
Abstract
Facial shape differences are one of the most significant phenotypes in humans. It is affected largely by skull shape. However, research into the genetic basis of the craniofacial morphology has rarely been reported. The present study aimed to identify genetic variants influencing craniofacial morphology in northern Han Chinese through whole-exome sequencing (WES). Phenotypic data of the volunteers’ faces and skulls were obtained through three-dimensional CT scan of the skull. A total of 48 phenotypes (35 facial and 13 cranial phenotypes) were used for the bioinformatics analysis. Four genetic loci were identified affecting the craniofacial shapes. The four candidate genes are RGPD3, IGSF3, SLC28A3, and USP40. Four single-nucleotide polymorphism (SNP) site mutations in RGPD3, IGSF3, and USP40 were significantly associated with the skull shape (p < 1×10−6), and three SNP site mutations in RGPD3, IGSF3, and SLC28A3 were significantly associated with the facial shape (p < 1×10−6). The rs62152530 site mutation in the RGPD3 gene may be closely associated with the nasal length, ear length, and alar width. The rs647711 site mutation in the IGSF3 gene may be closely associated with the nasal length, mandibular width, and width between the mental foramina. The rs10868138 site mutation in the SLC28A3 gene may be associated with the nasal length, alar width, width between tragus, and width between the mental foramina. The rs1048603 and rs838543 site mutations in the USP40 gene may be closely associated with the pyriform aperture width. Our findings provide useful genetic information for the determination of face morphology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human facial shapes are species-specific and have individual specificity. The individual specificity of human facial shapes plays an important role in human personal identity and social activities (Koblenzer 2003; Zhang and Gao 2009). Facial features are affected by genetic environmental factors and their interactions. It has been shown that the heritability of most facial features exceeds 60% (Crouch et al. 2018; Djordjevic et al. 2016). The craniofacial shape has been found to be mainly determined by genetic factors (Relethford 1994; Relethford and Harpending 1994), and the shape of the skull determines the basic shape of the face (Lieberman et al. 2000). The correlation between the skull and facial shapes has also been used in forensic practice by forensic anthropologists for processes such as facial image imposition and craniofacial approximation (Claes et al. 2010; Yoshino et al. 1995). Research into the genetic basis of the skull shape is, therefore, crucial for further studies of the genetic variations of facial shapes. However, there is a lack of research on the molecular genetic basis of facial features.
To date, the published papers have shown whole-genome association analyses of human facial shapes (Adhikari et al. 2016; Cha et al. 2018; Claes et al. 2018; Cole et al. 2016; Lee et al. 2017; Liu et al. 2012; Paternoster et al. 2012; Qiao et al. 2018; Shaffer et al. 2016). Researchers have successfully applied these results to human facial modeling predictions and achieved satisfactory results (Claes et al. 2014). The subjects of the published studies were Europeans, Latin Americans, Africans, and Koreans. Whole-genome association analyses of facial morphology have shown association with PAX3, PRDM16, TP63, C5orf50, COL17A1, DCHS2, RUNX2, GLI3, PAX1, EDAR, MAFB, PAX9, MIPOL1, ALX3, HDAC8, CACNA2D3, SCHIP1, PDE8A, OSR1-WDR35, HOXD1-MTX2, WDR27, SOX9, DHX35, etc. However, the results were not consistent among the above-studied population (except PAX3, which was associated with facial morphology in a few of published papers), indicating that ethnic factors had a large impact on facial features.
Therefore, the present study aimed to identify genetic variants influencing craniofacial morphology in northern Han Chinese through whole-exome sequencing (WES) and Sanger Sequencing.
Materials and methods
Study subjects 50 volunteers, including 13 males and 37 females, were recruited from three universities in the Jinzhou area, Liaoning, China. To minimize the impact of non-genetic confounding factors, such as region, ethnicity, age, and BMI, we strictly used the following criteria to select volunteers for this study: (1) no craniofacial surgery, trauma history and craniofacial deformity; (2) no genetic kinship each other; (3) normal Han adults who had been living in Liaoning for at least three generations and were between 20 and 30 years of age; and (4) normal body size with a BMI of 18.5–24. This study was approved by the Ethics Committee of Jinzhou Medical University. All participants provided written informed consent. Venous blood collection and a head CT scan were completed between March 2017 and July 2017 following the standard procedures.
Facial and skull phenotypic measurements using three-dimensional (3D) CT scanning of the head were used to collect craniofacial phenotype information and to reconstruct the 3D face and skull images based on the difference in gray values of the acquired CT data. The craniofacial landmarks on the CT were then marked by group discussion as previously described (Evison et al. 2010). The Euclidean distance between the landmarks was calculated to replace the directly measured distance, which avoided the inaccuracies of mapping, rotation, and other issues caused by selection of a coordinate system. The convenience and reliability of this morphometric method have been proven by numerous studies (Lele and Richtsmeier 1991, 1995; Subhash Lele 1993; Starbuck et al. 2015).
The VG Studio MAX 2.2 software (Volume Graphics Co., Ltd., Heidelberg, Germany) was used to perform 3D facial and skull reconstruction using CT images from the 50 subjects. This software was also used simultaneously to measure the 3D coordinates of the facial and skull landmarks.
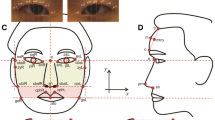
In this study, a total of 34 facial landmarks were measured (Ferrario et al. 1998), including 10 landmarks on the facial midline and 12 landmarks each on the left and right sides of the face. A total of 25 skull landmarks were measured (Adams et al. 2004; Kragskov et al. 1997; Moreddu et al. 2013), including 9 landmarks on the midline and 8 landmarks each on the left and right skull (Fig. 1). The WinEDMA 1.0.1 software (http://c.faculty.umkc.edu/colet) was used to calculate the Euclidean distances between landmarks. The above-mentioned reports (Adhikari et al. 2016; Cha et al. 2018; Claes et al. 2018; Cole et al. 2016; Lee et al. 2017; Liu et al. 2012; Paternoster et al. 2012; Qiao et al. 2018; Shaffer et al. 2016) concluded that gene loci of facial phenotypes were mostly associated with the shapes of the nose, mouth, and mandible, especially the nasal region. Therefore, this study selected phenotypes mostly from these regions. A total of 48 phenotypes were selected, including 35 facial phenotypes and 13 cranial phenotypes (the details are listed in Table 1).

Facial and skull landmarks used in this study. Facial landmarks: 1 glabella (g); 2 nasion (n); 3 sellion (sl); 4 pronasale (prn); 5 subnasale (sn); 6 labrale superius (ls); 7 stomion (sto); 8 labrale inferius (li); 9 supramentale (sm); 10 gnathion (gn); 11, 12 alare (al); 13, 14 subalare (sal); 15, 16 cheilion (ch); 17, 18 zygion (zy); 19, 20 gonion (go); 21, 22 entocanthion (en); 23, 24 ectocanthion (ex); 25, 26 otobasion superius (obs); 27, 28 superaurale (sa), 29, 30 tragion (t); 31, 32 subaurale (sba); 33, 34 otobasion inferius (obi). Skull landmarks: 1 glabella (G); 2 nasion (N); 3 rhinion (RHI); 4 nasal spine (SP); 5 subspinale (SS); 6 exoprothion (EPR); 7 infradentale anterius (IDA); 8 pogonion (PG); 9 gnathion (GN); 10, 11 maxillofrontale (MF); 12, 13 naso-maxillary junction (NMJ); 14, 15 lateral piriform point (LP); 16, 17 frontomalare orbitale (FMO); 18, 19 zygion (ZY) 20, 21 gonion (GO); 22, 23 mentale (ML); 24, 25 orbitale (OR)
WES and whole-genome association analysis
WES and whole-genome association analysis were performed by Beijing Novogene Technology Co., Ltd. (Beijing, China, http://www.novogene.com). The quality of DNA samples was evaluated by agarose gel electrophoresis analysis. The DNA samples that were accurately quantified as greater than 0.6 µg by Qubit were used for library construction. The Agilent’s liquid chip capture system was applied to efficiently enrich the DNA containing all human exon regions. High-throughput deep sequencing was subsequently performed on the Illumina platform. The Agilent SureSelect Human All Exon V6 Kit was used for library construction and the capture experiments. After library construction, Qubit 2.0 was used for the preliminary quantification. The Agilent 2100 system was subsequently used to verify the library insert size. The Illumina platform was used for sequencing according to the effective concentration of the library and the data output requirements. High-throughput paired-end sequencing was performed (paired-end 150 bp, PE150).
After WES was completed, a bioinformatics analysis was performed that included three main aspects: sequencing data quality assessment, SNP detection, and whole-genome association analysis. The sequencing data quality control requirements were as follows: the sequencing error rate of each base position was less than 1%, the average Q20 ratio was greater than 90%, the average Q30 ratio was greater than 80%, the average error rate was less than 0.1%, the sequencing reads achieved an alignment rate of 95% or greater, and the read depth of the base at one position reached 10X or more.
The SNP testing was performed as follows: high-quality sequences were aligned with the human reference genome (GRCh37/hg19) to detect variant information in the sample, and the detected variations were analyzed and annotated.
Sanger sequencing
Sanger sequencing validation in this study was performed by the BGI Beijing Branch (http://www.genomics.cn). Premier 3.0 was used for primer design; the primer sequences are listed in Table 5. The PCR products were purified using Millipore 96 purification plates. The sequencing reaction mixture was as follows: 2 μL of the buffer mix (Bigdye 3.1, 5× sequencing buffer in H2O, 2 μL of the purified PCR product, and 1 μL of the primer (5 mmol/L). The sequencing reaction program was as follows: 95 °C 15 s → (95 °C 15 s → 50 °C 5 s → 60 °C 90 s) × 35 cycles → stop reaction. Electrophoresis was used to detect the PCR-amplified products.
Statistical analysis
The GEMMA (Genome-wide Efficient Mixed Model Association algorithm) software was used to construct a linear mixed model and calculate the association between the 48 phenotypes and genotypes (SNP variant loci) (Zhou and Stephens 2012). A mixed linear model was constructed using GEMMA software in R studio: y = Wα + xβ + u + ε, y: n quantitative traits of individuals; W: covariate (gender); x: n individual genotypes; β: slope coefficient, the size of the genotype. A mixed linear model can eliminate confounding results more accurately, and can directly reflect the relationship between craniofacial phenotype and genotype. Combining other studies of GWAS correlation analysis with our stringent requirements for quality control, our p value threshold is set to 1E−6 to avoid the effects of other mixing factors on experimental results. Therefore, the p values of less than 1 × 10−6 were regarded as statistically different.
Results
Craniofacial phenotypic measurement
Using the facial photographs of 50 individuals, we accessed 35 facial features and 13 cranial features. The craniofacial phenotypic measurements of adult northern Han Chinese are shown in Table 2.
WES and whole-genome association analysis
A total of 23,011 SNP sites were detected by WES. The sequencing quality control is shown in Table 3 and Fig. 2. The association analysis between the 23,011 SNP loci and 48 phenotypes showed that a C > T missense mutation occurred at rs62152530 (NM_001144013) in RGPD3 (2q12). This SNP site mutation is closely associated with 4 facial phenotypes (sl–sn, obs_l–obi_l, sa_r–sba_r, sa_l–sba_l, and al_r–al_l). This mutation causes an Asp > Asn change in the protein amino acid sequence at position 111. A G > C missense mutation occurred at rs647711 (NM_001007237) in IGSF3 (1p13). This SNP site mutation is closely associated with one facial phenotype (sl–sn) and two skull phenotypes (GO_R–GO_L and ML_R–ML_L), and causes an Asp > Glu change in the protein amino acid sequence at position 1020. A T > C missense mutation occurred at rs10868138 (NM_001199638) in SLC28A3 (9q21). This SNP site mutation is closely associated with 4 facial phenotypes (n–sn, sl–sn, t_r–t_l, and al_r–al_l) and one skull phenotype (ML_R–ML_L). This mutation causes a Tyr > Cys change in the protein amino acid sequence at position 113. G > A missense mutations occurred at the rs1048603 (NM_018218) and rs838543 (NM_018218) sites in USP40 (2q37). These two SNP site mutations are closely associated with one skull phenotype (NMJ_R–NMJ_L). The mutations caused a Cys > Thr change in the protein amino acid sequence at position 1123 and a Thr > Cys change at position 2033 (Table 4, Figs. 3, 4).

Error rate distribution along the reads. a The distribution of the average quality value (Phred score) of the WES data. b The depth of the WES; the abscissa indicates the depth of sequencing, and the ordinate indicates the ratio of bases with a sequencing depth of X. c The coverage depth and coverage rate of each chromosome; the abscissa indicates the chromosome number, the left ordinate indicates the average coverage depth, and the right ordinate indicates the coverage rate

The whole-exome region showing genome-wide significant associations with facial traits. The SNP site mutations at 1p13 (rs647711), 9q21 (rs10868138), and 2q12 (rs62152530) have stronger associations with the facial morphology of the Han people of northern China. A G > C missense mutation occurred at rs647711 (IGSF3). This SNP site mutation is closely associated with one facial phenotype (nose length) (a). A T > C missense mutation occurred at rs10868138 (SLC28A3). This SNP site mutation is closely associated with four facial phenotypes (nose length, tragus width, nasal width n–sn, sl–sn, t_r–t_l, and al_r–al_l) (b, d, f, j). A C > T missense mutation occurred at rs62152530 (RGPD3). This SNP mutation is closely associated with four facial phenotypes (nose length, ear length, and nasal width) (c, h, i, e). Here, the LocusZoom plot used the linkage disequilibrium (LD) information in the hg19 1000 Genome Nov 2014 ASN (East Asia) database to obtain the r2. Since no LD information was available in the region of the database where the rs62152530 site was located, the image of this locus did not have r2 information

The whole-exome region showing genome-wide significant associations with the skull traits. A G > C missense mutation occurred at rs647711 (IGSF3). This SNP site mutation is closely associated with two skull phenotypes (width between the gonions and width between the mentales) (a, b). A T > C missense mutation occurred at rs10868138 (SLC28A3). This SNP site mutation is closely associated with one skull phenotype (width between the mentales) (c). Missense mutations of G > A occurred at both rs1048603 (USP40) and rs838543 (USP40). These two SNP site mutations are closely associated with one skull phenotype (pyriform aperture width) (d, e). Here, the LocusZoom plot was generated using the linkage disequilibrium (LD) information in the hg19 1000 Genome Nov 2014 ASN (East Asia) database
Sanger sequencing
Sanger sequencing validation showed that rs62152530, rs647711, rs10868138, rs1048603, and rs838543 all had mutations that were consistent with the WES results. The mutation rates of these sites were 100% (50/50), 24% (12/50), 14% (7/50), 22% (11/50), and 20% (10/50), respectively. All of the sites were heterozygous mutations (Fig. 5).

Sanger sequencing validated the mutations at the five SNP sites. A C > T missense mutation occurred at rs62152530 (RGPD3), a G > C missense mutation occurred at rs647711 (IGSF3), a T > C missense mutation occurred at rs10868138 (SLC28A3), and G > A missense mutations occurred at both rs1048603 (USP40) and rs838543 (USP40). The Sanger sequencing results confirmed that the five SNP sites identified by WES, indeed, had mutations
Discussion
We identified the SNP loci of four genes which were significantly associated with the craniofacial shape of northern Han Chinese. This result is the first report on the genetics of northern Han Chinese craniofacial shapes, which has significance for understanding craniofacial growth and development and improving the accuracy of human facial modeling based on DNA. Our findings could not replicate the previously published results (Table 5).
Five SNP sites at 2q12, 1p13, 9q21, and 2q37 were found to be associated with craniofacial phenotypes in northern Han Chinese. These SNP sites are located in the four candidate genes RGPD3 (RANBP2-Like and GRIP Domain Containing 3), IGSF3 (Immunoglobulin Superfamily Member 3), SLC28A3 (Solute Carrier Family 28 Member 3), and USP40 (Ubiquitin Specific Peptidase 40).
RGPD3 (Ciccarelli et al. 2005) is located in the Ran-binding protein-related gene cluster on chromosome 2 (cytogenetic location: 2q12.2; genomic coordinates (GRCh37/hg19) 107,021,136–107,084,832 and (GRCh38/hg38) 106,391,290–106,468,376). This gene is a protein-coding gene, and one of its related pathways is RNA transport. A study of new complex syndromes caused by the absence of 2q12.1–2q14.2 suggests that its absence may lead to a deformed appearance in child patients, which is manifested as a flat arcus supraorbitalis, wide alveolar ridge, high forehead, low ear, and other facial features (Stoeva et al. 2011). Because the RGPD3 gene is located within the range of 2q12.1–2q14.2, RGPD3 is highly likely to be associated with these special facial features. In this study, we found that a C > T missense mutation occurred at rs62152530. This SNP site mutation is closely associated with the facial nasal length (sl–sn), ear length (obs_l–obi_l, sa_r–sba_r, and sa_l–sba_l), and nasal width (al_r–al_l), which partially overlap with the special facial features caused by the loss of 2q12.1–2q14.2, suggesting that RGPD3 is associated with the morphology of the nose and ears.
IGSF3 is located on chromosome 1 (cytogenetic location: 1p13.1; genomic coordinates (GRCh38): 1: 116,574,397–116,667,754) and is a member of the immunoglobulin superfamily. However, studies have not detected the IGSF3 mRNA in peripheral blood leukocytes, the spleen, or the liver, suggesting that IGSF3 is not involved in immune functions (Saupe et al. 1998). Foster et al. revealed that loss of IGSF3 was associated with congenital nasolacrimal duct obstruction. RT-PCR analysis confirmed the presence of IGSF3 transcripts in the embryonic mouse lacrimal gland, and IGSF3 transcripts were detected in the lacrimal canaliculi and lacrimal glands 1 day after mice were born. Congenital lacrimal duct obstruction includes Hasner valve obstruction and congenital lacrimal duct atresia (including bony nasolacrimal duct atresia caused by bone malformation) (Foster et al. 2014). This finding suggests that IGSF3 may be involved in the regulation of craniofacial developmental. In this study, a G > C missense mutation was found at rs647711 in IGSF3, and this SNP mutation was closely associated with the facial nasal length (sl–sn), the width between the gonions, and the width between the mentales (GO_R–GO_L and ML_R–ML_L) of the skull. This result suggests that IGSF3 may not only participate in regulating development of the lacrimal ducts, but may also participate in the growth and development of other structures of the face and skull.
SLC28A3, also known as CNT3 (Ritzel et al. 2001), is a solute carrier family member located on chromosome 9 [cytogenetic location: 9q21.32–q21.33; genomic coordinates (GRCh38): 9: 84,273,122–84,368,633], and is involved in the regulation of various cellular processes, including neurotransmission, vascular tone, nucleoside transport and metabolism, and other processes. SLC28A3 has extensive specificity for the transport of pyrimidine and purine nucleosides (Barber et al. 2010). Studies have shown that SLC28A3 is associated with obesity, and it may be associated with the lipid-lowering response of statins (Comuzzie et al. 2012; Guallar et al. 2007). Body fat content has a great influence on facial morphological characteristics, and thus, SLC28A3 may have a certain correlation with facial morphology. In this study, a T > C missense mutation was found at rs10868138 in SLC28A3. This SNP site mutation is closely associated with the facial nasal length, the width between the tragus, the alar width (n–sn, sl–sn, t_r–t_l, and al_r–al_l), and the width between the mental foramina (ML_R–ML_L).
USP40 is located on chromosome 2 [cytogenetic location: 2q37.1; genomic coordinates (GRCh38): 2:233, 475,518–233,566,788] and belongs to a large family of cysteine proteases. USP40 modifies cellular proteins through ubiquitin by playing the role of a deubiquitinating enzyme (Quesada et al. 2004). USP40 is expressed in many organ tissues, and its biological functions, protein localization, and structural characterization are completely unknown. A few reports have linked this gene to Parkinson’s disease or glomerular function (Li et al. 2006; Takagi et al. 2017; Zhao et al. 2012). In this study, we found G > A missense mutations at rs1048603 and rs838543 in the USP40 gene; these two SNP site mutations were closely associated with the width of the pyriform aperture. This result suggests that USP40 has a wide range of biological functions and may participate in the regulation of craniofacial development.
Candidate genes for human facial variation and GWAS studies showed a few consistent results. Most of the novel genes identified in each study were different. In fact, racial factors are a major cause because the results come from different populations, such as Europeans, Americans, Africans and East Asians, etc. (Adhikari et al. 2016; Cha et al. 2018; Claes et al. 2018; Cole et al. 2016; Lee et al. 2017; Liu et al. 2012; Paternoster et al. 2012; Qiao et al. 2018; Shaffer et al. 2016). Even in the same race, such as in Europeans, few of the genes or loci identified in each study have been replicated (Adhikari et al. 2016; Lee et al. 2017; Paternoster et al. 2012). In addition, during the review process of this paper, Qiao et al. (2018) reported that, in the GWAS study of the discovery panel of Uyghurs, six significant loci were identified, four of which, rs1868752, rs118078182, s60159418 at or near UBASH3B, COL23A1, PCDH7, and rs17868256 were replicated in independent cohorts of Uyghurs or Southern Han Chinese, which are not insistent with our findings. In addition, the environment may be the likely cause of this situation, for example, even if the study participants in each study are Europeans, they are from different countries or from European descent/Americans. The inconsistency between Qiao’s and our findings may also be due to ethnic and/or environmental factors in the Uighur or South Han and the Northern Han Chinese. Detection of effective genes or loci affecting craniofacial morphology cannot ignore sample size. The authors acknowledge the limitation of a small sample size; only 50 subjects were selected due to funding limitation. However, the findings through WES and whole-genome association analysis in the Han Chinese are novel and significant in the understanding of the genetic basis of facial development.
Another possibility is that the measurement methods and phenotypic descriptions in each study are different. For phenotypic and genomic association analysis, the accuracy of phenotypic measurement is crucial. In the previous studies, 2D method was often used to measure craniofacial morphology. In the 2D method, even with 3D reconstructions, the linear distance between the two projection points is measured on a plane parallel to the coronal or sagittal plane when measuring the distance between two landmarks. However, the phenotypic accuracy of 2D image measurement is limited. Liu et al. (2012) indicated that 2D image noise, image size, and pixel resolution had an impact on phenotypic measurements, and some marker point coordinates cannot be measured on a 2D image. Cha et al. (2018) also showed that the main limitation of their research was that facial measurements were derived from 2D images rather than 3D images (they subsequently measured more facial features such as angle, ratio, curvature, width, height, etc.). In the present study, we used geometric morphometrics to extract the 3D coordinates of two landmarks and calculated the Euclidean distance between the two points. We believe that this new method improves the accuracy of our study. In addition, to minimize the measurement error, in this study, the two authors performed three geometric measurements on all coordinate points, respectively, using the average of the measured data as the final ones.
Overall, the present study not only identified four loci influencing craniofacial morphology in northern Han Chinese, but also established an approach for analyzing the genetic basis of human facial traits through WES combined with 3D CT scanning. Compared with GWAS, WES display is more accurate and cost-effective, and the sequencing depth can reach 120x and more, which can meet the research of large target and multi-target area. In this study, WES can more directly reflect the effect of protein-coding regions on craniofacial phenotypes.
Taken together, our results show that RGPD3, IGSF3, SLC28A3, and USP40 have significant associations with the facial morphology of the northern Han people and that the associated phenotypes are concentrated in the nose and mandible regions. Although not the same genes as reported, it is consistent with the relevant regions. Of these genes, both IGSF3 and SLC28A3 are significantly associated with facial and cranial morphology, suggesting that genes that affect cranial morphology likely affect facial morphology, as well. Cranial morphology, in turn, should be incorporated into genomics research of facial morphology.
References
Adams GL et al (2004) Comparison between traditional 2-dimensional cephalometry and a 3-dimensional approach on human dry skulls. Am J Orthod Dentofac Orthop 126(4):397–409
Adhikari K et al (2016) A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat Commun 7:10815
Barber MJ et al (2010) Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One 5(3):e9763
Cha S et al (2018) Identification of five novel genetic loci related to facial morphology by genome-wide association studies. BMC Genom 19(1):481
Ciccarelli FD et al (2005) Complex genomic rearrangements lead to novel primate gene function. Genome Res 15(3):343–351
Claes P et al (2010) Computerized craniofacial reconstruction: conceptual framework and review. Forensic Sci Int 201(1–3):138–145
Claes P, Hill H, Shriver MD (2014) Toward DNA-based facial composites: preliminary results and validation. Forensic Sci Int Genet 13:208–216
Claes P et al (2018) Genome-wide mapping of global-to-local genetic effects on human facial shape. Nat Genet 50(3):414–423
Cole JB et al (2016) Genomewide association study of African children identifies association of SCHIP1 and PDE8A with facial size and shape. PLoS Genet 12(8):e1006174
Comuzzie AG et al (2012) Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One 7(12):e51954
Crouch DJM et al (2018) Genetics of the human face: identification of large-effect single gene variants. Proc Natl Acad Sci USA 115(4):E676–E685
Djordjevic J, Zhurov AI, Richmond S (2016) Genetic and environmental contributions to facial morphological variation: a 3D population-based twin study. PLoS One 11(9):e0162250
Evison Martin, Ren Fu, Guimarães Marco (2010) Predicting face shape from the skull using a combined NMR and stereophotographic image database of living individuals. Sci Justice 50:36
Ferrario VF et al (1998) Preliminary evaluation of an electromagnetic three-dimensional digitizer in facial anthropometry. Cleft Palate Craniofac J 35(1):9–15
Foster J 2nd et al (2014) Identification of an IGSF3 mutation in a family with congenital nasolacrimal duct obstruction. Clin Genet 86(6):589–591
Guallar JP et al (2007) Altered expression of nucleoside transporter genes (SLC28 and SLC29) in adipose tissue from HIV-1-infected patients. Antivir Ther 12(6):853–863
Koblenzer CS (2003) Psychosocial aspects of beauty: how and why to look good. Clin Dermatol 21(6):473–475
Kragskov J et al (1997) Comparison of the reliability of craniofacial anatomic landmarks based on cephalometric radiographs and three-dimensional CT scans. Cleft Palate Craniofac J 34(2):111–116
Lee MK et al (2017) Genome-wide association study of facial morphology reveals novel associations with FREM1 and PARK2. PLoS One 12(4):e0176566
Lele Subhash (1993) Euclidean distance matrix analysis (EDMA): estimation of mean form and mean form difference. Math Geol 25(5):573–602
Lele S, Richtsmeier JT (1991) Euclidean distance matrix analysis: a coordinate-free approach for comparing biological shapes using landmark data. Am J Phys Anthropol 86(3):415–427
Lele S, Richtsmeier JT (1995) Euclidean distance matrix analysis: confidence intervals for form and growth differences. Am J Phys Anthropol 98(1):73–86
Li Y et al (2006) Genetic evidence for ubiquitin-specific proteases USP24 and USP40 as candidate genes for late-onset Parkinson disease. Hum Mutat 27(10):1017–1023
Lieberman DE, Ross CF, Ravosa MJ (2000) The primate cranial base: ontogeny, function, and integration. Am J Phys Anthropol Suppl 31:117–169
Liu F et al (2012) A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet 8(9):e1002932
Moreddu E et al (2013) Morphometric measurements and sexual dimorphism of the piriform aperture in adults. Surg Radiol Anat 35(10):917–924
Paternoster L et al (2012) Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am J Hum Genet 90(3):478–485
Qiao L et al (2018) Genome-wide variants of Eurasian facial shape differentiation and a prospective model of DNA based face prediction. J Genet Genom 45(8):419–432
Quesada V et al (2004) Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem Biophys Res Commun 314(1):54–62
Relethford JH (1994) Craniometric variation among modern human populations. Am J Phys Anthropol 95(1):53–62
Relethford JH, Harpending HC (1994) Craniometric variation, genetic theory, and modern human origins. Am J Phys Anthropol 95(3):249–270
Ritzel MW et al (2001) Molecular identification and characterization of novel human and mouse concentrative Na + -nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J Biol Chem 276(4):2914–2927
Saupe S et al (1998) Molecular cloning of a human cDNA IGSF3 encoding an immunoglobulin-like membrane protein: expression and mapping to chromosome band 1p13. Genomics 52(3):305–311
Shaffer JR et al (2016) Genome-wide association study reveals multiple loci influencing normal human facial morphology. PLoS Genet 12(8):e1006149
Starbuck JM, Ghoneima A, Kula K (2015) Bilateral cleft lip and palate: a morphometric analysis of facial skeletal form using cone beam computed tomography. Clin Anat 28(5):584–592
Stoeva I et al (2011) Primary congenital hypothyroidism (CH) due to 2q12.1-2q14.2 deletion: a new complex syndrome. Horm Res Paediatr 76:124–125
Takagi H et al (2017) USP40 gene knockdown disrupts glomerular permeability in zebrafish. Am J Physiol Renal Physiol 312(4):F702–F715
Yoshino M et al (1995) Evaluation of anatomical consistency in craniofacial superimposition images. Forensic Sci Int 74(1–2):125–134
Zhang X, Gao Y (2009) Face recognition across pose: a review. Pattern Recogn 42(11):2876–2896
Zhao B et al (2012) Association analysis of single-nucleotide polymorphisms of USP24 and USP40 with Parkinson’s disease in the Han Chinese population. Eur Neurol 68(3):181–184
Zhou X, Stephens M (2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44(7):821–824
Acknowledgements
We thank Professor Martin Evison of Northumbria University for the anthropometric guidance and the volunteers for their enthusiastic support for this research project. We also thank Beijing Novogene Technology Co., Ltd., for WES performing and the BGI Beijing Branch for Sanger sequencing validation.
Funding
This work is supported by Biological Anthropology Innovation Team Project of JZMU (Grant no. JYLJ201702 to F. R.) and Liaoning Distinguished Professor Project (Grant no. LNTP20183501 to F, R.).
Author information
Authors and Affiliations
Contributions
WW and FR conceived and designed the study. WW, GYZ, ZJX, BH, and DHL performed the experiments. WW, GYZ, ZJX, BH, DHL, and FR analyzed and validated the data. WW, GYZ, TYL, WL, and FR were the major contributors in writing the paper. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
This study was approved by the Ethics Committee of Jinzhou Medical University.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wu, W., Zhai, G., Xu, Z. et al. Whole-exome sequencing identified four loci influencing craniofacial morphology in northern Han Chinese. Hum Genet 138, 601–611 (2019). https://doi.org/10.1007/s00439-019-02008-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02008-6