
Abstract
Rare variants are thought to contribute to the genetics of inflammatory bowel disease (IBD), which is more common amongst the Ashkenazi Jewish (AJ) population. A family-based approach using exome sequencing of AJ individuals with IBD was employed with a view to identify novel rare genetic variants for this disease. Exome sequencing was performed on 960 Jewish individuals including 513 from 199 multiplex families with up to eight cases. Rare, damaging variants in loci prioritized by linkage analysis and those shared by multiple affected individuals within the same family were identified. Independent evidence of association of each variant with disease was assessed. A number of candidate variants were identified, including in genes involved in the immune system. The ability to achieve statistical significance in independent case/control replication data was limited by power and was only achieved for variants in the well-established Crohn’s disease gene, NOD2. This work demonstrates the challenges of identifying disease-associated rare damaging variants from exome data, even amongst a favorable cohort of familial cases from a genetic isolate. Further research of the prioritized rare candidate variants is required to confirm their association with the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are the two major forms of the inflammatory bowel diseases (IBD), a heterogeneous group of chronic and debilitating disorders involving inflammation of the gastrointestinal tract. The etiology of IBD involves an aberrant immune response to commensal microflora in genetically susceptible individuals (Malik 2015; Segal 2016). A positive family history remains the strongest risk factor for IBD, evidenced by epidemiological and genetic studies. The first gene associated with CD was NOD2 (Hugot et al. 2001; Ogura et al. 2001). To date, genome-wide association studies (GWAS) have identified over 240 risk loci for IBD (Anderson et al. 2011; Barrett et al. 2008; Franke et al. 2010; Jostins et al. 2012; Liu et al. 2015; Mirkov et al. 2017; de Lange et al. 2017). While the identified risk loci and underlying genetic associations have informed our understanding of the etiopathogenesis of IBD, the variance explained by these risk loci does not account for the estimated heritability of the disease.
Missing heritability is posited to be found in rare, high-impact coding variants (Manolio et al. 2009). Such variants are difficult to identify in population-based exome sequencing studies due to limited power and the high baseline rate of rare, neutral variants (Zuk et al. 2014; Kosmicki et al. 2016). In the context of IBD, studying the Ashkenazi Jewish (AJ) population has the potential to facilitate rare variant identification because this population has a relatively high prevalence of IBD, at least fourfold that of non-Jewish Europeans (Calkins and Mendeloff 1986; Odes et al. 1989; Mayberry et al. 1986; Roth et al. 1989; Bernstein et al. 2006). Furthermore, the AJs are a genetically isolated population (Carmi et al. 2014), characterized by repeated bottlenecks, expansions and endogamy with a consequential reduction in genetic heterogeneity (Ostrer 2001). Additional advantage can be gained by studying cases which are familial (Zielinski et al. 2012) or of an extreme phenotype, e.g., early onset cases (Uhlig and Schwerd 2016) as they are thought to be enriched for functional causal variants with stronger effects.
The increased incidence of CD amongst AJs has been genetically interrogated through GWAS (Kenny et al. 2012), which highlighted five novel CD-associated loci, and more recently by the analysis of exome sequence data from 1855 AJ CD cases and 3044 AJ non-IBD controls (Rivas et al. 2018). AJ enriched CD risk alleles were observed in the well-established CD risk gene NOD2 and in the gene LRRK2. Furthermore, AJ CD cases and controls were found to have a greater CD polygenic risk score (incorporating 124 CD risk alleles but not those in NOD2 or LRRK2) compared with non-Jewish European individuals. The authors conclude the presence of a coordinated selection for both rare and common CD risk alleles in the AJ population (Rivas et al. 2018).
We recently demonstrated the utility of an AJ family-based approach for identifying possible causal rare variants for IBD (Levine et al. 2016) through the study of the two largest AJ multiplex families described to date (54 and 26 CD cases, respectively). A novel frameshift mutation in CSF2RB was identified which replicated in an independent AJ case/control cohort and was shown to cause a loss of function in vitro (Chuang et al. 2016).
With a view to further delineating the genetics of IBD, and in particular, to search for high-impact rare variants, we performed whole exome sequencing on a newly established cohort of 960 Jewish individuals from 199 small to medium sized multiplex families with IBD and sporadic cases. We employed linkage analysis and the prioritization of rare variants shared by multiple affected individuals within the same family. Independent evidence of replication of the association of each variant with IBD was assessed with correction for multiple testing. The well-established gene, NOD2, was prioritized along with a number of other candidates. Despite the theoretical advantages afforded by studying AJ families, the identification of rare variants for IBD proves challenging.
Methods
Ethics
As per previously (Levine et al. 2016), ethical research governance approval was provided by the National Research Ethics Service London Surrey Borders Committee (10/H0906/115) and the University College London Research Ethics Committee (6054/001). Written informed consent was obtained from all participants.
Cohort summary
A large cohort of AJ individuals with IBD were recruited, primarily in the United Kingdom, through advertisements, hospitals and primary care (Schiff et al. 2018). Additional family members were recruited through the probands. Participants were interviewed by telephone to ascertain their Jewish ancestry, IBD phenotype, age of diagnosis and family history of IBD both in first-degree and more distant relatives. Written confirmation of each affected individual’s diagnosis was obtained from his or her doctor in the majority of cases. Saliva samples were collected by post and DNA was isolated according to standard procedures (Quinque et al. 2006).
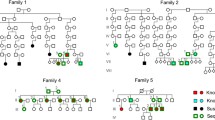
Whole exome sequencing was performed on 960 individuals comprising 513 cases from 199 multiplex IBD families, 364 sporadic cases and 83 unaffected individuals (predominantly relatives of affected individuals). The familial cases consisted of 340 individuals with CD, 160 with UC and 13 with an unknown or unclassified IBD subtype (IBD-U). The sporadic cases consisted of 205 individuals with CD, 153 with UC and 6 with IBD-U. Pedigrees for the 26 largest families with at least four affected individuals (lfams) are shown in Fig. 1. The number of exome sequenced affected individuals with IBD or CD per family are shown in Supplementary Table 1.


Pedigrees for the 26 largest families (lfams) with four or more exome sequenced affected individuals (WES). Affected individuals are indicated by filled symbols. Deceased individuals are indicated by a diagonal line. Phenotype (IBD, CD or UC) is shown. a Five families with two or more exome sequenced unaffected individuals. b 21 families with no exome sequenced unaffected individuals
Whole-exome sequencing
Data generation
Indexed paired-end libraries were prepared using the BGI 59 Exome Enrichment Kit (BGI, China) or the Agilent SureSelect Exome v4 Kit (Agilent, USA), and 2 × 150 bp sequencing was performed by BGI or Macrogen (Macrogen, South Korea) on the Illumina HiSeq 2000 system (Illumina, USA). Two samples were sequenced by both platforms and variant calls were 99.8% concordant (Supplementary Fig. 1).
Read alignment and variant calling
Sequence read alignment and variant calling was performed alongside 4325 other exomes (UCLex) using an in-house next-generation sequencing analysis pipeline (Pontikos et al. 2017). Reads were aligned to the human reference genome build 37/hg19 by Novoalign (version 3.02.08) and variants were called using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010) according to best practices with local realignment around indels, followed by joint variant calling and variant quality score recalibration (VQSR) (DePristo et al. 2011). VQSR uses machine learning to calculate quality scores for variants considering a number of quality parameters and employing a training set of highly validated variants as likely true positives. Further requirements for quality assurance included: genotyping quality (GQ) ≥ 30, reference/alternate read depth for heterozygote calls approximates to a 1:1 ratio (Chi-squared test p ≥ 0.001), alternate read depth for heterozygote and homozygote calls ≥ 3 and VQSR truth tranche ≤ 99.5% for SNPs and ≤ 99.0% for indels. In addition, variants were required to have a call rate ≥ 0.75 and to approximate to Hardy Weinberg Equilibrium (HWE) (chi-squared test, p ≥ 5 × 10− 8).
Variant annotation and filtering
The variant effect predictor (VEP version 76) (McLaren et al. 2016) was used to annotate all variants for their predicted impact on Ensembl gene transcripts and their frequency in Ashkenazi Jewish (gnomAD-AJ) individuals (n = 4925) and non-Finnish European (gnomAD-NFE) individuals (n = 55,860) from the gnomAD exome database (Lek et al. 2016) (http://gnomad.broadinstitute.org). Missense variants were annotated for their predicted deleteriousness according to CAROL (Lopes et al. 2012). Custom-made scripts were used to further annotate each variant with their CADD score (Kircher et al. 2014) and their frequency in a cohort of 500 non-IBD unrelated individuals from amongst the jointly processed UCLex samples that were most proximal to the sequenced AJs based on principal component analysis (UCLex-pAJ), as defined below. A variant was considered potentially pathogenic, and hence included, if it was a frameshift, start loss, stop gain, splice site acceptor/donor variant, or a missense variant which was predicted to be damaging by CAROL and had a CADD score ≥ 20. Variants were filtered based on their population frequency using all of gnomAD-AJ, gnomAD-NFE and UCLex-pAJ; variants were considered as either very rare (AF < 0.005), rare (AF < 0.05) or common (AF > 0.05). If the reference genome carried the minor allele, then genotypes and population allele frequencies were flipped.
Quality control
Sex
The ascertained sex of each sample was verified by the homozygosity rate of common variants on the non-pseudoautosomal regions of the X chromosome (Supplementary Fig. 2).
Relatedness
KING (Manichaikul et al. 2010) was used to calculate a kinship coefficient for each pair of samples based on common variants to identify duplicates and verify the ascertained family pedigrees. Kinship coefficients were mapped to relationships using default thresholds as follows: > 0.354 for duplicate samples/monozygotic twins, 0.177–0.354 for first-degree relatives, 0.0884–0.177 for second-degree relatives, 0.0442–0.0884 for third-degree relatives, and < 0.0442 for unrelated. In addition, a set of unrelated cases and a set of unrelated non-IBD UCLex samples were identified for use in ancestry quality control. A subset of the latter (UCLex-pAJ) was used for variant prioritization and gene burden testing (see “Ancestry” section).
Ancestry
The ascertained ancestry of samples was examined using principal component analysis (PCA) (Supplementary Fig. 3) with the R package FactoMineR (Lê et al. 2008). PCs were calculated from 5409 common independent exome-wide SNPs (Purcell et al. 2014) in 4602 unrelated UCLex samples. These samples include 1092 individuals from the 1000 Genomes Project (1000 Genomes Project Consortium et al. 2015) and 582 unrelated individuals from our cohort (as defined above). The remaining individuals in our cohort were projected onto the calculated PCs.
A subset of the recruited subjects was genetically defined as being of AJ ancestry as follows. The 920 individuals of self-declared AJ ancestry (excluding 40 from the recruited subjects of Sephardi, Middle Eastern (Ostrer 2001) or mixed Jewish ancestry) were defined by the first five PCs, multivariate outliers were removed and a five-dimensional Gaussian model fitted. The number of PCs and the confidence region used were varied and the false positive rate tested. A 90% ellipsoid confidence region captured 878 AJ individuals with only 17 false positives: sixteen that self-declared as being of Sephardi or Middle Eastern Jewish ancestry and one non-Jewish individual. The Mahalanobis distance from each non-IBD UCLex sample to these now genetically defined AJs was calculated to identify the 500 most proximal ancestry-matched controls (UCLex-pAJ).
Analysis approach
The prioritization of candidate variants for IBD in AJ multiplex families was performed by employing two principal strategies. The first sought to identify variants present in affected individuals across multiple families. For this, linkage analysis of all the families in the cohort was performed separately for the phenotypes of CD and IBD (encompassing CD, UC and IBD-U). UC was not examined separately owing to the smaller number of families with this phenotype. Loci with suggestive evidence of linkage were prioritized (LOD ≥ 1.5) and variants within these loci that were shared by the families contributing to the linkage signal were identified. As variants present across multiple families were, by definition, not expected to be exceedingly rare in the population, an allele frequency (AF) threshold of < 0.05 (5%) was employed. Independent evidence of association with IBD, CD or UC was assessed for in a replication cohort of unrelated AJ cases and controls. The second strategy sought to identify rarer variants across the whole exome that may only be found in a smaller subset of the families via a family-based rare variant prioritization analysis. For this, an AF threshold of < 0.005 (0.5%) was employed. As previously, independent evidence of association with IBD, CD or UC was assessed. Statistical evidence of an enrichment of the variant with disease amongst all the sequenced affected individuals in the families was further examined by gene dropping. Replication of such rare variants was limited by restrictive power (Kosmicki et al. 2016).
Linkage analysis
The SNP map used for linkage was generated as follows. First, all common (AF > 0.05) SNPs were extracted, assigned a genetic map position from the Rutgers map (Matise et al. 2007) and pruned for linkage disequilibrium (LD) (window size 50 SNPs, step size 5, r2 threshold 0.2) in UCLex-pAJ using PLINK (Purcell et al. 2007). The remaining SNPs were assigned an AF using gnomAD-AJ. In each 0.3 centiMorgan (cM) window, only the SNP with maximum heterozygosity was retained using the lowest minor allele frequency (MAF) SNP to break ties. The resulting SNP map contained 5945 SNPs.
Only affected individuals within the families were considered. The pedigrees were trimmed to remove non-informative individuals using the R package kinship2 (Sinnwell et al. 2014) leaving 150 informative families for IBD and 88 for CD. A non-parametric linkage analysis was performed using MERLIN (Abecasis et al. 2002) employing NPL ALL which places additional weight on families with three or more affected individuals (Whittemore and Halpern 1994). The maximum theoretical LOD scores for IBD and CD were 65.6 and 38.4, respectively. Genes partially or fully within a region with a log odds (LOD) score ≥ 1.5 were selected. Although not at genome-wide significance (Lander and Kruglyak 1995), such loci demonstrate suggestive evidence of linkage and may harbor disease-related variants. The per-family LOD scores at these loci were computed. For each of the linkage prioritized genes, rare (AF < 0.05) damaging variants seen in at least two affected family members in at least two separate families that were contributing to the linkage (with mean LOD > 0 across the region of interest) were identified.
Family-based rare variant prioritization and segregation analysis
With a view to identifying potentially pathogenic variants enriched in one or more family with the disease, 26 families with four or more CD or IBD (CD, UC and IBD-U) sequenced affected individuals (lfams) were considered (Fig. 1). Variants that were very rare in the population (AF < 0.005) and that were predicted to be damaging (as above) were included. A variant was retained if it was observed in at least 75% of the affected individuals in at least one family (CD or IBD separately).
The segregation of variants to unaffected individuals within five families with at least two sequenced unaffected carrier siblings or offspring for either CD or IBD was examined (Fig. 1a). Potential obligate carriers (parents of affected individuals) were excluded. The probabilities of the genotypes observed in these unaffected individuals, conditional on the observed genotypes in the affected individuals (assuming heterozygosity and only one carrier founder) were calculated. Variants observed in less than one-third of the unaffected individuals were prioritized.
Case/control association for replication
Variants prioritized from both loci identified by linkage analysis and the family-based rare variant and segregation analyses were examined for evidence of association with IBD (n = 1867), CD (n = 1286) and UC (n = 544) as compared with controls (n = 3035–3616) in an independent cohort of genetically confirmed AJ individuals (Rivas et al. 2018). For each phenotype in this independent dataset (IBD, CD and UC), identity by descent filtering was employed to remove related samples, prioritizing the cases, resulting in a slightly different number of controls for each. Per variant quality control of these data was undertaken using gnomAD indicators including call rate and random forest based filtering. No data were available for variants for which the alternate allele was seen in only one case or control. Given the cohort sizes for each phenotype and alpha (p value threshold after correction for multiple testing, see below), power calculations were performed for a range of population AFs and effect sizes via a simulation approach which sampled log odds ratios and applied Fisher’s exact test.
Gene dropping for replication
Gene dropping (MacCluer et al. 1986) was used to assess for an enrichment of variants prioritized from the family-based rare variant analysis in additional families in the study cohort relative to the control population. In each family, a genotype (number of copies of the alternate allele) was assigned to each founder using a Binomial distribution where the number of trials was two and the success probability in each trial was the control AF (maximum of gnomAD-AJ, gnomAD-NFE and UCLex-pAJ). If data were not available from gnomAD-AJ (exomes), the gnomAD AJ whole-genome dataset was utilized (n = 151). A depth-first traversal starting from each founder (one per spousal pair) was performed, and in the case of heterozygotes, a random number generator determined which parental allele was transmitted to each child. Thus, every individual in the family was assigned a genotype. Having performed this gene dropping across all the families, an overall simulated AF was calculated amongst all the cases excluding those from the family in which the variant was initially identified. The whole process was repeated 100,000 times, thus producing 100,000 simulated AFs. The probability of the variant being at a greater frequency in the family disease cohort relative to the simulated frequency under the null, and hence independent evidence of association with disease, was calculated.
Given the alpha value (p value threshold after correction for multiple testing, see below), power calculations were performed for CD and IBD for a range of population and cohort AFs as follows. First, cohort AFs were simulated using the population AF (n = 100,000), then the number of simulated frequencies less than the cohort frequency was modeled as a binomial distribution, and finally this binomial distribution was approximated to a normal distribution to extract power.
Correction for multiple testing
A Bonferroni threshold correcting for multiple testing was calculated based on the number of variants prioritized from both the linkage analysis (n = 11) and the family-based rare variant analysis (n = 413). For the former, three datasets were examined (IBD, CD and UC case/control replication). For the latter, two additional CD and IBD gene dropping datasets were examined. Examining all 424 variants, there was a highly significant correlation (Spearman’s rank test) between the CD and IBD case/control replication p values (p = 3.7 × 10− 8) but not between CD and UC (p = 0.234) or UC and IBD (p = 0.094). Amongst the 413 variants for which gene dropping was performed, there was also a highly significant correlation between the CD and IBD p values (p < 2 × 10− 16). There was no significant correlation between the case/control replication and the gene dropping p values (p = 0.53 for IBD and p = 0.71 for CD). As a consequence of the observed significant correlations, the number of datasets examined for the purpose of calculating the Bonferroni threshold was defined as two from the case/control replication and one from the gene dropping. Thus, the total number of tests performed was considered 11 × 2 + 413 × 3 yielding a Bonferroni threshold of p < 3.97 × 10− 5.
Gene burden testing
Gene burden analysis was performed to test for a statistically significant excess burden of rare damaging variants on a gene by gene basis amongst cases relative to controls. The cases utilized were the unrelated genetically defined AJ individuals (330 for CD and 550 for IBD). The controls were drawn from the UCLex-pAJ subjects with a case–control ratio of one (hence for IBD, the next 50 most proximal controls were added to UCLex-pAJ). For each gene, a combined multivariate and collapsing (CMC) test (Li and Leal 2008) was applied. Variants with AF < 0.01 in gnomAD-AJ were aggregated into one binary variable and variants with an AF > 0.01 and < 0.05 into another. If the variant was absent in the gnomAD-AJ population, then gnomAD-NFE or UCLex-pAJ frequencies were used. Only damaging variants were considered. The multivariate test was a log-likelihood ratio (LLR) test in which the null and alternative hypothesis models included covariates for the first five ancestry PCs. Given the observed inflation (Supplementary Fig. 4), these results were only used to provide supplementary data for prioritized variants.
Results
Linkage analysis
Non-parametric linkage analysis of IBD identified two loci achieving LOD ≥ 1.5 on chromosomes 9 and 13 (Supplementary Fig. 5A) with maximum LOD scores of 1.741 and 1.873, respectively. Linkage analysis of CD identified only one locus achieving LOD ≥ 1.5, on chromosome 16 with a maximum LOD score of 2.074 (Supplementary Fig. 5B). Supplementary Table 2 summarizes these linkage regions in terms of their genomic position and the genes they contain. Of note, NOD2 is within the chromosome 16 locus.
The distribution of previously described CD-associated NOD2 variants amongst the cohort is shown in Supplementary Table 3. The variant G908R (rs2066845) failed quality control owing to a poor call rate. All of the other well-established NOD2 variants (Huang et al. 2017; Rivas et al. 2018) were identified in at least one family and their AF in the unrelated CD cases approximated to that in both the independent AJ case/control dataset and the literature (e.g., L1007insC/rs2066847 was observed at an AF of 0.075 in CD cases compared with 0.072 in CD cases in the AJ case/control dataset).
A total of 51 rare (AF < 0.05) damaging variants were observed in at least two individuals in at least one family in the genes within the linkage-defined loci. Of these, 30 variants were seen in a family contributing to the linkage (per-family LOD > 0) and 11 were seen in at least two such families (Table 1 and Supplementary Table 4) representing all three of the linkage loci. Replication data in unrelated AJ cases and controls were available for all of these variants. Examining IBD, CD and UC and employing the Bonferroni threshold of p < 3.97 × 10− 5 (see “Methods”), three variants, all within the gene NOD2, were significantly associated with disease. One additional NOD2 variant approached the significance threshold (p = 3.7 × 10− 4). Of the eight remaining variants, none achieved p < 0.05 with a positive odds ratio. Their population control AFs ranged from 0.04 down to 0.004. At this AF range, OR of 1.7–3.6, 2.1–3.8 and 2.1–5.5 would be required for 80% power to achieve significance at the Bonferroni threshold in IBD, CD or UC, respectively.
Examining the gene burden results for the eight prioritized unique genes in CD and IBD, only NOD2 in CD was significant at p < 0.05 with p = 7.7 × 10− 6.
Family-based rare variant prioritization
Examining very rare (AF < 0.005) damaging variants that were observed in at least 75% of the affected individuals in 26 families with four or more affected individuals (lfams) identified 413 variants (Supplementary Table 5). One variant was observed in two lfams, a missense in NLRP9 (rs143301793). Of the 413 variants, 362 were seen in an lfam with IBD and 233 were seen in an lfam with CD (182 in common). The IBD variants were identified in 24 families with a median of 11.5 per family and a range of 1–51. The CD variants were identified in 13 families with a median of 12 per family and a range of 5–51.
Replication data in unrelated AJ cases and controls were available for 207 of the 413 variants; the remainder were absent owing to failure to pass quality control filtering or due to an alternate allele count of zero or one in the cases and controls examined. As expected, these 207 variants were more common than those for which replication data were missing (mean UCLex-pAJ AF 9.4 × 10− 4 versus 2.9 × 10− 4, Wilcoxon p = 9 × 10− 11). Examining IBD, CD and UC and employing the Bonferroni threshold of p < 3.97 × 10− 5, no variants were associated with disease. The variant with the smallest p value was a missense variant in the gene DNAH3 (rs140821281, p = 0.0016). The population AFs for these variants ranged from zero to 4.99 × 10− 3. At an AF range of 4.99 × 10− 3 down to 9 × 10− 6 (the smallest non-zero value observed), OR of 3.3–20.5, 3.4–24.6 and 4.8–33.5 would be required for 80% power to achieve significance at the Bonferroni threshold in IBD, CD or UC, respectively.
In the gene dropping data, which examines for an excess of the variant in the familial cohort with the index family removed, two variants achieved the Bonferroni threshold. These were a missense variant in the gene ZNF366 (S332R) and a splice donor variant in the gene MDGA1 (rs202070332). They were each seen in affected individuals in one additional family within the cohort yielding maximum disease cohort AFs of approximately 0.0015, enriched from the population frequency (zero) at p < 1 × 10− 5 (100,000 simulations). No case/control replication data were available for either of these variants owing to their rarity.
At an AF range of 4.99 × 10− 3 down to 9 × 10− 6, the extent to which the AF in the family cohort would have to exceed the population control AF for 80% power to achieve significance at the Bonferroni threshold would be 0.005–0.0167 for IBD. Examining only CD cases in the families, the equivalent increase would have to be 0.006–0.0203.
The 233 and 362 variants seen in a CD or IBD lfam, respectively, were in 226 and 353 unique genes. Gene burden results were available for 187 and 305 of these, respectively. No gene achieved p < 1.4 × 10− 4 (correcting for 353 genes).
Segregation analysis
Amongst five families each with two or more unaffected offspring or siblings of affected individuals, 68 variants were present in ≥ 75% of the cases (CD or IBD). Of these variants, nine were present in ≤ 1/3 of the relevant unaffected family members (Table 2) from four families. Pedigrees of these families showing the segregation of the prioritized variants are in Supplementary Fig. 6.
None of these four families had power to detect a significant lack of transmission of variants to ≤ 1/3 of the unaffected family members conditional on the observed genotypes amongst the affected individuals at p < 0.05.
Replication data were available for six of these variants, none of which were significant at p < 0.05. The minimum gene dropping p value achieved was p = 0.016 for a missense variant in CYB561A3. The power to detect an association is as given above.
Discussion
In this study, we have searched for rare coding variants for IBD using whole exome sequencing. This is a challenging endeavor because of the complex and polygenic nature of genetic susceptibility to IBD, itself a heterogeneous phenotype. To help address this complexity, we studied familial cases from the AJ population which, due to its population history, may harbor a smaller number of pathogenic variants compared with an outbred population. This is the first time a familial AJ IBD cohort of this size has been subjected to exome sequencing.
To identify rare, damaging variants potentially implicated in the aetiopathogenesis of the disease, we employed linkage analysis and selected variants observed in multiple members of the same family. The resulting prioritized variants were assessed for statistical evidence of association with IBD in an independent AJ case/control cohort and using gene dropping simulations. Through this approach, variants in the genes NOD2, ZNF366 and MDGA1 achieved statistical significance after correcting for multiple testing. ZNF366 is of particular interest as it is a dendritic cell-specific transcription factor implicated in the regulation of IL10 (Søndergaard et al. 2018). A large number of additional very rare variants were identified as being enriched in familial cases but the replication cohort and gene dropping simulations lacked the power to confirm or refute an association given their low frequency. It is also relevant to note that the gene dropping significance relies heavily upon the observed control AF, the accuracy of which is limited by the sample size of the studied cohorts (Qiao et al. 2017) and furthermore that the observation of a second family sharing a very rare variant (such that the gene dropping is significant) could be confounded by cryptic relatedness.
Variants were further prioritized based on their lack of transmission to unaffected siblings and offspring; although of limited power to achieve statistical significance, this provided suggestive evidence. A number of these variants were of interest as the genes were either previously described in relation to IBD [such as THEMIS (Chabod et al. 2012)] or were involved in the regulation of the innate immune system which is of increasingly recognized importance in the pathogenesis of IBD (Sewell et al. 2012; Smith et al. 2009). These genes include MCOLN2 (Cuajungco et al. 2016) and NLRP2 (Bruey et al. 2004). Interestingly, we previously identified a different variant in NLRP2 in another large AJ CD family (Levine et al. 2016).
Even with the advantages afforded by our study design, focusing on an isolated population and multiplex families, the robust identification of rare, damaging candidate variants proved challenging. This is likely secondary to technical/analytic deficiencies, and importantly, insufficient power. Given the AF thresholds imposed in our study, the number of variants prioritized and the size of the replication cohort, 80% power to achieve significance would only be observed for a variant with an OR exceeding 2 or 3.5 at the higher limit of population AFs for rare (AF < 0.05) and very rare (AF < 0.005) variants, respectively, with these numbers increasing substantially as the AF decreases towards zero. Considerably larger cohort sizes would be required to test for association at smaller effect sizes for such rare variants. Furthermore, when examining the lack of transmission of candidate variants to unaffected individuals within families, there was an insufficient number of meiosis and larger numbers of unaffected individuals within these families would be required.
The unequivocal identification of NOD2 not only confirms its undoubtedly strong association with CD in this population but more importantly, it validates the methods used. The results also suggest that further damaging exonic single nucleotide risk variants for IBD in AJ multiplex families at AF > 0.01 and < 0.05 that are of comparable effect size to that of the NOD2 variants are unlikely to be present.
This study has not examined the role of known IBD-associated common variants (AF > 0.05) (Mirkov et al. 2017) which are recognized to contribute substantially to the heritability of the disease, including in families (Stittrich et al. 2016). To fully evaluate the genetic architecture of IBD in this familial AJ cohort, such variants will need examination.
This study has demonstrated the challenges of identifying disease-associated rare damaging variants from exome data, even amongst a favorable cohort of familial cases from a genetic isolate. Further research of the prioritized rare candidate variants is required to test their association with the disease. A combination of environmental factors, common variants, and rare variants (some of which we may have identified) are likely to contribute to the familial aggregation of IBD in AJ families.
References
1000 Genomes Project Consortium, The 1000 Genomes Project, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74. https://doi.org/10.1038/nature15393
Abecasis GR, Stacey S, Cherny WO, Cookson, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30(1):97–101. https://doi.org/10.1038/ng786
Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC et al (2011) Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 43(3):246–252. https://doi.org/10.1038/ng.764
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR et al (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40(8):955–962. https://doi.org/10.1038/ng.175
Bernstein CN, Rawsthorne P, Cheang M, Blanchard JF (2006) A population-based case control study of potential risk factors for IBD. Am J Gastroenterol 101(5):993–1002. https://doi.org/10.1111/j.1572-0241.2006.00381.x
Bruey JM, Bruey-Sedano N, Newman R, Chandler S, Stehlik C, Reed JC (2004) PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and Caspase-1 activation in macrophages. J Biol Chem 279(50):51897–51907. https://doi.org/10.1074/jbc.M406741200
Calkins BM, Mendeloff AI (1986) Epidemiology of inflammatory bowel disease. Epidemiol Rev 8:60–91. http://www.ncbi.nlm.nih.gov/pubmed/3533585
Carmi S, Hui KY, Kochav E, Liu X, Xue J, Grady F, Guha S et al (2014) Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nat Commun 5:4835. https://doi.org/10.1038/ncomms5835
Chabod M, Pedros C, Lamouroux L, Colacios C, Bernard I, Lagrange D, Balz-Hara D et al (2012) A spontaneous mutation of the rat themis gene leads to impaired function of regulatory T cells linked to inflammatory bowel disease. PLoS Genet 8(1):e1002461. https://doi.org/10.1371/journal.pgen.1002461
Chuang L-S, Villaverde N, Hui KY, Mortha A, Rahman A, Levine AP, Haritunians T et al (2016) A frameshift in CSF2RB predominant among Ashkenazi Jews increases risk for Crohn’s disease and reduces monocyte signaling via GM-CSF. Gastroenterology 151(4):710–723.e2. https://doi.org/10.1053/j.gastro.2016.06.045
Cuajungco MP, Silva J, Habibi A, Jessica AV (2016) The mucolipin-2 (TRPML2) ion channel: a tissue-specific protein crucial to normal cell function. Pflugers Archiv Eur J Physiol 468(2):177–192. https://doi.org/10.1007/s00424-015-1732-2
de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L et al (2017) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 49(2):256–261. https://doi.org/10.1038/ng.3760
DePristo MA, Banks E, Poplin RE, Garimella KV, Maguire JR, Hartl C, Philippakis AA et al (2011) A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806.A
Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW et al (2010) Genome-wide meta-analysis increases to 71 the number of confirmed crohn’s disease susceptibility loci. Nat Genet 42(12):1118–1125. https://doi.org/10.1038/ng.717
Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, Barrett JC (2017) Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547(7662):173–178. https://doi.org/10.1038/nature22969
Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cézard J-P, Belaiche J, Almer S et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411(6837):599–603. https://doi.org/10.1038/35079107
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC et al (2012) Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491(7422):119–124. https://doi.org/10.1038/nature11582
Kenny EE, Pe’er I, Karban A, Ozelius L, Mitchell AA, Ng SM, Erazo M, Ostrer H, Abraham C, Abreu MT, Atzmon G, Barzilai N, Brant SR, Bressman S, Burns ER, Chowers Y, Clark LN, Darvasi A, Doheny D, Duerr RH, Eliakim R, Giladi N, Gregersen PK, Hakonarson H, Jones MR, Marder K, McGovern DP, Mulle J, Orr- Urtreger A, Proctor DD, Pulver A, Rotter JI, Silverberg MS, Ullman T, Warren ST, Waterman M, Zhang W, Bergman A, Mayer L, Katz S, Desnick RJ, Cho JH, Peter I (2012) A genome-wide scan of Ashkenazi Jewish Crohn's disease suggests novel susceptibility loci. PLoS Genet. 8(3):e1002559. https://doi.org/10.1371/journal.pgen.1002559
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315. https://doi.org/10.1038/ng.2892
Kosmicki JA, Churchhouse CL, Rivas MA, Benjamin MN (2016) Discovery of rare variants for complex phenotypes. Hum Genet 135:625–634. https://doi.org/10.1007/s00439-016-1679-1
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11(3):241–247. https://doi.org/10.1038/ng1195-241
Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25(1):1–18. https://doi.org/10.18637/jss.v025.i01
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce- Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536(7616):285–91. https://doi.org/10.1038/nature19057
Levine AP, Pontikos N, Schiff ER, Jostins L, Speed D, Lovat LB, Barrett JC, Grasberger H, Plagnol V, Segal AW (2016) Genetic complexity of Crohn’s disease in two large Ashkenazi Jewish families. Gastroenterology. https://doi.org/10.1053/j.gastro.2016.06.040
Li B, Leal SM (2008) Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83:311–321. https://doi.org/10.1016/j.ajhg.2008.06.024
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S et al (2015) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47(9):979–986. https://doi.org/10.1038/ng.3359
Lopes MC, Joyce C, Ritchie GRS, John SL, Cunningham F, Asimit J, Zeggini E (2012) A combined functional annotation score for non-synonymous variants. Hum Hered 73(1):47–51. https://doi.org/10.1159/000334984
MacCluer JW, VandeBerg JL, Read B, Ryder OA (1986) Pedigree analysis by computer simulation. Zoo Biol 5(2):147–160. https://doi.org/10.1002/zoo.1430050209
Malik TA (2015) Inflammatory bowel disease. Historical perspective, epidemiology, and risk factors. Surg Clin N Am. https://doi.org/10.1016/j.suc.2015.07.006
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Wei-Min Chen (2010) Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England) 26(22):2867–2873. https://doi.org/10.1093/bioinformatics/btq559
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI et al (2009) Finding the missing heritability of complex diseases. Nature 461(7265):747–753. https://doi.org/10.1038/nature08494
Matise TC, Chen F, Chen W, De La Vega FM, Hansen M, He C, Hyland FCL et al (2007) A second-generation combined linkage physical map of the human genome. Genome Res 17(12):1783–1786. https://doi.org/10.1101/gr.7156307
Mayberry JF, Judd D, Smart H, Rhodes J, Calcraft B, Morris JS (1986) Crohn’s disease in Jewish people—an epidemiological Study in South-East Wales. Digestion 35(4):237–240. https://doi.org/10.1159/000199374
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K et al (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17(1):122. https://doi.org/10.1186/s13059-016-0974-4
Mirkov MU, Verstockt B, Cleynen I (2017) Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol 2(3):224–234. https://doi.org/10.1016/S2468-1253(16)30111-X
Odes HS, Fraser D, Hollander L (1989) Epidemiological data of Crohn’s disease in Israel: etiological implications. Public Health Rev 17(4):321–335. http://www.ncbi.nlm.nih.gov/pubmed/2491514
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H et al (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411(6837):603–606. https://doi.org/10.1038/35079114
Ostrer H (2001) A genetic profile of contemporary Jewish populations. Nat Rev Genet 2(11):891–898. https://doi.org/10.1038/35098506
Pontikos N, Yu J, Moghul I, Withington L, Blanco-Kelly F, Vulliamy T, Wong TLE et al (2017) Phenopolis: an open platform for harmonization and analysis of genetic and phenotypic data. Bioinformatics 33(15):2421–2423. https://doi.org/10.1093/bioinformatics/btx147
Purcell S, Neale B, Todd-Brown K, Thomas L, Manuel AR, Ferreira D, Bender J, Maller et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. https://doi.org/10.1086/519795
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C et al (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506(7487):185–190. https://doi.org/10.1038/nature12975
Qiao D, Lange C, Laird NM, Won S, Hersh CP, Morrow J, Cho MH (2017) Gene-based segregation method for identifying rare variants in family-based sequencing studies. Genet Epidemiol 41(4):309–319. https://doi.org/10.1002/gepi.22037
Quinque D, Kittler R, Kayser M, Stoneking M, Nasidze I (2006) Evaluation of saliva as a source of human DNA for population and association studies. Anal Biochem 353(2):272–277. https://doi.org/10.1016/j.ab.2006.03.021
Rivas MA, Avila BE, Koskela J, Huang H, Stevens C, Pirinen M, Haritunians T et al (2018) Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet 14(5):e1007329. https://doi.org/10.1371/journal.pgen.1007329
Roth M-P, Petersen GM, McElree C, Feldman E, Rotter JI (1989) Geographic origins of Jewish patients with inflammatory bowel disease. Gastroenterology 97(4):900–904. https://doi.org/10.1016/0016-5085(89)91495-9
Schiff ER, Frampton M, Semplici F, Bloom SL, McCartney SA, Vega R, Lovat LB, Wood E, Hart AL, Crespi D, Furman MA, Mann S, Murray CD, Segal AW, Levine AP (2018) A new look at familial risk for inflammatory bowel disease in the Ashkenazi Jewish population. Dig Dis Sci. https://doi.org/10.1007/s10620-018-5219-9
Segal AW (2016) Making sense of the cause of Crohn’s—a new look at an old disease. F1000Research 5:2510. https://doi.org/10.12688/f1000research.9699.2
Sewell GW, Rahman FZ, Levine AP, Jostins L, Smith PJ, Walker AP, Bloom SL, Segal AW, Smith AM (2012) Defective tumor necrosis factor release from Crohn’s disease macrophages in response to toll-like receptor activation: relationship to phenotype and genome-wide association susceptibility loci. Inflamm Bowel Dis 18(11):2120–2127. https://doi.org/10.1002/ibd.22952
Sinnwell JP, Terry M, Therneau, Daniel JS (2014) The kinship2 R Package for pedigree data. Hum Hered 78(2):91–93. https://doi.org/10.1159/000363105
Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJB, Sewell GW, Segal AW (2009) Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med 206(9):1883–1897. https://doi.org/10.1084/jem.20091233
Søndergaard JN, van Heeringen SJ, Looman MWG, Tang C, Triantis V, Louche P, Janssen-Megens EM et al (2018) Dendritic cells actively limit interleukin-10 production under inflammatory conditions via DC-SCRIPT and dual-specificity phosphatase 4. Front Immunol 9:1420. https://doi.org/10.3389/fimmu.2018.01420
Stittrich AB, Ashworth J, Shi M, Robinson M, Mauldin D, Brunkow ME, Glusman G (2016) Genomic architecture of inflammatory bowel disease in five families with multiple affected individuals. Hum Genome Var 3:15060. https://doi.org/10.1038/hgv.2015.60
Uhlig HH, Schwerd T (2016) From genes to mechanisms. Inflamm Bowel Dis 22(1):202–212. https://doi.org/10.1097/MIB.0000000000000614
Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50(1):118–127. http://www.ncbi.nlm.nih.gov/pubmed/8086596
Zielinski D, Gymrek M, Erlich Y (2012) Back to the family: a renewed approach to rare variant studies. Genome Med 4(12):97. https://doi.org/10.1186/gm398
Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, Daly MJ, Neale BM, Sunyaev SR, Eric SL (2014) Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 111(4):E455–E464. https://doi.org/10.1073/pnas.1322563111
Acknowledgements
The authors kindly acknowledge all participants and their referring clinicians. Funding for this study was provided by the Charles Wolfson Charitable Trust and the Medical Research Council. The authors would also like to thank the Helmsley IBD Exomes Program and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://ibd.broadinstitute.org/about.
Author information
Authors and Affiliations
Corresponding author
Additional information
E. R. Schiff and M. Frampton jointly served as first authors.
A.W. Segal and A. P. Levine jointly served as senior authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schiff, E.R., Frampton, M., Ben-Yosef, N. et al. Rare coding variant analysis in a large cohort of Ashkenazi Jewish families with inflammatory bowel disease. Hum Genet 137, 723–734 (2018). https://doi.org/10.1007/s00439-018-1927-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1927-7