniofacial surgeons and was surgically corrected by cranial vault remodeling. Patient 5 did not undergo surgery. Malformations of other organs were rare and restricted to individual patients.

Facial profiles of patients with CHD5 variants. a–c Patient 1 age 6 months. d, e Patient 2 age 11 years 4 months. f Patient 4 age 1 year. g, h Patient 7 age 24 years. i, j Patient 10 age 3 (i) and 5 years (j). k–n Patient 13 age 9 months (k), 9 years (l) and 22 years (m, n). o, p Patient 14 age 3 years six months (o) and 6 years (p). Facial dysmorphism was related to craniosynostosis in Patients 1 and 4. Other patients displayed subtle facial features, such as high forehead, but no consistent facial dysmorphism emerges from the whole panel
Inherited variants are associated with intra-familial variability and incomplete penetrance
Intellectual disability was not diagnosed in any of Patients 1 (family 1), 4 (family 2), and 6 (family 3), and the clinical features were variable within families (Fig. 1). Patient 1, corresponding to individual 1-III-3 (Family 1, Generation III, Individual 3), presented with speech delay, normal motor development, and craniosynostosis. Speech delay was observed also in her cousins 1-III-1 and 1-III-2. Individual 1-III-2 also displayed craniosynostosis, while Individual 1-III-1 was additionally characterized by motor delay and oppositional defiant disorder (detailed clinical data of these two individuals are available in Online Resource Table 2). The p.(Arg193Trp) variant was inherited from the respective mothers, who are sisters. Detailed clinical features were not available for these subjects, but delayed speech was reported for 1-II-2, while her sister 1-II-3 was described as asymptomatic. Suspected craniosynostosis was reported in the maternal grand-mother 1-I-2, who was deceased.
Patient 4 (Individual 2-III-1) had normal development and intellectual abilities, but presented with a sensory processing disorder, obsessive–compulsive tendencies, and was diagnosed with Asperger-like syndrome. He had hypotonia, metopic craniosynostosis, and tonic–clonic seizures over a period of 2 years that spontaneously resolved. His sister (2-III-2) displayed dyslexia, dysorthographia, dyscalculia, mood disorder, anxiety, and depression. The p.(Glu314*) variant present in the two siblings was inherited from their father (2-II-1), who showed dyslexia and borderline intellectual disability.
Patient 6 (3-III-1) presented with mild motor delay and speech impairment, several behavioral problems, mild facial dysmorphism, and strabismus. This family comprises three additional affected members: the proband’s sister (3-III-2) and his paternal uncle (3-II-1) displayed learning difficulties, while his father (3-II-2) had severe psychiatric issues and died from suicide.
Altogether, these data point to the existence of an intra-familial phenotypic variability associated with inherited variants. Moreover, unaffected carriers were also reported in each family, indicating an incomplete penetrance. The lack of a thorough clinical history of each individual could also account for the reported differences.
Discussion
In this study, we report 13 sporadic cases and 3 families with predicted damaging variants altering highly conserved amino acids of CHD5. Patients with these genetic alterations display a broad spectrum of developmental disturbances, recurrently including developmental delay, learning difficulties or intellectual disability, behavioral problems, seizures, hypotonia, and craniosynostosis. Variants identified in patients include both missense substitutions altering highly conserved amino acids mainly located in functional domains and variants predicted to lead to haploinsufficiency by nonsense-mediated mRNA decay (i.e., frameshift and nonsense variants). The probability that CHD5 is intolerant to haploinsufficiency, calculated by a recent study including 753,994 individuals, is 0.93 (Collins et al. 2021). CHD5 is also catalogued among haploinsufficient genes by the Genome Aggregation Database (gnomAD), with a probability to be LOF intolerant (pLI) of 1 and an LOF observed/expected upper bound fraction (LOEUF) of 0.16. Furthermore, its missense Z-score (referring to the number of observed and expected missense variants within the same database) indicates that this gene is highly missense-constrained (Z-score = 5.32). These metrics indicate that variants disrupting the coding sequence of CHD5 are counter-selected in human populations, and also suggest that they likely are disease-causing (Karczewski et al. 2020). Nevertheless, 25 out of the 141,456 individuals present in gnomAD harbor CHD5 variants predicted to be associated with a LOF of the corresponding allele. Incomplete penetrance and variable expressivity, as observed in the familial cases herein reported, could at least partially account for this finding. Furthermore, several of these truncating variants display an allelic imbalance lower than the 0.5 expected for heterozygous variants, suggesting that some of them could be present only at the somatic state in older individuals.
CHD5 is known to play an important role in the context of chromatin remodeling, which it achieves by means of its intrinsic ATPase activity and of its presence within the NuRD complex. Additionally, CHD5 is involved in the regulation of the expression of a subgroup of Polycomb target genes through the maintenance of the repressive H3K27me3 histone methylation mark (Egan et al. 2013). Hence, variants that disrupt CHD5 activity may impact the epigenetic landscape of cells in a way that results in transcriptional disturbances and possibly generates one or several episignatures that are unique for CHD5-related disorders. The pathogenic mechanism(s) by which the variants described in this study contribute to different neurodevelopmental disturbances remains to be defined. Truncating and missense variants could alter the activity of CHD5 and of the NuRD complex in different ways, i.e., by either loss- or gain-of-function, and affect distinct aspects of the epigenetic processes related to the NuRD complex. These mechanisms might include haploinsufficiency of CHD5 within the complex, impaired assembly or composition of the NuRD complex, impaired binding to nucleosomes, or impaired ability to carry out ATP-dependent nucleosome remodeling. The sample size of our cohort and in particular the number of LOF variants was unfortunately too small to establish significant genotype–phenotype correlations based on type and position of the variants. However, from this small cohort, we anticipate that missense substitutions might be more prone to cause epileptic phenotypes. Indeed, approximately half of the patients with missense variants (6/11) had developmental and epileptic encephalopathy and/or an ongoing antiepileptic treatment. In particular, three out of four patients with variants within the helicase domain displayed severe epilepsy (all three patients with West syndrome), while only one patient with variants outside this domain had severe epilepsy (suppression-burst) and two had controlled seizures. Thus, it seems that missense variants, particularly those located in the helicase domain, predispose to early onset epilepsy with a higher likelihood than LOF or missense variants outside this domain. However, this observation needs to be confirmed on larger sample sizes.
CHD5 is located on chromosome 1p36.31. Patients with CHD5 variants share nonspecific clinical features with the 1p36 deletion syndrome, a disorder characterized by moderate-to-severe intellectual disability, language deficits, hypotonia, seizures, and distinctive facial features. Depending on the extent of the chromosomal deletion, CHD5 haploinsufficiency could contribute to the clinical features of this disorder or worsen the severity of intellectual disability, as previously suggested (Shimada et al. 2015). Furthermore, the genes responsible for epilepsy, a frequent feature of the 1p36 deletion syndrome, have not yet been fully characterized. GABRD and KCNAB2 are considered likely candidates for the epileptic phenotype, because patients with a deletion of these genes are more frequently epileptic than those without (Heilstedt et al. 2002; Shimada et al. 2015). However, CHD5 might also be held accountable for different reasons. With the exception of a single patient (Shimada et al. 2015), CHD5 was always reported to be deleted together with KCNAB2, as CHD5 is adjacent and proximal to KCNAB2. Additionally, point variants in KCNAB2 have never been described thus far in association with epilepsy. Finally, 16/21 patients with a 1p36 deletion encompassing CHD5 were reported to display epilepsy, while 19/29 with retained CHD5 copy do not have epilepsy (Shimada et al. 2015). RERE haploinsufficiency might also play a role in the epilepsy of some patients with the 1p36 deletion (Fregeau et al. 2016), but this gene is located proximally to CHD5 and deleted only in patients with very large deletions [8/50 in (Shimada et al. 2015)]. Thus, RERE haploinsufficiency would not explain the epilepsy of most patients with 1p36 deletion. A similar reasoning applies to SPEN, a newly identified ID-associated gene with rare seizures, which is proximal to RERE (Radio et al. 2021). Taken together, these data suggest that several genes may be involved in the epilepsy phenotype of the 1p36 deletion syndrome and that CHD5 represents one of its potential modifiers. Notably, seizures are a frequent feature of patients with CHD5 point variants as well, hence supporting the epileptogenic role of CHD5 in the context of the 1p36 deletion syndrome.
Patients described in this study also show overlapping features with other neurodevelopmental disorders caused by de novo heterozygous variants in other CHD genes, which show intolerance to LOF and missense variants similar to that of CHD5. Pathogenic variants in CHD1 lead to a developmental disorder associated with developmental delay, speech apraxia, autism, hypotonia, and facial dysmorphic features (Pilarowski et al. 2018). CHD2 pathogenic variants cause a developmental and epileptic encephalopathy (Suls et al. 2013; Chen et al. 2020). Disease-causing variants in CHD7 and CHD8 cause CHARGE syndrome and a syndromic form of autism spectrum disorder, respectively (Vissers et al. 2004; Zentner et al. 2010; O’Roak et al. 2011; Merner et al. 2016). Finally, pathogenic variants in CHD3 and CHD4 have recently been described in patients with developmental delay, intellectual disability, macrocephaly, impaired speech, and dysmorphic features (Weiss et al. 2016, 2020; Blok et al. 2018; Drivas et al. 2020). Specifically, CHD3 mutations cause Snijders Blok–Campeau syndrome, which is frequently characterized by autism and signs of connective tissue laxity (Blok et al. 2018; Drivas et al. 2020). CHD4 mutations cause Sifrim–Hitz–Weiss syndrome, frequently associated with heart malformations as well as numerous other findings (Chiari malformation, Moyamoya disease, hypogonadism, deafness, and limb malformation) (Weiss et al. 2016, 2020). Interestingly, there is an important clinical variability for most CHD-related disorders, which makes recognition of these syndromes complicated but yet possible. Notably, seizures are rarely observed in patients with CHD4 variants and occur only in a minority of patient with CHD3 alterations. Also craniosynostosis has been rarely reported in association with variants in other CHD genes (Siakallis et al. 2019; Tønne et al. 2020). In our cohort, craniosynostosis was observed in two individuals belonging to different families and in one patient with a de novo variant. The specific association of CHD5 defects with craniosynostosis remains puzzling based on the reported preferential expression of this gene in brain and testis, but possibly suggests that CHD5 might be expressed more broadly at some stages of embryonic development or that craniosynostosis is linked to an indirect effect of CHD5 alterations on gene expression programs that coordinate boundary formation or differentiation of overlying cranial neural crest. Interestingly, the knockdown of chd5, which shows an expression pattern in adult zebrafish resembling that of CHD5 in adult human individuals, results in craniofacial development defects including reduced head size and decreased cartilage formation in the head, raising the possibility of additional conserved roles of CHD5 during vertebrate embryogenesis (Bishop et al. 2015). The splicing analysis performed in the present study led to the detection of CHD5 transcripts also in blood and fibroblasts, suggesting that CHD5 expression might not be restricted to brain and testis. Hence, a CHD5 expression pattern that is broader than previously reported could account for the non-brain-related phenotypes observed in this cohort of patients.
CHD5 is also a known tumor suppressor gene frequently deleted or silenced in diverse human cancers (Bagchi et al. 2007). None of the patients included in this study have had tumors so far, suggesting that germline alterations of CHD5, contrary to somatic alterations, do not predispose to a higher risk of tumorigenesis, as previously reported for other tumor suppressor genes, including for instance genes encoding subunits of the SWI–SNF complex or ASXL1 (Romero and Sanchez-Cespedes 2014; Carlston et al. 2017). However, considering the relatively young age of this cohort, we cannot rule out an increased risk to develop tumors in adult life.
In conclusion, we describe the first cohort of patients with heterozygous variants in CHD5, associated with a new syndrome mainly characterized by developmental delay, intellectual disability, behavioral symptoms, and epilepsy. Elaborated functional studies are required to understand the impact of the variants reported in this study on CHD5 protein levels and the NuRD complex during brain development.
Data availability
The raw data supporting the results presented in this study are available upon request from the corresponding authors Christel Depienne or Cyril Mignot.
References
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Bagchi A, Papazoglu C, Wu Y et al (2007) CHD5 is a tumor suppressor at human 1p36. Cell 128:459–475. https://doi.org/10.1016/j.cell.2006.11.052
Bishop B, Ho KK, Tyler K et al (2015) The chromatin remodeler chd5 is necessary for proper head development during embryogenesis of Danio rerio. Biochimica et Biophysica Acta (BBA) Gene Regul Mech 189:1040–1050. https://doi.org/10.1016/j.bbagrm.2015.06.006
Carlston CM, O’Donnell-Luria AH, Underhill HR et al (2017) Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum Mutat. https://doi.org/10.1002/humu.23203
Chen J, Zhang J, Liu A et al (2020) CHD2-related epilepsy: novel mutations and new phenotypes. Dev Med Child Neurol 62:647–653. https://doi.org/10.1111/dmcn.14367
Collins RL, Glessner JT, Porcu E et al (2021) A cross-disorder dosage sensitivity map of the human genome. BMJ. https://doi.org/10.1101/2021.01.26.21250098
Delmas V, Stokes DG, Perry RP (1993) A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci 90:2414–2418. https://doi.org/10.1073/pnas.90.6.2414
Drivas TG, Li D, Nair D et al (2020) A second cohort of CHD3 patients expands the molecular mechanisms known to cause Snijders Blok-Campeau syndrome. Eur J Hum Genet 28:1422–1431. https://doi.org/10.1038/s41431-020-0654-4
den Dunnen JT, Dalgleish R, Maglott DR et al (2016) HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37(6):564–569. https://doi.org/10.1002/humu.22981
Egan CM, Nyman U, Skotte J et al (2013) CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev Cell 26:223–236. https://doi.org/10.1016/j.devcel.2013.07.008
Fregeau B, Kim BJ, Hernández-García A et al (2016) De novo mutations of RERE cause a genetic syndrome with features that overlap those associated with proximal 1p36 deletions. Am J Hum Genet 98:963–970. https://doi.org/10.1016/j.ajhg.2016.03.002
Havrilla JM, Pedersen BS, Layer RM, Quinlan AR (2019) A map of constrained coding regions in the human genome. Nat Genet 51:88–95. https://doi.org/10.1038/s41588-018-0294-6
Heilstedt HA, Burgess DL, Anderson AE et al (2002) Loss of the potassium channel β-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia 42:1103–1111. https://doi.org/10.1046/j.1528-1157.2001.08801.x
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Marfella CGA, Imbalzano AN (2007) The Chd family of chromatin remodelers. Mutat Res Fundam Mol Mech Mutagen 618:30–40. https://doi.org/10.1016/j.mrfmmm.2006.07.012
Merner N, Forgeot d’Arc B, Bell SC et al (2016) A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): a case report and literature review. Am J Med Genet 170:1225–1235. https://doi.org/10.1002/ajmg.a.37566
Ng PC (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814. https://doi.org/10.1093/nar/gkg509
Nitarska J, Smith JG, Sherlock WT et al (2016) A Functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep 17:1683–1698. https://doi.org/10.1016/j.celrep.2016.10.022
O’Roak BJ, Deriziotis P, Lee C et al (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43:585–589. https://doi.org/10.1038/ng.835
Pierson TM, Otero MG, Grand K et al (2019) The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am J Med Genet 181:548–556. https://doi.org/10.1002/ajmg.c.31752
Pilarowski GO, Vernon HJ, Applegate CD et al (2018) Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J Med Genet 55:561–566. https://doi.org/10.1136/jmedgenet-2017-104759
Radio FC, Pang K, Ciolfi A et al (2021) SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am J Hum Genet 108(3):502–516. https://doi.org/10.1016/j.ajhg.2021.01.015
Rentzsch P, Witten D, Cooper GM et al (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894. https://doi.org/10.1093/nar/gky1016
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med 17:405–423. https://doi.org/10.1038/gim.2015.30
Romero OA, Sanchez-Cespedes M (2014) The SWI/SNF genetic blockade: effects in cell differentiation, cancer and developmental diseases. Oncogene 33:2681–2689. https://doi.org/10.1038/onc.2013.227
Samocha KE, Kosmicki JA, Karczewski KJ et al (2017) Regional missense constraint improves variant deleteriousness prediction. BioRxiv. https://doi.org/10.1101/148353
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362. https://doi.org/10.1038/nmeth.2890
Shimada S, Shimojima K, Okamoto N et al (2015) Microarray analysis of 50 patients reveals the critical chromosomal regions responsible for 1p36 deletion syndrome-related complications. Brain Dev 37:515–526. https://doi.org/10.1016/j.braindev.2014.08.002
Siakallis L, Tan AP, Chorbachi R, Mankad K (2019) A unique case of CHARGE syndrome with craniosynostosis. Childs Nerv Syst 35:11–13. https://doi.org/10.1007/s00381-018-4005-5
Siepel A (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15:1034–1050. https://doi.org/10.1101/gr.3715005
Snijders Blok L, Rousseau J, Twist J et al (2018) CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun 9:4619. https://doi.org/10.1038/s41467-018-06014-6
Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36:928–930. https://doi.org/10.1002/humu.22844
Suls A, Jaehn JA, Kecskés A et al (2013) De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 93:967–975. https://doi.org/10.1016/j.ajhg.2013.09.017
Tønne E, Due-Tønnessen BJ, Mero I-L et al (2020) Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis. Eur J Hum Genet. https://doi.org/10.1038/s41431-020-00788-4
Tyagi M, Imam N, Verma K, Patel AK (2016) Chromatin remodelers: we are the drivers!! Nucleus 7:388–404. https://doi.org/10.1080/19491034.2016.1211217
Vissers LELM, van Ravenswaaij CMA, Admiraal R et al (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36:955–957. https://doi.org/10.1038/ng1407
Weiss K, Terhal PA, Cohen L et al (2016) De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet 99:934–941. https://doi.org/10.1016/j.ajhg.2016.08.001
Weiss K, Lazar HP, Kurolap A et al (2020) The CHD4-related syndrome: a comprehensive investigation of theclinical spectrum, genotype–phenotype correlations, and molecular basis. Genet Med 22:389–397. https://doi.org/10.1038/s41436-019-0612-0
Woodage T, Basrai MA, Baxevanis AD et al (1997) Characterization of the CHD family of proteins. Proc Natl Acad Sci 94:11472–11477. https://doi.org/10.1073/pnas.94.21.11472
Zentner GE, Layman WS, Martin DM, Scacheri PC (2010) Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet 152A:674–686. https://doi.org/10.1002/ajmg.a.33323
Zhuang T, Hess RA, Kolla V et al (2014) CHD5 is required for spermiogenesis and chromatin condensation. Mech Dev 131:35–46. https://doi.org/10.1016/j.mod.2013.10.005
Acknowledgements
We thank the patients and their family for their participation in this study. The research generating these results was financially supported by Fondation Maladies Rares, Bio-Psy Labex, “Investissements d’avenir” program ANR-10-IAIHU-06 (IHU-A-ICM), the Japan Agency for Medical Research and Development (AMED) (JP20ek0109486, JP20dm0107090, JP20ek0109301, JP20ek0109348, and JP20kk0205012 to N. M.); JSPS KAKENHI (JP17H01539 to N. M. and JP20K08164 to T. M.); Intramural research grants for Neurological and Psychiatric Disorders of NCNP from the Ministry of Health, Labour and Welfare (30-6 and 30-7 to N. M.), the Takeda Science Foundation (to T. M. and N. M.) and the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG007672 (Duke University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Members of the Undiagnosed Diseases Network include, Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Mercedes E. Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg-Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope*, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell'Angelica, Shweta U. Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Laure Fresard, William A. Gahl, Ian Glass, Bernadette Gochuico, Rena A. Godfrey, Katie Golden-Grant, Alica M. Goldman, Madison P. Goldrich, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A. Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Laryssa Huryn, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Lefkothea Karaviti, Jennifer Kennedy, Dana Kiley, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Grace L. LaMoure, Brendan C. Lanpher, Ian R. Lanza, Lea Latham, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, John MacDowall, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, Bryan C. Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Colleen E. McCormack, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Deborah Mosbrook-Davis, John J. Mulvihill, David R. Murdock, Anna Nagy, Mariko Nakano-Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Bradley Power, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Susan L. Samson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Vandana Shashi*, Jimann Shin, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica-Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Amelia L. M. Tan, Queenie K.-G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Audrey Thurm, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Shinya Yamamoto, John Yang, Muhammad Yousef, Diane B. Zastrow, Wadih Zein, Chunli Zhao, Stephan Zuchner. *Authors meeting authorship criteria.
Funding
Open Access funding enabled and organized by Projekt DEAL. The research generating these results was financially supported by Fondation Maladies Rares, Bio-Psy Labex, “Investissements d’avenir” program ANR-10-IAIHU-06 (IHU-A-ICM), the Japan Agency for Medical Research and Development (AMED) (JP20ek0109486, JP20dm0107090, JP20ek0109301, JP20ek0109348, and JP20kk0205012 to N. M.); JSPS KAKENHI (JP17H01539 to N. M. and JP20K08164 to T. M.); Intramural research grants for Neurological and Psychiatric Disorders of NCNP from the Ministry of Health, Labour and Welfare (30-6 and 30-7 to N. M.), the Takeda Science Foundation (to T. M. and N. M.), and the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG007672 (Duke University).
Author information
Authors and Affiliations
Consortia
Contributions
Funding acquisition: CD and CM; supervision: CD and CM; conceptualization: CD, CM, DL, and IP; writing—original draft: CD, CM, DL, and IP; investigation, formal analysis, and writing—review and editing: all authors.
Corresponding authors
Ethics declarations
Conflict of interests
Dr. Louie is a clinical laboratory director in molecular genetics and the Greenwood Genetic Center receives fee income from clinical laboratory testing. Erin Torti and Richard Person are employees of GeneDx, Inc. All other authors declare no conflict of interest.
Ethics approval
This research study was approved by INSERM (RBM C12-06). Ethics approval were locally obtained for genetic analyses and/or data sharing for additional patients. Genetic analyses and patient inclusion were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments.
Animal research
Not applicable.
Consent to participate
Informed consent for genetic analyses was obtained from all individual participants included in this study or their legal guardians.
Consent for publication
Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The members of the Undiagnosed Diseases Network are present in the Acknowledgements section.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parenti, I., Lehalle, D., Nava, C. et al. Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy. Hum Genet 140, 1109–1120 (2021). https://doi.org/10.1007/s00439-021-02283-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02283-2