
Abstract
Retinitis pigmentosa (RP) is a blinding eye disease affecting nearly two million people worldwide. Dogs are affected with a similar illness termed progressive retinal atrophy (PRA). Lapponian herders (LHs) are affected with several types of inherited retinal dystrophies, and variants in PRCD and BEST1 genes have been associated with generalized PRA and canine multifocal retinopathy 3 (cmr3), respectively. However, all retinal dystrophy cases in LHs are not explained by these variants, indicating additional genetic causes of disease in the breed. We collected DNA samples from 10 PRA affected LHs, with known PRCD and BEST1 variants excluded, and 34 unaffected LHs. A genome-wide association study identified a locus on CFA20 (praw = 2.4 × 10–7, pBonf = 0.035), and subsequent whole-genome sequencing of an affected LH revealed a missense variant, c.3176G>A, in the intraflagellar transport 122 (IFT122) gene. The variant was also found in Finnish Lapphunds, in which its clinical relevancy needs to be studied further. The variant interrupts a highly conserved residue, p.(R1059H), in IFT122 and likely impairs its function. Variants in IFT122 have not been associated with retinal degeneration in mammals, but the loss of ift122 in zebrafish larvae impaired opsin transport and resulted in progressive photoreceptor degeneration. Our study establishes a new spontaneous dog model to study the role of IFT122 in RP biology, while the affected breed will benefit from a genetic test for a recessive condition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Retinitis pigmentosa (RP) is the most common group of inherited retinal degenerations affecting 1 in 4000 people worldwide (Verbakel et al. 2018). RP is characterized by progressive loss of vision resulting from abnormalities in rod and cone photoreceptor cells or the retinal pigment epithelium (RPE). Most cases are isolated, but approximately 20–30% of RP patients have accompanying extra-ocular manifestations (Ferrari et al. 2011). According to the Retinal Information Network RetNet (http://sph.uth.edu/retnet/, accessed in November 2020), over 70 genes and loci have been implicated in nonsyndromic RP to date, with the majority of them associated with autosomal recessive inheritance (Daiger et al. 1998). Still, many RP patients have a disease of an unknown genetic cause, highlighting the need for new genetic studies and discoveries (Daiger et al. 2013). As we move to the era of personalized medicine with successful gene therapy and gene editing, the knowledge of the causal variants becomes increasingly important.
The domestic dog (Canis lupus familiaris) has become a powerful model to study genetics of human inherited diseases over the past 15 years. Currently, more than 280 likely causal variants have been reported to Mendelian traits or disorders in dogs (Online Mendelian Inheritance in Animals database; www.OMIA.org). The discoveries have been facilitated by the unusual genomic architecture of the dog breeds, the abundance of naturally occurring inherited diseases in different dog breeds, improved genomic tools, and expanding dog DNA banks worldwide (Lindblad-Toh et al. 2005; Lequarré et al. 2011; Groeneveld et al. 2016; Hytonen and Lohi 2016; Bunel et al. 2019).
As an example, over 100 dog breeds are affected with progressive retinal atrophy (PRA), an inherited retinal degeneration similar to RP (Miyadera et al. 2012). Findings in ophthalmoscopic examinations of PRA affected dogs typically include diffuse tapetal hyperreflectivity resulting from retinal thinning, attenuation of the retinal blood vessels, and at later stages, atrophy of the optic nerve head (Parry 1953). Over 20 genes have been implicated to date, with the most recent reported from the Lhasa Apso dogs with a LINE-1 insertion variant in the IMPG2 gene (Miyadera et al. 2012; Hitti-Malin et al. 2020). Another PRA affected breed is the Lapponian Herder (LH), in which the autosomal recessive PRCD variant explains a subset of cases (Zangerl et al. 2006). The LH breed also suffers from another inherited retinal disorder termed canine multifocal retinopathy (cmr3), a canine equivalent of bestrophinopathy that results from BEST1 variants (Zangerl et al. 2010). Typical clinical findings in cmr3 include multiple elevated subretinal brown-gray lesions with fluid accumulation, but generalized PRA has also been proposed as a rare outcome in some older dogs (Zangerl et al. 2010). A third genetic form of inherited retinopathy is suspected in LH, as not all cases of inherited retinal dystrophies have been explained by the known PRCD and BEST1 variants.
With a series of clinical, genetic, and bioinformatics studies, we describe here a recessive missense variant in IFT122 as the likely cause of PRA in LH. IFT122 represents a novel candidate gene for mammalian RPs.
Materials and methods
Study cohort
Samples from 563 purebred LHs and 577 FLs, donated to our canine biobank at the University of Helsinki, were included in this study. The initial discovery cohort consisted of 44 LHs and the breed screening cohort of 519 LHs, respectively. All dogs in the initial cohort had undergone thorough eye examinations performed by veterinary ophthalmologists board-certified by the European College of Veterinary Ophthalmologists. The examination included basic neuro-ophthalmic assessment followed by slit-lamp biomicroscopy for adnexa and anterior segment evaluation and indirect ophthalmoscopy to examine the fundus. Mydriasis was achieved using topical tropicamide (Oftan Tropicamid 1%, Santen, Tampere, Finland). The inclusion criterion for cases was generalized PRA, while controls were examined healthy at the age of ten years or older. EDTA blood samples were collected from all the dogs and genomic DNA extracted from the white blood cells using the semi-automated Chemagen extraction robot (PerkinElmer Chemagen Technologie GmbH, Baeswieler, Germany) following the manufacturer’s instructions. DNA concentration and purity were assessed by using the Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and Nanodrop ND-1000 UV/Vis Spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) and samples were stored at − 20 °C.
Pedigree analysis and genome-wide association study
A pedigree was drawn around the affected individuals in the initial cohort with the GenoPro 2.5.4.1 software. To perform GWAS, samples from eight cases and 12 controls were genotyped by using the Illumina’s CanineHD BeadChip array with 220,853 markers (San Diego, CA, USA) at the GeneSeek Laboratory (Neogen Genomics, Lincoln, NE, USA). All genotyped samples had a call rate of > 95%. Only markers with > 95% call rate and minor allele frequency of > 5% were included in the analysis, resulting in the total number of 145,387 markers. No markers were removed due to Hardy–Weinberg exact test (threshold p = 0.0001). Allele frequency differences between cases and controls were calculated using the Plink v1.90 software (Chang et al. 2015) and multiple testing correction implemented by using the Bonferroni method, which set the genome-wide significance level to 3.4 × 10–7. MDS plot and QQ plot analysis and genomic inflation factor lambda were utilized to assess population stratification. The CanFam 3.1 reference genome assembly was used in all genetic studies.
Whole-genome sequencing
Sample of an affected LH was whole-genome sequenced using the Illumina HiSeq X ultra-high-throughput sequencing platform with paired-end reads (2 × 150 bp) at the Novogene Bioinformatics Institute (Beijing, China). The reads were aligned to dog reference genome CanFam3.1 using the SpeedSeq open-source software with Burrows-Wheeler Aligner (BWA) (v0.7.17), sorting and BAM compression was done using the Sambamba and duplicate reads were marked using SAMBLASTER (Chiang et al. 2015). Post-alignment processing included local realignment around known INDELs and base quality scores recalibration to reduce erroneous variant calls with Genome Analysis Tool Kit GATK (McKenna et al. 2010). Variant calling was done using the HaplotypeCaller in gVCF mode. CombineGVCs was used to combine gVCFs and joint genotyping was done with GenotpeGVCFs using GATK version 4.1.0. Mobile element insertions were analyzed using the Mobile Element Locator Tool (MELT) (Gardner et al. 2017), with reference sequences of the transposons for MEI discovery retrieved from the Repbase database (Jurka et al. 2005). Structural variants were identified with the DELLY2 software (Rausch et al. 2012). Insertions, deletions, duplications and inversions were called independently in DELLY2. Functional annotation of the variants was done in ANNOVAR using Ensembl release-100 and NCBI Canis lupus familiaris Annotation Release 105. Variant filtering was performed using webGQT server deployed on dog variant datasets, assuming recessive mode of inheritance, i.e. the case was set to be homozygous for the alternative allele in SNV filtering and controls wild-type (Arumilli et al. 2020). When filtering the SVs and MEIs, the case was allowed to be either homozygous or heterozygous for the alternate allele to prevent erroneous zygosity calling hampering the results, while controls were wild-type. Variant data from exome (n = 100) and whole-genome sequenced (n = 227) dogs from our other ongoing projects without the studied phenotype and from various breeds were used in filtering as controls (Online Resource 1). Filtering the SNV variants included all 327 dogs, while only the whole-genome sequenced individuals could be utilized in MEI and SV filterings.
Variant screening
We analyzed PRCD-PRA and cmr3 variants, known to affect LHs, in all cases before their inclusion in any further genetic analysis to exclude known inherited retinal disease variants as a cause of disease (Zangerl et al. 2006, 2010). Primer sequences were obtained from the original study for cmr3 (Zangerl et al. 2010) and designed with Primer3 software for the PRCD variant (Koressaar and Remm 2007) and were, in 5′ > 3′ orientation, TCCTAATCCAGTGGCAGCAG (F) and ATCAGCTTCTCACGGTTGGA (R) for the PRCD variant and AAGGAGGGAAAAGATAGGGT (F) and AGGTGGAAGGAGGGTAGAAT (R) for the two cmr3 variants. Initial segregation analysis for the IFT122 variant was made using PCR primers TCACCTTGTCCTATGAGCCC (F) and GCAGAGAAGACGAATGGCTG (R), designed with Primer3 software. All PCR products were amplified using Biotools DNA Polymerase (Biotools B&M Labs, S.A., Valle de Tobalina, Madrid, Spain) and treated with exonuclease I and shrimp alkaline phosphatase before capillary sequencing in the Institute for Molecular Medicine Finland (Helsinki, Finland). The sequence data were analyzed using Unipro UGENE v35.0 (UniPro, Novosibirsk, Russia). Variant screening in the LH breed cohort and additional FLs was performed using Custom TaqMan SNP Genotyping Assay (ThermoFisher Scientific, Waltham, MA, USA) and CFX96 Touch Real-Time PCR Detection System (BioRad, Hercules, CA, USA) with the following primers, in 5′ > 3′ orientation, TGTGGTTCTTGTGCTTTTGTAGGA (F) and TGCCCAGGTTGTTCAGCAA (R) and probes VIC-TGCTACCGCTGCTCC (wild-type) and FAM-TGCTACCACTGCTCC (variant). Finally, large-scale screening of the IFT122 variant was performed in a sample set of 6023 dogs from 183 breeds and 130 mixed-breed dogs (Online Resource 2) submitted for commercial testing at Genoscoper Laboratories Oy, Helsinki, during 2019–2020. Genotyping in this sample set was carried out according to manufacturer-recommended standard protocols on a custom-designed Illumina Infinium XT genotyping bead chip (Illumina, San Diego, CA, USA) commercially available as the MyDogDNA/Optimal Selection canine DNA test (Wisdom Health, Vancouver, WA, USA) (Donner et al. 2016, 2018).
SD-OCT imaging
To examine the retinal phenotype associated with the IFT122 variant in greater resolution, SD-OCT imaging was performed using the Heidelberg Spectralis HRA + OCT instrument (Heidelberg Engineering, Heidelberg, Germany) in the Department of Ophthalmology, Helsinki University Hospital, Helsinki, Finland. Imaging was performed to a PRA affected dog aged 13 years with the homozygous c.3176G > A variant and a clinically healthy wild-type LH aged 11 years. Thicknesses of the whole retina (including the RPE) and the presumed photoreceptor cell layer (including the RPE, the outer nuclear layer, and the inner and outer segments of the photoreceptor cells) were compared between the case and the control dog.
Additional clinical measurements
To examine any extra-ocular manifestations that the IFT122-variant might have, three PRA affected LHs (one male, two females, aged 12–13 years), homozygous for the IFT122 variant, were recruited for further clinical examinations, including general examination, complete blood count, serum biochemistry, abdominal ultrasound and thoracic radiographs. In addition, blood gas analysis, serum symmetric dimethylarginine (SDMA) and urinalysis including urine protein/creatinine ratio were studied in two of these dogs.
IFT122 expression in human tissues and cells
Promoter-level expression data measured by cap analysis of gene expression (CAGE) (Forrest et al. 2014) was downloaded from FANTOM5 SSTAR (Abugessaisa et al. 2016) (https://fantom.gsc.riken.jp/5/sstar/Main_Page). The normalized expression values of genes with multiple CAGE peaks (promoters; IFT122 and IFT140) were summed across all the peaks. The RNA-sequencing data of LUHMES differentiation was obtained from and processed as described in (Lauter et al. 2020). Briefly, read counts were normalized by the sum of spike-in reads for each sample (Katayama et al. 2013) and divided by the minimum non-zero value (5.78 × 10–6).
Results
Slowly progressing late-onset PRA in LHs
Over the years, several LH owners and breeders have contacted us as their dogs have been diagnosed with PRA that is not explained by the previously reported autosomal recessive PRCD and cmr3 variants (Zangerl et al. 2006, 2010). We analyzed LH samples donated to our canine biobank at the University of Helsinki to find the genetic cause of this new type of PRA. An initial study cohort of 44 LHs was included in the study, with readily available phenotype information as comprehensive eye examinations had been carried out on each individual dog as a part of breeding programs and health check-ups. These examinations had revealed clinical findings compatible with generalized PRA in 10 LHs (five males, five females), as these dogs presented with bilateral, diffuse tapetal hyperreflectivity and vessel attenuation. Two of the 10 cases were only examined as young or middle-aged adults (at 1.9 and 5.1 years of age) when they exhibited only mild fundus changes and were therefore diagnosed to have “PRA suspected”. The remaining eight dogs were also examined later in life and had been diagnosed as “PRA affected” at an average age of 9.0 years (± SD 2.9). Typical early findings included night blindness and diffuse tapetal hyperreflectivity. Disease progression was slow as some of the affected dogs still had some visual capacity left at 13 years. All the cases were genotyped for the PRCD p.C2Y and the cmr3 (p.P463fs, p.G489V) variants (Zangerl et al. 2006, 2010). Of the 10 cases, all were wild-type for the PRCD variant, while four were wild-type and six heterozygous for the two cmr3 variants, excluding these recessive variants as the genetic cause of their phenotype. The rest of the initial cohort, 34 LHs (16 males, 18 females), were regarded as controls, as they were examined healthy at the age of 10 years or older. The two cmr3 variants were also screened in these control dogs, indicating 25 wild-type and nine heterozygous dogs.
GWAS indicates a PRA locus on chromosome 20
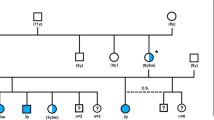
We constructed a pedigree around the affected individuals to assess the mode of inheritance. This analysis suggested that the new type of PRA in LHs is likely to be an autosomal recessive condition, as the affected dogs were born to unaffected parents, there were multiple cases in one of the affected litters, and males and females were equally affected (Fig. 1).

Pedigree analysis suggested autosomal recessive PRA in LH, as affected dogs were born to unaffected individuals, there were multiple cases in one of the affected litters, and both sexes were equally affected. The black symbol denotes affected dogs; squares indicate males and circles females, respectively. Dogs included in the GWAS are marked with a yellow background and the whole-genome sequenced case with a black arrow
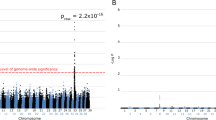
We then performed a genome-wide association study (GWAS) with eight cases and 12 controls and with 145,387 single nucleotide polymorphism (SNP) markers to map the disease locus. Comparison of allele frequencies between cases and controls indicated a PRA associated locus on canine chromosome 20 with nine genome-wide significant markers (praw = 2.4 × 10–7, pBonf = 0.035, λ = 1, Fig. 2a, b). An in-depth analysis of the genotypes in the locus revealed an 8.7 Mb homozygous region at 19,145–8,763,312 bp in the affected LHs, not observed in the controls (Fig. 2c). As there were no informative markers upstream of the first homozygous marker at 19,145 bp, the subsequent variant search was performed from the beginning of CFA20 until the first heterozygous marker in a case at 8,894,743 bp.

GWAS reveals a PRA locus on chromosome 20. a, b Results from the GWAS with 8 cases and 12 controls indicate a PRA locus on the canine chromosome 20 (praw = 2.4 × 10–7, pBonf = 0.035, λ = 1), where 9 markers meet genome-wide significance (threshold p = 3.4 × 10–7 marked with red line). c In-depth analysis of the genotypes in the associated locus reveals a shared homozygous haplotype block of 8.7 Mb in the cases absent in control dogs. Rows represent individual dogs and columns each SNP with light gray denoting affected genotypes, intermediate heterozygotes, and dark gray opposite homozygotes. The locations of the nine markers with genome-wide significance are marked with red arrows
WGS reveals a recessive missense variant in IFT122
To find candidate variants in the associated chromosomal region, we next performed whole-genome sequencing on a PRA affected LH (Fig. 1, black arrow). In total, 498,114,393 reads were collected of which 99.37% were mapped to the reference sequence. The mean coverage was 30.3X. Comparing to the reference sequence, altogether 11,332 single nucleotide variants (SNV) and small deletions and insertions were homozygous in the associated chromosomal region. In addition, 49 mobile element insertions (MEIs) and 356 structural variants (SVs) were detected in the locus. After filtering against control dogs (Online Resource 1), 79 SNVs, one MEI, and one SV were case-specific (Online Resource 3). Only one non-synonymous variant was predicted to alter protein structure and was therefore chosen for additional screening: c.3176G>A in the intraflagellar transport 122-gene (IFT122, XM_533734.6, Fig. 3a, b) was predicted to lead to an amino acid substitution from arginine to histidine (p.R1059H, XP_533734.2). In silico analyses with PROVEAN and PolyPhen-2 predicted the variant to be deleterious (score = − 4.054, cut-off = − 2.5) and probably damaging (score = 0.961), respectively, which was in line with the Clustal Omega analysis showing conservation in the changed amino acid position (Fig. 3c). Both the dog and human IFT122 consist of 1,241 amino acids with 93% similarity at the amino acid level.

The IFT122 sequence and protein structure. a Schematic presentation of the IFT122 gene with exons marked in red, UTR sequences in orange and introns in gray. A missense variant c.3176G>A in exon 26 was identified in whole-genome sequencing data. b Chromatograms of the variant site (highlighted with orange background) and its surrounding sequence in wild-type, carrier and affected LH. c The variant site p.(R1059H) and its surrounding amino acids are highly conserved among different species. d Schematic presentation of the IFT122 protein with the canine PRA associated missense variant p.(R1059H) highlighted in red. The previously reported variants in human CED patients, all in relation to NP_443715.1, are marked with black arrows (Walczak-Sztulpa et al. 2010; Alazami et al. 2014; Tsurusaki et al. 2014; Moosa et al. 2016). The human IFT122 contains seven WD repeat domains, marked with light green circles, and a C-terminal TPR domain, marked with yellow
Validation of the IFT122 variant in the initial cohort of 10 PRA cases and 34 controls revealed complete segregation and full penetrance as all controls were either wild-type (n = 21) or heterozygous (n = 13), while all cases were homozygous (n = 10), supporting a recessive mode of inheritance. Subsequently, the variant was screened in all available LH samples in our biobank (n = 519), revealing 362 wild-type, 143 heterozygous and 14 homozygous LHs and a carrier frequency of 28%. Of the 14 new homozygotes, 6 had been diagnosed with PRA by veterinary ophthalmologists board-certified by the European College of Veterinary Ophthalmologists. Two dogs that were examined at the age of two and four years presented with tapetal hyperreflectivity. One dog had been eye examined at the age of eight years and diagnosed with mild cortical cataract. The owner reported a reduced vision in this dog but had thought it resulted from the cataract and did not take it to eye examination again before its death at 12 years of age. The remaining five dogs were not available for eye examinations, but a general veterinarian had suspected PRA in one dog. In addition, owners reported complete blindness in one dog while night blindness was reported in another dog. Of the 14 new IFT122 variant homozygotes, 11 were wild-type and three heterozygous for the PRCD p.C2Y variant and eight wild-type and five heterozygous for the two cmr3 variants (p.P463fs, p.G489V), while DNA sample of one dog had run out and was not available for cmr3 testing.
Breed-specificity of the IFT122 c.3176G>A variant
To study the breed-specificity of the IFT122 variant, we analyzed the genomic data available from additional 55 dogs from 16 breeds and 8 wolves available through the Dog Biomedical Variant Database Consortium (Online Resource 2). This analysis revealed one heterozygous Finnish Lapphund (FL), while other screened samples were wild-type.
To further study the segregation and prevalence of the variant in FLs, we genotyped samples from 577 FLs available in our biobank, including seven PRA affected dogs with the PRCD variant excluded. Of the tested dogs, 508 were wild-type, 68 heterozygous and 1 homozygous for the IFT122 variant, indicating a carrier frequency of 12%. The only homozygote FL had been eye examined healthy at the age of five years, but no clinical information after that was available. Importantly, all the PRA affected FLs were wild-type for the IFT122 variant, indicating that another genetic cause of PRA is evident in the breed.
To further investigate the breed-specificity of the variant, we gathered results from 6023 dogs from 183 breeds and 130 mixed breed dogs submitted for routine commercial gene panel screening at Genoscoper Laboratories Ltd, Helsinki during 2019–2020. These dogs were all wild-type for the variant.
OCT imaging reveals retinal and photoreceptor cell layer thinning
To examine the retinal phenotype associated with the homozygous IFT122 c.3176G > A variant in a greater detail, spectral-domain optical coherence tomography (SD-OCT) was performed for two dogs: a PRA affected, IFT122 variant homozygote LH aged 13 years, and a clinically healthy wild-type LH aged 11 years. The image was acquired using the optic disc as a landmark. Due to limited visual perception, the affected dog could not fixate properly, and the image was obtained more peripherally. The thicknesses of the whole retina with the retinal pigment epithelium (RPE) and outer retinal layers, including outer nuclear layer, inner segments and outer segments of photoreceptors, and RPE, were severely reduced in the affected dog (113 μm and 53 μm, respectively) when compared to the control dog (185 μm and 92 μm, respectively) (Fig. 4). Interestingly, the photoreceptor layer was not completely lost despite the dog’s old age, supporting the observed slow progression rate in the other affected dogs, and also the owner’s assessment that the dog had reduced but not entirely lost vision.

SD-OCT images of a healthy control (left-hand side) and a PRA affected IFT122 variant homozygote (right-hand side) indicate reduced thickness of the whole retina and RPE (a) and the outer retinal layers, including the outer nuclear layer, inner and outer segments of photoreceptors, and RPE (b) in the affected dog (113 μm and 53 μm, respectively) compared to the control dog (185 μm and 92 μm, respectively)
Clinical examinations to detect extra-ocular manifestations
IFT122 forms a part of the protein complex that regulates ciliary function (Rosenbaum and Witman 2002; Hirano et al. 2017; Takahara et al. 2018). Therefore, to understand the possible role of the IFT122 variant in extra-ocular manifestations, we performed clinical examinations including thoracic radiographs, abdominal ultrasound, complete blood count and serum biochemistry for three homozygous LHs (aged 12–13 years). All the three LHs had been diagnosed with generalized PRA. One of the females was completely blind, while the other two had reduced, but not totally lost ability to see. Apart from the ocular signs, all three dogs were clinically healthy. In laboratory tests, one of the dogs had mildly elevated creatinine value (118 μmol/l, reference < 116 μmol/l) and there was mild hematuria in that dog, while other renal parameters were normal in all of the dogs. Abdominal ultrasonography findings previously associated with IFT122 variants in humans (Walczak-Sztulpa et al. 2010), including renal and liver abnormalities, were not detected. Thoracic radiographs were unremarkable in all dogs. None of the dogs had situs inversus.
IFT122 expression in human
To explore the IFT122 expression pattern across human tissues and cells, we utilized the Functional Annotation of Mammalian Genome 5 (FANTOM5) expression atlas, which catalogs promoter activities in a wide variety of human tissues and cells (Forrest et al. 2014). IFT122 is most highly expressed in testis (Online Resources 4 and 5a). High expression is also observed in brain tissues, such as the pituitary gland, as well as the fetal kidney. Notably, it is also highly expressed in retina. As IFT122 forms the intraflagellar transport A (IFT-A) complex together with IFT144, IFT140, IFT43, IFT121 and IFT139 (Takahara et al. 2018), their expression levels were also studied and found to be generally comparable in retina, chondrocyte, and osteoblast (Online Resource 5b). We further investigated the expression changes of IFT122 in Lund human mesencephalic (LUHMES) cell line which can recapitulate the neuronal cell development process in vitro (Lauter et al. 2020). Here we found that IFT122 is strongly upregulated from day 2 to day 5 (Online Resource 5c), along with many other genes related to axonogenesis and ciliogenesis, indicating that IFT122 plays an important role in neuronal systems including retina.
Discussion
Nonsyndromic RP is a significant cause of blindness in man, and over 70 genes and loci have been implicated in the disease (Daiger et al. 1998). However, the genetic background, in many cases, remains still unknown. We report here a recessive missense variant in IFT122 as a candidate causal variant for a novel canine RP model and provide a new spontaneous large animal model to study the pathogenesis of RP and retinal biology.
IFT122 encodes a cytoplasmic member of the WD-repeat protein family (Gross et al. 2001). IFT122 has seven N-terminal WD repeat domains and a C-terminal tetratricopeptide repeat domain (TPR) (Fig. 3d) (Takahara et al. 2018; Gross et al. 2001). Interaction in the TPR domain and IFT144 and IFT140 form the core subcomplex of the IFT-A complex, whereas the interaction between the WD repeat domain and IFT43 and IFT121 form its peripheral subcomplex together with IFT139 (Takahara et al. 2018). The core and the peripheral subcomplexes are connected by the interaction of IFT122, IFT43, and IFT121 (Takahara et al. 2018). IFT-A is essential for the assembly and maintenance of cilia as it mediates retrograde trafficking of ciliary proteins (Rosenbaum and Witman 2002; Hirano et al. 2017). The photoreceptor cells are initially formed from primary cilia, and in the adult photoreceptor cells, the outer and inner segments are connected to each other only by a connecting cilium (De Robertis 1956). The connection between these two segments is crucial for proper photoreceptor function, as the photopigments and light-transducing machinery are located in the outer segments, whereas protein synthesis occurs in the inner segments of the photoreceptor cells (Young 1976).
It is estimated that around 2,000 opsin molecules are required every minute to maintain the rod outer segments in mammals and the IFT machinery presumably has an important role in its transport (Rosenbaum and Witman 2002). Concordantly, studies on zebrafish larvae carrying a null mutation in ift122 show that ift122 is essential for efficient opsin transport and the distal elongation of the outer segments (Boubakri et al. 2016). The mutant larvae presented with progressive photoreceptor degeneration with slow onset—a phenotype that resembles the disease in the affected LHs. To our knowledge, variants in IFT122 have not previously been associated with RP in humans. Still, variants in IFT144 and IFT140, the other two components of the IFT-A core subcomplex, have been implicated in both syndromic and nonsyndromic RP (Coussa et al. 2013; Hull et al. 2016). On the other hand, several variants in IFT122 have been reported to cause cranioectodermal dysplasia (CED) in humans (Walczak-Sztulpa et al. 2010; Alazami et al. 2014; Tsurusaki et al. 2014; Moosa et al. 2016). The disorder, also called the Sensenbrenner syndrome, is characterized by craniofacial, skeletal and ectodermal abnormalities, but defects in liver, kidneys, teeth and skin have also been reported in patients with different IFT122 variants (Walczak-Sztulpa et al. 2010; Alazami et al. 2014; Tsurusaki et al. 2014; Moosa et al. 2016). A report describing two CED-affected siblings without a molecular diagnosis also found evidence of photoreceptor dystrophy; the patients, examined at the age of four years, exhibited reduced scotopic and photopic responses in electroretinography, while fundus examination detected no abnormalities (Eke et al. 1996). These findings mimic the early phase of IFT122-PRA in LHs, where night blindness precedes ophthalmoscopic fundus changes. The lack of ocular signs in the CED patients with confirmed IFT122 variants may be due to the affected site of the IFT122 protein. All but one of the reported variants in human patients reside in the N-terminal WD repeat domain, whereas the canine missense variant is located in the C-terminal region (Walczak-Sztulpa et al. 2010; Alazami et al. 2014; Tsurusaki et al. 2014; Moosa et al. 2016). Additionally, most of the CED affected individuals were examined only in early infancy, and the retinal dystrophy might develop later in life. As the IFT122 was highly expressed in the human retina samples, we propose late-onset RP as a potential finding in the CED patients too.
In contrast to the IFT122-affected CED patients with skeletal abnormalities and other defects as well as to the ift122 null zebrafish larvae model where the development of cystic kidneys was detected, similar extra-ocular manifestations were not observed in the LHs homozygous for the p.(R1059H) variant. The limited retinal phenotype in dogs may result from the affected site of the IFT122 protein or reflect species-specific differences in the IFT122 biology. We propose the former to be more plausible, as the discovered canine variant resides near the C-terminus, whereas all but one of the reported variants in CED patients are close to the N-terminal WD repeat domain (Fig. 3d). Interestingly, different variants in IFT144, another IFT-A core subcomplex component, have been reported to result both in syndromic and nonsyndromic RP (Coussa et al. 2013) and the same might also apply to IFT122 variants. Furthermore, the canine IFT122 variant is located in the C-terminal region that is known to interact with IFT144 (Takahara et al. 2018), and the functional IFT122-IFT144 interaction might be of particular importance for normal photoreceptor function. Only one of the reported variants in CED patients, the p.L712P, is located in the TPR domain (Moosa et al. 2016). The variant was found in compound heterozygous state from an Argentinian girl together with a splice-site variant in IFT122 (Moosa et al. 2016). Retinal abnormalities were not reported, but seemingly, clinical data were available only from early infancy.
Additionally, studies on hTERT-RPE1 cells indicate that total abolition of IFT122 in knockout cells results in cilia absence, whereas even exogenous expression of CED-associated missense variants, located in the WD repeat domain, restored ciliogenesis, but not ciliary protein trafficking (Takahara et al. 2018). Therefore, we hypothesize that ciliary protein trafficking might be more susceptible to disturbances than ciliogenesis, and the IFT122 C-terminal interaction with IFT144 may be essential to protein, including opsin, trafficking in the photoreceptor cells. While we cannot make final conclusions of the canine IFT122 variant’s exact pathological mechanism due to lack of suitable tissue material, it seems likely that the missense variant in the C-terminal region might lead to insufficient opsin transport in the photoreceptor cells leading to their degeneration, while ciliogenesis would remain normal. Previously, variants in cilia-associated genes TTC8, BBS4, FAM161A and CCDC66 have been implicated in both syndromic and nonsyndromic PRA in several dog breeds (Dekomien et al. 2010; Downs et al. 2014; Downs and Mellersh 2014; Chew et al. 2017; Makelainen et al. 2020).
The IFT122 variant reported here was also found in Finnish Lapphunds (FLs), a related breed, with a carrier frequency of 12%. Unfortunately, the only identified homozygous FL had been eye examined only at the age of five years and was not available for re-examination. As the average age at diagnosis in LHs was nine years, variant causality in the FLs remains to be determined. Amongst the screened 577 FLs there were seven PRA affected dogs with the known PRCD variant excluded, but all of them were wild-type for the IFT122 variant. These results indicate that there is yet another genetic cause of PRA in FLs to be found.
In conclusion, we report here a missense variant in IFT122 as a likely cause of autosomal recessive PRA in LHs. Our study establishes a new spontaneous large animal model to study retinal biology and disease and implicates IFT122 as a potential candidate gene for human RP. Further studies are needed to establish the exact pathological mechanism. Meanwhile, a gene test can be developed to aid veterinary diagnostics and breeding programs, which is of particular importance as the affected breed also suffers from other genetic forms of inherited retinal dystrophies (Zangerl et al. 2006, 2010).
Availability of data and material
WGS data from an affected LH have been submitted to the NCBI SRA under the accession number PRJNA682002 (BioProject), SAMN16976490 (BioSample).
References
Abugessaisa I, Shimoji H, Sahin S, Kondo A, Harshbarger J, Lizio M, Hayashizaki Y, Carninci P, Forrest A, Kasukawa T (2016) FANTOM5 transcriptome catalog of cellular states based on Semantic MediaWiki. Database
Alazami AM, Seidahmed MZ, Alzahrani F, Mohammed AO, Alkuraya FS (2014) Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol Genet Genom Med 2:103–106
Arumilli M, Layer RM, Hytönen MK, Lohi H (2020) webGQT: a shiny server for genotype query tools for model-based variant filtering. Front Genet 11:152
Boubakri M, Chaya T, Hirata H, Kajimura N, Kuwahara R, Ueno A, Malicki J, Furukawa T, Omori Y (2016) Loss of ift122, a retrograde intraflagellar transport (IFT) complex component, leads to slow, progressive photoreceptor degeneration due to inefficient opsin transport. J Biol Chem 291:24465–24474
Bunel M, Chaudieu G, Hamel C, Lagoutte L, Manes G, Botherel N, Brabet P, Pilorge P, André C, Quignon P (2019) Natural models for retinitis pigmentosa: progressive retinal atrophy in dog breeds. Hum Genet 138:441–453. https://doi.org/10.1007/s00439-019-01999-6
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4
Chew T, Haase B, Bathgate R, Willet CE, Kaukonen MK, Mascord LJ, Lohi HT, Wade CM (2017) A Coding variant in the gene bardet-biedl syndrome 4 (BBS4) is associated with a novel form of canine progressive retinal atrophy. G3 Genes Genomes Genet 7:2327–2335
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, Hall IM (2015) SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 12:966–968
Coussa RG, Otto EA, Gee HY, Arthurs P, Ren H, Lopez I, Keser V, Fu Q, Faingold R, Khan A, Schwartzentruber J, Majewski J, Hildebrandt F, Koenekoop RK (2013) WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet 84:150–159. https://doi.org/10.1111/cge.12196
Daiger S, Rossiter B, Greenberg J, Christoffels A, Hide W (1998) Data services and software for identifying genes and mutations causing retinal degeneration. Invest Ophthalmol Vis Sci 39:S295
Daiger S, Sullivan L, Bowne S (2013) Genes and mutations causing retinitis pigmentosa. Clin Genet 84:132–141
De Robertis E (1956) Morphogenesis of the retinal rods; an electron microscope study. J Biophys Biochem Cytol 2:209–218. https://doi.org/10.1083/jcb.2.4.209
Dekomien G, Vollrath C, Petrasch-Parwez E, Boevé MH, Akkad DA, Gerding WM, Epplen JT (2010) Progressive retinal atrophy in Schapendoes dogs: mutation of the newly identified CCDC66 gene. Neurogenetics 11:163–174
Donner J, Kaukonen M, Anderson H, Moller F, Kyostila K, Sankari S, Hytonen M, Giger U, Lohi H (2016) Genetic panel screening of nearly 100 mutations reveals new insights into the breed distribution of risk variants for canine hereditary disorders. PLoS ONE 11:e0161005. https://doi.org/10.1371/journal.pone.0161005
Donner J, Anderson H, Davison S, Hughes AM, Bouirmane J, Lindqvist J, Lytle KM, Ganesan B, Ottka C, Ruotanen P, Kaukonen M, Forman OP, Fretwell N, Cole CA, Lohi H (2018) Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs. PLoS Genet 14:e1007361. https://doi.org/10.1371/journal.pgen.1007361
Downs LM, Mellersh CS (2014) An intronic SINE insertion in FAM161A that causes exon-skipping is associated with progressive retinal atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS ONE 9:e93990
Downs LM, Wallin-Håkansson B, Bergström T, Mellersh CS (2014) A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever. Canine Genet Epidemiol 1:4
Eke T, Woodruff G, Young ID (1996) A new oculorenal syndrome: retinal dystrophy and tubulointerstitial nephropathy in cranioectodermal dysplasia. Br J Ophthalmol 80:490–491. https://doi.org/10.1136/bjo.80.5.490
Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino F, Parmeggiani F (2011) Retinitis pigmentosa: genes and disease mechanisms. Curr Genom 12:238–249
Forrest AR, Kawaji H, Rehli M, Baillie JK, De Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M (2014) A promoter-level mammalian expression atlas. Nature 507:462
Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, Mills RE, Devine SE, 1000 Genomes Project Consortium (2017) The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res 27:1916–1929. https://doi.org/10.1101/gr.218032.116
Groeneveld LF, Gregusson S, Guldbrandtsen B, Hiemstra SJ, Hveem K, Kantanen J, Lohi H, Stroemstedt L, Berg P (2016) Domesticated animal biobanking: land of opportunity. PLoS Biol 14:e1002523
Gross C, De Baere E, Lo A, Chang W, Messiaen L (2001) Cloning and characterization of human WDR10, a novel gene located at 3q21 encoding a WD-repeat protein that is highly expressed in pituitary and testis. DNA Cell Biol 20:41–52. https://doi.org/10.1089/10445490150504684
Hirano T, Katoh Y, Nakayama K (2017) Intraflagellar transport-A complex mediates ciliary entry and retrograde trafficking of ciliary G protein-coupled receptors. Mol Biol Cell 28:429–439. https://doi.org/10.1091/mbc.E16-11-0813
Hitti-Malin RJ, Burmeister LM, Ricketts SL, Lewis TW, Pettitt L, Boursnell M, Schofield EC, Sargan D, Mellersh CS (2020) A LINE-1 insertion situated in the promoter of IMPG2 is associated with autosomal recessive progressive retinal atrophy in Lhasa Apso dogs. BMC Genet 21:1–17
Hull S, Owen N, Islam F, Tracey-White D, Plagnol V, Holder GE, Michaelides M, Carss K, Raymond FL, Rozet JM, Ramsden SC, Black GC, Perrault I, Sarkar A, Moosajee M, Webster AR, Arno G, Moore AT (2016) Nonsyndromic retinal dystrophy due to bi-allelic mutations in the ciliary transport gene IFT140. Invest Ophthalmol Vis Sci 57:1053–1062. https://doi.org/10.1167/iovs.15-17976
Hytonen MK, Lohi H (2016) Canine models of human rare disorders. Rare Dis 4:e1241362. https://doi.org/10.1080/21675511.2016.1241362
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Katayama S, Töhönen V, Linnarsson S, Kere J (2013) SAMstrt: statistical test for differential expression in single-cell transcriptome with spike-in normalization. Bioinformatics 29:2943–2945
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291
Lauter G, Coschiera A, Yoshihara M, Sugiaman-Trapman D, Ezer S, Sethurathinam S, Katayama S, Kere J, Swoboda P (2020) Differentiation of ciliated human midbrain-derived LUHMES neurons. bioRxiv
Lequarré A, Andersson L, André C, Fredholm M, Hitte C, Leeb T, Lohi H, Lindblad-Toh K, Georges M (2011) LUPA: a European initiative taking advantage of the canine genome architecture for unraveling complex disorders in both human and dogs. Vet J 189:155–159
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819
Makelainen S, Hellsand M, van der Heiden AD, Andersson E, Thorsson E, Holst BS, Haggstrom J, Ljungvall I, Mellersh C, Hallbook F, Andersson G, Ekesten B, Bergstrom TF (2020) Deletion in the Bardet-Biedl syndrome gene TTC8 results in a syndromic retinal degeneration in dogs. Genes (Basel). https://doi.org/10.3390/genes11091090
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Miyadera K, Acland GM, Aguirre GD (2012) Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within-and across-breed studies. Mamm Genome 23:40–61
Moosa S, Obregon MG, Altmüller J, Thiele H, Nürnberg P, Fano V, Wollnik B (2016) Novel IFT122 mutations in three Argentinian patients with cranioectodermal dysplasia: expanding the mutational spectrum. Am J Med Genet Part A 170:1295–1301
Parry HB (1953) Degenerations of the dog retina. II. Generalized progressive atrophy of hereditary origin. Br J Ophthalmol 37:487–502
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28:i333–i339
Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3:813–825. https://doi.org/10.1038/nrm952
Takahara M, Katoh Y, Nakamura K, Hirano T, Sugawa M, Tsurumi Y, Nakayama K (2018) Ciliopathy-associated mutations of IFT122 impair ciliary protein trafficking but not ciliogenesis. Hum Mol Genet 27:516–528. https://doi.org/10.1093/hmg/ddx421
Tsurusaki Y, Yonezawa R, Nishimura G, Pooh RK, Nakashima M, Saitsu H, Miyake N, Saito S, Matsumoto N (2014) Whole exome sequencing revealed biallelic IFT122 mutations in a family with CED1 and recurrent pregnancy loss. Clin Genet 85:592–594. https://doi.org/10.1111/cge.12215
Verbakel SK, van Huet RA, Boon CJ, den Hollander AI, Collin RW, Klaver CC, Hoyng CB, Roepman R, Klevering BJ (2018) Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 66:157–186
Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A (2010) Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 86:949–956
Young RW (1976) Visual cells and the concept of renewal. Invest Ophthalmol Vis Sci 15:700–725
Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling SE, Mullins RF, Graphodatsky AS, Ripoll D, Felix JS, Stone EM (2006) Identical mutation in a novel retinal gene causes progressive rod–cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 88:551–563
Zangerl B, Wickström K, Slavik J, Lindauer SJ, Ahonen S, Schelling C, Lohi H, Guziewicz KE, Aguirre GD (2010) Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3). Mol Vis 16:2791
Acknowledgements
We thank DVM Elina Rautala at the Department of Equine and Small Animal Medicine, University of Helsinki for the help in interpreting ultrasonographic examination results and CSC-IT Center for Science, Finland, for computational resources. We thank the Dog Biomedical Variant Database Consortium (Gus Aguirre, Catherine André, Danika Bannasch, Doreen Becker, Brian Davis, Cord Drögemüller, Kari Ekenstedt, Kiterie Faller, Oliver Forman, Steve Friedenberg, Eva Furrow, Urs Giger, Christophe Hitte, Marjo Hytönen, Vidhya Jagannathan, Tosso Leeb, Hannes Lohi, Cathryn Mellersh, Jim Mickelson, Leonardo Murgiano, Anita Oberbauer, Sheila Schmutz, Jeffrey Schoenebeck, Kim Summers, Frank van Steenbeck, Claire Wade) for providing access to genome sequence variants from control dogs and wolfs. Veterinarians, dog breeders, dog owners and breed clubs are warmly thanked for sending samples and providing phenotype and pedigree information.
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This study was financially supported by the Jane and Aatos Erkko Foundation (http://jaes.fi), the Academy of Finland (http://www.aka.fi), HiLife (http://www.helsinki.fi/en/helsinki-institute-of-life-science), Wisdom Health (http://www.wisdompanel.com) and the Sigrid Jusélius Foundation (http://www.sigridjuselius.fi) to HL; by the Mary and Georg C. Ehrnrooth Foundation (http://www.marygeorg.fi/saatio.html), the Evald and Hilda Nissi Foundation (http://www.nissinsaatio.fi), the Orion Research Foundation (http://www.orion.fi/en/rd/orion-research-foundation/) and the Canine Health Research Fund (http://www.helsinki.fi/en/support-us/funds-and-campaigns-you-can-support/canine-health-research-fund) to MK and by the Scandinavia–Japan Sasakawa Foundation (https://sjsf.se/), Japan Eye Bank Association (http://www.j-eyebank.or.jp/kaigai.htm), Astellas Foundation for Research on Metabolic Disorders (https://www.astellas-foundation.or.jp/), Japan Society for the Promotion of Science (JSPS) Overseas Research Fellowships (https://www.jsps.go.jp/english/e-ab/index.html) to MY. The funders had no role in study design, data collection, analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
MK and HL contributed to the study conception and design. Material preparation, data collection and data analysis were performed by MK, I-TP, KW, MA, JD, I-JJ, SH, MY, JAT and JK. MK wrote the first draft of the manuscript and HL and all other authors commented on it. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
HL is a paid consultant to, and JD an employee of Genoscoper Laboratories Ltd, which will provide a genetic test for IFT122-PRA in LHs.
Ethics approval
All dogs included in this study were privately owned pet dogs, of which EDTA blood samples (1–3 mL) were collected under the permission of the Animal Experimental Board of Regional State Administrative Agency of Southern Finland (ESAVI/7482/04.10.07/2015, ESAVI/25696/2020). All experiments were performed following relevant regulations and guidelines and with the owners’ written consent.
Consent to participate
All experiments were performed following relevant regulations and guidelines and with the owners’ written consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
439_2021_2266_MOESM1_ESM.xlsx
Supplementary file1 Online Resource 1 List of the control dogs used for filtering the whole-genome sequencing data (numbers per breed) (XLSX 10 KB)
439_2021_2266_MOESM3_ESM.xlsx
Supplementary file3 Online Resource 3 List of case-specific variants from the whole-genome sequenced affected LH (XLSX 16 KB)
439_2021_2266_MOESM4_ESM.xlsx
Supplementary file4 Online Resource 4 Promoter-level expression of IFT122 in various human tissues and cells in FANTOM5 (XLSX 59 KB)
439_2021_2266_MOESM5_ESM.tif
Supplementary file5 Online Resource 5 IFT122 expression pattern in human tissues and cells as well as neuronal differentiation process. a IFT122 expression in various human tissues and cell samples in FANTOM5. Samples are sorted from left to right in the order of the expression level from highest to lowest. b Expression of the six subunit genes of the IFT-A complex in retina, chondrocyte, and osteoblast. c IFT122 expression changes during the LUHMES cell differentiation. The error bars represent the standard error of the mean of replicates. tpm: tags per million (TIF 756 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaukonen, M., Pettinen, IT., Wickström, K. et al. A missense variant in IFT122 associated with a canine model of retinitis pigmentosa. Hum Genet 140, 1569–1579 (2021). https://doi.org/10.1007/s00439-021-02266-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02266-3