
Abstract
Isolated familial hypoparathyroidism is an extremely rare disorder, which to date has been linked to several loci including mutations in CASR, GCM2, and PTH, as well as a rare condition defined as X-linked recessive hypoparathyroidism, previously associated with a 1.5 Mb region on Xq26-q27. Here, we report a patient with hypocalcemia-induced seizures leading to the diagnosis of primary hypoparathyroidism. Mutations in CASR, GCM2, and PTH were ruled out, while whole exome sequencing of the family suggested FHL1, located on chromosome Xq26, as the most likely causative gene variant (FHL1, exon 4, c.C283T, p.R95W). Since FHL1 has not been linked to calcium regulation before, we provide evidence for its functional role in hypoparathyroidism by: (i) bioinformatics analysis coupling its action to known modulators of PTH function; (ii) observing strong expression of fhl1b in Corpuscles of Stannius, gland-like aggregates in zebrafish that function in calcium regulation similar to mammalian PTH; and (iii) implicating fhl1b and FHL1 as regulators of calcium homeostasis in zebrafish and human cells, respectively. Altogether, our data suggest that FHL1 is a novel regulator of calcium homeostasis and implicate it as the causative gene for X-linked recessive hypoparathyroidism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
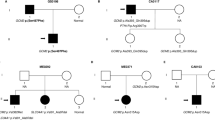
Deep Sequencing, or Next Generation Sequencing (NGS), is a powerful tool to identify causative variants in clinical cases, where other methods have been exhausted. Here, we describe a 4-year-old male, born at full term following an uneventful pregnancy and delivery to non-consanguineous parents, and four siblings (Fig. 1) presented with generalized seizure at the age of 9 days. Upon ruling out all other major etiologies for seizures, initial evaluation identified severe hypocalcemia of 5.7 mg/dl [9–11], hyperphosphatemia of 11.5 mg/dl [5–9], and inappropriately low PTH of 20.7, alongside normal albumin, magnesium, potassium, chloride, blood gases, and vitamin D levels. Urinary calcium levels were low at 0.18 mg/dl [5.2–35.7]. Physical examination revealed no dysmorphic features, short stature, malformations, or developmental delay. Additional studies included chest X-ray, brain US, echocardiogram, renal ultrasound, electroencephalogram, bone age study, and audiogram were all normal. Following a clinical geneticist consultation, 22q11.2 deletion was ruled out via Fluorescent In-Situ Hybridization (FISH). Sanger Sequencing detected no genomic variants in PTH and GCM2 genes. Since no additional loci were related to the phenotype, array-CGH was not performed.

Family pedigree. Squares denote male family members, circles female members, and shaded symbols affected members; the arrow points to the proband
At the age of 4 years, the patient continued to exhibit persistent hypocalcemia alongside inappropriately low PTH levels. In addition, mild orolingual muscle weakness was detected in the patient. Notably, a younger, sixth female sibling was also found to have mild hypoparathyroidism albeit without any electrolyte abnormality, while all other family members exhibit normal PTH and calcium levels. The patient’s father, as well as all other male siblings, demonstrates slight hypotonicity, requiring occupational therapy.
Taken together, these findings indicate that the patient suffers from isolated familial hypoparathyroidism of unknown origin. Exome sequencing of the affected male, his parents, and siblings was performed to detect a causal gene.
Results
Genomic analysis uncovers FHL1 as the causative gene
Whole exome sequencing (WES) of the patient generated 1,065,106 variants passing initial filters, 58,977 of which were rare variants (allele frequency <1% in all databases), and 1031 were found inside exons and resulted in an amino acid change. When searching for de novo mutations found in the patient or in his parents/siblings, only one variant with low allele coverage (<5×) was detected. When focusing on homozygous recessive/X-linked variants, four variants remained. Prioritization of these variants by combining variant severity and gene information revealed an X-linked nonsynonymous mutation in FHL1, ‘Four and a Half LIM domains 1’ gene (FHL1, exon 4, c.C283T, p.R95W) to be the top candidate variant. The variant was predicted to be deleterious by the highest number of prediction tools (8.5/10; see “Materials and methods”). It is a very rare variant with prevalence of 0.0005/45 in the Exome Aggregation Consortium, 0.0006/6 in the NHLBI exome sequencing project and is not present in the 1000 genomes project, or is it found in our personal database of over 900 Israeli exomes. Sanger sequencing confirmed the variant is present in the patient, heterozygous in his mother, and not detected in other family members.
The FHL1 c.C283T variant was predicted by the majority of our employed tools to be deleterious, it is found in a conserved region of the gene, and alternate allele coverage was >30×. Manually reviewing the three other homozygous recessive/X-linked variants (ZNF366 c.A58G p.K20E, CORO1B c.G1252A p.A418T, and ZNF208 c.T986C p.I329T), all three variants were not predicted to be deleterious by any of the tools we employed.
FHL1 (also known as SLIM, SLIM1, or SLIMMER) is a member of the gene family encoding LIM domain containing proteins. An LIM domain is mainly constituted of two cysteine-rich zinc-finger motifs, which coordinately bind zinc atoms to mediate protein–protein interactions (Kadrmas and Beckerle 2004). FHL1 expression is highly enriched in striated muscles (Fig. 2), and has, therefore, been suggested to play an important role in skeletal muscle growth and remodeling (Cowling et al. 2008). FHL1 was demonstrated to have an important role in muscle development and disease. A number of genetic studies linked FHL1 missense mutations to congenital myopathies previously recognized by particular structural features, including Reducing body myopathy (RBM) and Emery-Dreifuss Muscular Dystrophy (EDMD) (Cowling et al. 2011). Mutations in the FHL1 gene have also been associated with arrhythmias (San Román et al. 2016), HCM (Xu et al. 2015), and dilated cardiomyopathy (Christodoulou et al. 2014) in several patients affected by skeletal muscle disorders as well.

Expression of FHL1 protein in human tissues, estimated by immunohistochemistry (IHC) using FHL1 antibody. Height of the bars represents IHC staining strength. Taken from the Human Protein Atlas (Uhlén et al. 2015)
Although the patient exhibits minimal muscular symptoms (see above), the phenotype differs significantly than previously ascribed to FHL1 mutations. Nonetheless, due to the clinical assessment that the patient’s severe phenotype and early onset strongly suggest a genetic cause, we have decided to further pursue the connection between FHL1 and hypocalcemia.
FHL1 interacts with calcium-regulatory proteins
Using gene ontology, we tested the biological pathways that were enriched by some of the 11 core genes plus FHL1. Several pathways associated with molecules transport, including ion transport, transmembrane transport, and regulation of transport, were enriched with at least four of the manually curated targets plus FHL1 (Supplementary Table 1). Next, to test the interactions between FHL1 and hypocalcemia-related proteins, we have manually assembled a list of 11 genes (PTH, CALC1, PTHRP, TBX1, GCM2, CASR, AIRE, GNA11, GATA3, GNAS, and TRPM6), which code for proteins known to be involved in calcium sensing/metabolism with direct connection to primary hypoparathyroidism.
We have assembled an induced network module, consisting of the 11 proteins coded by hypocalcemia-related genes and FHL1 (Fig. 3). The induced network demonstrated an indirect connection between FHL1 and GATA3 (via STAT4) and CASR (via AKAP12 and FLNA).

Induced module of FHL1 and connected proteins of interest. Nodes with black labels are seed proteins, and nodes with gray labels are intermediate nodes. Taken from ConsensusPathDB (Kamburov et al. 2011)
STAT4 is a transcription factor belonging to the STAT protein family that is expressed mostly in immune cells and binds to hundreds of sites in the genome. It was shown that FHl1 promotes both the degradation and the dephosphorylation of STAT4 (Tanaka et al. 2005), presumably affecting GATA3 expression via this mechanism.
AKAP12 (A-kinase anchor proteins) is a member of a structurally diverse protein group, which has the common function of binding to the regulatory subunit of protein kinase A (PKA) and confining the holoenzyme to discrete locations within the cell. A connection with FHL1 was noted in direct protein interaction (Vinayagam et al. 2011) and FLNA (Malovannaya et al. 2011). FLNA, coded for Filamin A protein, is known to interact with CASR (calcium-sensing receptor) and likely act as part of a scaffold that binds signaling components and other key cellular elements (e.g., the cytoskeleton) to facilitate the interaction of the receptor with its signaling pathways (Ray 2015).
fhl1b regulates calcium levels in zebrafish
The zebrafish (Danio rerio) is a powerful model organism for studying vertebrate biology, being well suited to both developmental and genetic analysis (Dooley and Zon 2000). Regulation of Ca2+ levels in vertebrates requires the ability to sense extracellular Ca2+ concentrations, a process carried out by the transmembrane calcium-sensing receptor (CASR) (Loretz 2008). Low Ca2+ levels sensed by CASR in the parathyroid gland, the main organ responsible for calcium homeostasis, lead to expression and secretion of PTH, the key hypercalcemic hormone, which acts on the kidneys, bones, and intestine to increase calcium levels. In contrast, lower vertebrates, like fish rely mostly on gills (or skin, during embryonic stages) as the primary organs for Ca2+ uptake (Hwang et al. 2011; Liao et al. 2007). Nonetheless, although fish do not harbor parathyroid glands, similar calcium-regulating pathways have been uncovered, involving mainly the Corpuscles of Stannius (CS), gland-like aggregates adjacent to the fish kidneys. CS strongly express CASR, which mediates the expression of PTH1 and STC1 (Stanniocalcin), the main hypercalcemic, and hypocalcemic hormones in fish, respectively (Hwang and Chou 2013). Thus, zebrafish represent an important model for studying calcium homeostasis in both physiological and disease states.
Similar to many other genes, due to teleost genome duplication, FHL1 is represented in fish by two paralogs: fhl1a and fhl1b (Glasauer and Neuhauss 2014). In light of its human phenotype, most studies to date have focused on the role of fhl1a and fhl1b on muscular and cardiac function (Bührdel et al. 2015; Chauvigné et al. 2005; Li et al. 2013) and more recently (Xu et al. 2016) on its role in pancreas-liver fate decision during development. Accordingly, in-situ hybridization experiments have demonstrated (Thisse et al. 2015) expression of fhl1a and fhl1b in the zebrafish muscular system, heart and pancreas, among others. Interestingly, fhl1b has been shown to be expressed strongly and specifically in the CS. Hence, we hypothesized that fhl1b might be involved in calcium regulation in fish.
To test this hypothesis, we injected fish embryos with morpholinos to inhibit the translation of zebrafish fhl1b and compared free plasma calcium levels between morpholinos-injected (fhl1b ATG MO) and uninjected (UI) fish. fhl1b expression was decreased by 45% in ATG MO group (SEM—3.2%, P < 0.01). The analysis was carried out in fish grown in media containing both low and high physiologic (25 μg/ml) calcium levels, so as to evaluate the potential relevance of fhl1b to calcium homeostasis across a wide range of calcium levels.
We detected an increase in plasma calcium levels in fhl1b ATG MO fish compared UI fish in both low and high calcium media (P < 0.05 for fish grown in low calcium media and P = 0.12 for 25 μg/ml Calcium media) (Fig. 4). Taken together, these results indicate a role for fhl1b in regulating fish plasma calcium levels.

Dysregulation of Fhl1b KD on calcium levels. Fhl1b ATG MO was used for Fhl1b KD. Calcium levels were measured in larvae with fhl1b KD vs. UI and in low calcium medium levels vs. 25 μg/ml CaCl2 medium levels (high physiologic calcium level). Data show the mean ± SEM (n = 3). *P < 0.05
fhl1b expression affects other calcium-regulatory genes
Having demonstrated the significant effect of fhl1b down-regulation on zebrafish plasma calcium levels, we were next interested in determining the mediators of this effect. Towards this end, we compared the expression of key calcium-regulatory genes (parathyroid hormone paralog 1—PTH1, calcium-sensing receptor—CASR, Stanniocalcin—STC1, epithelial calcium channel—ECaC, and GATA Binding Protein 3-Gata3) in fhl1b ATG MO fish to UI fish via quantitative RT-PCR (qPCR) (Fig. 5). Consistent with the increased calcium levels observed in fhl1b ATG MO fish, we detected in the latter significant up-regulation of PTH1, the main hypercalcemic hormone in both fish and mammals, compared to UI fish. Interestingly, we also found induction of CASR transcript levels in fhl1b ATG MO fish. All other genes were not affected by fhl1b down-regulation (Fig. 5). Hence, increased calcium levels detected in fhl1b-deficient fish are associated with increased PTH1/CASR expression.

Regulation of several calcium homeostasis-related genes by fhl1b KD. Fish were injected with fhl1b MO and were exposed to regular water (a)/high Ca2+ water levels (b). RT-PCR was used for expression level calculation of parathyroid hormone paralog 1 (PTH1), calcium-sensing receptor (CASR), Stanniocalcin (Stc1), epithelial calcium channel (ECaC), and GATA Binding Protein 3 (Gata3). Beta-actin expression was used as control gene. Data show the mean ± SEM (n = 3). *P < 0.05
fhl1b is a positive regulator of PTH expression in human cells
To assess a potential direct effect of FHL1 on calcium regulation via PTH expression in human cells, we used a luciferase reporter assay, whereby human embryonic kidney (HEK293) cells were transfected with a construct containing the luciferase reporter under the control of the human PTH promoter. Activation of PTH locus was measured by bioluminescence in the presence of empty vector or FHL1, with and without CASR presence, under both normal (1.8 mM calcium) and high calcium conditions (3.6 mM calcium). We detected a strong and significant increase in activation of the PTH promoter when FHL1 was introduced into the cells, in both normal and high calcium conditions (Fig. 6). Next, activation of PTH locus was measured by bioluminescence in the presence of siRNA against FHL1 or control siRNA. A consistent decrease in PTH activation was noted in the FHL1 siRNA group which was more significant in the high calcium conditions (Fig. 7). Taken together, these results indicate that FHL1 might function as a positive regulator of PTH expression in human cells.

HEK293 cells were co-transfected with PTH under Luciferase promoter with co-expression of CASR and FHL1. Effect was measured as changes in luminescence. a Effect of CASR and FHL1 co-transfection on luminescence at 1 h of high Ca2+. b Effect of CASR and FHL co-transfection on luminescence at 3 h of high Ca2+. Data show the mean ± SEM (n = 3). *P < 0.05, **P < 0.01

HEK293 cells were co-transfected with PTH under Luciferase promoter with siRNA against FHL1. Effect was measured as changes in luminescence. a Effect of FHL1 siRNA on luminescence at 1 h of high Ca2+. b Effect of FHL1 siRNA on luminescence at 3 h of high Ca2+. Data show the mean ± SEM (n = 3). *P < 0.05
Conclusions
The human FHL1 gene, located on chromosome Xq26, consists of eight exons, giving rise to three protein isoforms: FHL1A, FHL1B, and FHL1C. FHL1A is the predominant isoform in skeletal and cardiac muscle, and comprises an N-terminal half LIM domain followed by four complete LIM domains. FHL1B and FHL1C, the minor isoforms, share the same N-terminal two and a half LIM domains as FHL1A; however, alternative splicing results in unique C-termini: FHL1B contains nuclear import and export sequences and an RBP-J-binding domain, while the FHL1C C-terminus only consists of an RBP-J-binding domain (Cowling et al. 2011). The protein-binding LIM domains demonstrate a unique structure, consisting of highly conserved cysteine and histidine residues which bind Zn2+ and are required for fold and stability of each LIM domain structure. The FHL1 protein has an established role in mammalian skeletal muscle, where it localizes to the nucleus and focal adhesions and is able to shuttle between these compartments. FHL1A has been shown to play a role in promoting myoblast fusion and muscle growth via NFATc1 activation (Cowling et al. 2008).
Accordingly, a wide range of FHL1 mutations have been associated with various muscle- and cardiac-related disease phenotypes in humans. FHL1 mutations were first reported in 2008 and linked to three phenotypes: X-linked myopathy with postural muscle atrophy and generalized hypertrophy (XMPMA),
X-linked dominant scapuloperoneal myopathy (X-SM), and classical reducing body myopathy (RBM) (Windpassinger et al. 2008; Schessl et al. 2008; Quinzii et al. 2008). Many reports later described additional mutations that broadened the phenotypic spectrum further, adding Emery–Dreifuss muscular dystrophy (EDMD) and hypertrophic cardiomyopathy (HCM) with no or minimal musculo-skeletal involvement to the clinical presentations of FHL1 mutations (Gueneau et al. 2009; Friedrich et al. 2012).
Herein, we report a male patient, demonstrating primary hypoparathyroidism, manifesting in significant hypocalcemia from birth. Isolated familial hypoparathyroidism is an extremely rare disorder, which to date has been linked to four different loci. These include mutations in CASR, GCM2, and PTH, as well as a rare condition defined as “X-linked recessive hypoparathyroidism (XLHPT)” (Whyte and Weldon 1981; Winter et al. 1983). The latter condition was described in two multi-generation families, where affected males demonstrated neonatal idiopathic hypoparathyroidism with consequent hypocalcemia and were also sterile. Post-mortem analysis of one of the patients demonstrated aplasia of the parathyroid gland. Carrier females were normo-calcemic and asymptomatic. The XLHPT locus was mapped to a 906-kb region on Xq27 that contains three genes (ATP11C, U7snRNA, and SOX3) (Trump et al. 1998). STS and SNP-based analyses identified a 23–25-kb deletion, which did not contain genes. The deleted region was replaced by a 340-kb insertion that originated from 2p25 and contained a segment of the SNTG2 gene that lacked an open reading frame. Hence, the authors speculated that this insertion could have a position effect on SOX3 expression, which was shown to be expressed in the developing mouse parathyroid between E10.5 and E15.5 (Bowl et al. 2005). Notably, however, SOX3 mutations have never been reported to cause hypocalcemia nor have SOX3 been linked functionally to calcium homeostasis in any organism. Mutations in CASR were ruled out in our patient on a clinical basis (i.e., lack of calciuresis) and GCM2 and PTH mutations were ruled out via Sanger sequencing. Therefore, we carried out WES of the family, which identified FHL1 as the most likely causative gene. Although FHL1 has never been linked to calcium regulation, the strong bioinformatic evidence, the mild muscular phenotype that developed in the patient and the close proximity (~3 Mb) of the FHL1 locus to the XLHPT locus, convinced us to pursuit a potential role for FHL1 in calcium homeostasis using the zebrafish model. While XLHPT prevalence is less than 1 in 1 000 000 live births, the FHL1 c.C283T variant is reported in the Exome Aggregation Consortium with a prevalence of 0.0005. There are also 11 hemizygotes reported in the Exome Aggregation Consortium. Sequencing coverage bias, incomplete penetrance, and variable phenotype severity may account for this discrepancy in genotype–phenotype correlation.
Teleost genome duplication resulted in many mammalian genes being represented by two variants in fish, which often serve non-identical functions. Indeed, while fhl1a was reported to be expressed and regulates muscular and cardiac function in fish, we noted that large scale in-situ hybridization experiments pointed towards strong and specific expression fhl1b in the CS, which functions as the main calcium-regulating hormone in fish. CS produce and secrete PTH1 and STC-1 which act as the main hyper- and hypocalcemic hormones, respectively. Bioinformatic gene interaction analysis revealed that Fhl1 interacts with many calcium-regulatory proteins. Next, through the use of Fhl1b MO, we were able to demonstrate a role for fhl1b in calcium regulation in fish, which exhibited aberrantly high calcium levels in response to fhl1b downregulation. Gene expression assays revealed that this effect is probably linked to enhanced PTH1 expression in fhl1b morphants. While these results establish a direct role for fhl1b in calcium regulation in fish, this phenotype is clearly opposite than that seen in the patient. Nonetheless, it should be noted that calcium metabolism in mammals and fish is significantly different, reflecting not only the evolutionary distance but also the different environments in which the two organisms function. In particular, fish rely on calcium influx via the skin and gills as the main source of calcium, whereas humans depend on dietary calcium and bone metabolism to increase circulating calcium levels. Indeed, when we over-expressed FHL1b in human cells, we observed strong and significant increase in activation of the human PTH gene, confirming that FHL1 loss of function in the patient might well be the etiologic factor leading to hypoparathyroidism.
Of note, fhl1b was recently demonstrated to play a major role in organ differentiation in fish, where it has been shown to dictate the pancreas–liver fate decision during development. Therefore, it is possible that FHL1 could regulate not only PTH expression but also the development of the parathyroid gland. In fact, while this obviously would require further experimentation to prove, we propose that FHL1 might be the disease-causing gene of XLHPT.
In summary, we have uncovered FHL1 as a novel potential regulator of calcium homeostasis in both fish and humans and have implicated it in isolated hypoparathyroidism. This effect is probably mediated by the control over PTH expression and potentially over parathyroid development. While the exact mechanism linking FHL1 to PTH still needs to be elucidated, this report contributes another important layer to the calcium-regulatory network.
Materials and methods
Library preparation, exome capture, and sequencing
Genomic DNA was isolated from peripheral blood leukocytes. Library preparation for NGS was performed according to the TruSeq (Illumina) sample-preparation protocol. DNA libraries were then hybridized to exome-capture probes with NimbleGenSeqCap EZ Human Exome Library, version 2.0 (Roche NimbleGen). Exome-enriched libraries were sequenced on the HiSeq 2000 (Illumina) with an average coverage of 80-fold per each sample.
Variant calling and prioritization
Sequence reads were aligned to the reference human genome (GRCh37/hg19), using the Burrows Wheeler Aligner (BWA) (Li and Durbin 2009). Variants were called following the Genome Analysis Toolkit (GATK) (McKenna et al. 2010) best practices. Briefly, duplicate reads were marked using Picard (http://picard.sourceforge.net). Reads were realigned around detected insertions and deletions (indels) and base qualities recalibrated using GATK. Variant calling was performed using the GATK UnifiedGenotyper tool. Variants with low base or mapping qualities, demonstrating strand bias, aberrant read position distribution, or reference vs. alternate quality score discrepancy, were marked and filtered from subsequent analysis. Variants were annotated using ANNOVAR (Wang et al. 2010) with frequency information gathered from dbSNP138 (Sherry et al. 2001), European and general population from the 1000 genomes project (http://www.1000genomes.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and a personal database of one hundred Ashkenazi Jews exomes. Insertions and deletions (Indels) found adjacent to homopolymers longer than five bases or repeats were marked and were not considered as true candidates. For monogenic diseases diagnosis, variants with allele frequencies higher than 1% in any of the databases were considered prevalent and were excluded from downstream analysis. For polygenic diseases and pharmacologic-related variants, variants with frequency higher than 10% were excluded. Variant severity was predicted using ten different prediction tools (Supplementary Table 3) gathered by dbNSFP (Liu et al. 2013). A variant was considered to be deleterious if more than half of the prediction tools mark it as such. Variants were prioritized by combining the aforementioned annotations with information regarding their affected gene. Gene disease associations were collected from OMIM (http://omim.org/), Orphanet (http://www.orpha.net) and the Human Phenotype Ontology (Robinson and Mundlos 2010). Tissue expression and developmental stages were collected from Uniprot knowledge base (Magrane and Consortium 2011). Genic intolerance to functional variation (RVIS) were retrieved from (Petrovski et al. 2013). Combination of variant and gene annotation data was performed using in-house scripts. Coverage was calculated using the Ensembl coding sequence regions. CNV analysis from exome data was not performed, since current tools have very low recall, low reproducibility, and high error rate (Hong et al. 2016; Kadalayil et al. 2015).
Sanger validation
FHL1 variant was tested for all family members via Sanger sequencing with 100% concordance noted between Sanger results and exome analysis.
Induced network and gene ontology analysis
ConsensusPathDB (Kamburov et al. 2011), which agglomerated the contents of 32 major public repositories for human molecular interactions of heterogeneous types including Gene Ontology, pathway annotations, and protein–protein interaction (PPI) network was used for functional analysis and correlation of gene function. Relevant data integrated for all input genes or a prioritized subset of molecules and mapped onto interaction networks.
Experimental animals
Zebrafish were reared in local tap water at 28.5 °C under a 14:10-h light–dark photoperiod at the Sheba Cancer Research Center, Ramat Gan, Israel. Experimental protocols were approved by the Tel Aviv University Institutional Animal Care and Utilization Committee.
Whole-body Ca2+ content
The clean embryo medium of 6dpf embryos pool (~20 embryos) was used for calcium extraction using nitric acid as previously described (Tseng et al. 2009). CaCl2 was used to generate the standard curves. We calibrated the concentration of environmental calcium that will not lead to increase in blood calcium in normal fish (25 μg/ml) and tested for linear correlation between the number of fish in pool to the total Ca2+ measured from their grouped extraction (Supplementary Figs. 1 and 2).
RNA extraction and RT-PCR
6dpf embryos pool (~20 embryos) were digested with 50 μl trypsin (0.17%) for 60 min in 37C followed by phase separation using chloroform and precipitation using isopropanol. Finally, RNA was washed with 80% of cold ethanol and resuspended in DEPC-treated water. The final RNA concentration and purity were measured using a NanoDrop Spectrophotometer (ND-1000; Thermo Scientific, USA).
Reverse transcription reaction for mRNA was done using random-primer and SuperScript III reverse transcriptase (Invitrogen). SYBR green PCR master mix (Applied Biosystems) was used for qPCR reaction using StepOnePlus real-time PCR system (Applied Biosystems). Expression values are calculated based on the comparative threshold cycle (Ct) method. mRNA expression levels are normalized to B-actin
Luciferase reporter assay
HEK 293 cells (1 × 105 cells/ml) were seeded into 96-well Opti-plates (Packard Instrument, Inc., Meriden, CT). After overnight incubation, the cells were transfected with PTH::luciferase construct and empty vector/CASR/fhl1b/CASR + Fhl1/FHL1 siRNA/control siRNA. Transfection was performed using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h, the cells were incubated with normal/high Ca2+ for 1 or 3 h. Luciferase activities were determined using the Dual Luciferase kit (Promega) according to the manufacturer’s recommendations. Results were normalized to the constitutively expressed Renilla luciferase.
Microinjection of antisense morpholino oligonucleotides (MOs)
The morpholino oligonucleotide (MO) was obtained from Gene Tools (Philomath, OR). The fhl1b morpholino (5′-TTGGACCGGCTTGCCATAGTCAGTC-3′) was prepared with 1 × Danieau solution [in mM: 58 NaCl, 0.7 KCl, 0.4 MgSO4, 0.6 Ca(NO3)2, 5.0 HEPES pH 7.6]. Uninjected fish were used as the control. fhl1b MO (0.3 M) was injected to zebrafish embryos at one cell stage. No significant morphological phenotype was found.
References
Bowl MR, Nesbit MA, Harding B, Levy E, Jefferson A, Volpi E et al (2005) An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest 115(10):2822–2831
Bührdel JB, Hirth S, Kessler M, Westphal S, Forster M, Manta L et al (2015) In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem Biophys Res Commun 461(2):217–223
Chauvigné F, Cauty C, Rallière C, Rescan PY (2005) Muscle fiber differentiation in fish embryos as shown by in situ hybridization of a large repertoire of muscle-specific transcripts. Dev Dyn Off Publ Am Assoc Anat 233(2):659–666
Christodoulou DC, Wakimoto H, Onoue K, Eminaga S, Gorham JM, DePalma SR et al (2014) 5′RNA-Seq identifies Fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest 124(3):1364–1370
Cowling BS, Cottle DL, Wilding BR, D’Arcy CE, Mitchell CA, McGrath MJ (2011) Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord NMD. 21(4):237–251
Cowling BS, McGrath MJ, Nguyen M-A, Cottle DL, Kee AJ, Brown S et al (2008) Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J Cell Biol 183(6):1033–1048
Dooley K, Zon LI (2000) Zebrafish: a model system for the study of human disease. Curr Opin Genet Dev 10(3):252–256
Friedrich FW, Wilding BR, Reischmann S, Crocini C, Lang P, Charron P et al (2012) Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet 21(14):3237–3254
Glasauer SMK, Neuhauss SCF (2014) Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol Genet Genomics MGG 289(6):1045–1060
Gueneau L, Bertrand AT, Jais J-P, Salih MA, Stojkovic T, Wehnert M et al (2009) Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet 85(3):338–353
Hong CS, Singh LN, Mullikin JC, Biesecker LG (2016) Assessing the reproducibility of exome copy number variations predictions. Genome Med [cited 10 Mar 2017];8. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4976506/
Hwang P-P, Chou M-Y (2013) Zebrafish as an animal model to study ion homeostasis. Pflugers Arch 465(9):1233–1247
Hwang P-P, Lee T-H, Lin L-Y (2011) Ion regulation in fish gills: recent progress in the cellular and molecular mechanisms. Am J Physiol Regul Integr Comp Physiol 301(1):R28–R47
Kadalayil L, Rafiq S, Rose-Zerilli MJJ, Pengelly RJ, Parker H, Oscier D et al (2015) Exome sequence read depth methods for identifying copy number changes. Brief Bioinform 16(3):380–392
Kadrmas JL, Beckerle MC (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5(11):920–931
Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R (2011) ConsensusPathDB: toward a more complete picture of cell biology. Nucl. Acids Res 39:D712–D717
Li M, Andersson-Lendahl M, Sejersen T, Arner A (2013) Knockdown of desmin in zebrafish larvae affects interfilament spacing and mechanical properties of skeletal muscle. J Gen Physiol 141(3):335–345
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinforma Oxf Engl 25(14):1754–1760
Liao B-K, Deng A-N, Chen S-C, Chou M-Y, Hwang P-P (2007) Expression and water calcium dependence of calcium transporter isoforms in zebrafish gill mitochondrion-rich cells. BMC Genomics 4(8):354
Liu X, Jian X, Boerwinkle E (2013) The dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 34(9):2393–2402
Loretz CA (2008) Extracellular calcium-sensing receptors in fishes. Comp Biochem Physiol A: Mol Integr Physiol 149(3):225–245
Magrane M, Consortium U (2011) UniProt Knowledgebase: a hub of integrated protein data. Database J Biol Databases Curation. doi:10.1093/database/bar009
Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW et al (2011) Analysis of the human endogenous coregulator complexome. Cell 145(5):787–799
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9(8):e1003709
Quinzii CM, Vu TH, Min KC, Tanji K, Barral S, Grewal RP et al (2008) X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet 82(1):208–213
Ray K (2015) Calcium-sensing receptor: trafficking, endocytosis, recycling, and importance of interacting proteins. Prog Mol Biol Transl Sci 132:127–150
Robinson PN, Mundlos S (2010) The human phenotype ontology. Clin Genet 77(6):525–534
San Román I, Navarro M, Martínez F, Albert L, Polo L, Guardiola J et al (2016) Unclassifiable arrhythmic cardiomyopathy associated with Emery-Dreifuss caused by a mutation in FHL1. Clin Genet 90(2):171–176
Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS et al (2008) Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 118(3):904–912
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM et al (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29(1):308–311
Tanaka T, Soriano MA, Grusby MJ (2005) SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity 22(6):729–736
Thisse, B, Pflumio, S, Fürthauer, M, B Loppin, Heyer, V, Degrave, A, et al (2015) Expression of the zebrafish genome during embryogenesis. ZFIN Direct Data Submission (http://zfin.org) [cited 15 Nov 2016]. https://zfin.org/ZDB-PUB-010810-1
Trump D, Dixon PH, Mumm S, Wooding C, Davies KE, Schlessinger D et al (1998) Localisation of X linked recessive idiopathic hypoparathyroidism to a 1.5 Mb region on Xq26-q27. J Med Genet 35(11):905–909
Tseng D-Y, Chou M-Y, Tseng Y-C, Hsiao C-D, Huang C-J, Kaneko T et al (2009) Effects of stanniocalcin 1 on calcium uptake in zebrafish (Danio rerio) embryo. Am J Physiol Regul Integr Comp Physiol 296(3):R549–R557
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A et al (2015) Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419
Vinayagam A, Stelzl U, Foulle R, Plassmann S, Zenkner M, Timm J et al (2011) A directed protein interaction network for investigating intracellular signal transduction. Sci Signal. 4(189):8
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl Acids Res 38(16):e164
Whyte MP, Weldon VV (1981) Idiopathic hypoparathyroidism presenting with seizures during infancy: X-linked recessive inheritance in a large Missouri kindred. J Pediatr 99(4):608–611
Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, Lohberger B et al (2008) An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet 82(1):88–99
Winter WE, Silverstein JH, Maclaren NK, Riley WJ, Chiaro JJ (1983) Autosomal dominant hypoparathyroidism with variable, age-dependent severity. J Pediatr 103(3):387–390
Xu J, Cui J, Del Campo A, Shin CH (2016) Four and a half LIM domains 1b (Fhl1b) is essential for regulating the liver versus pancreas fate decision and for β-cell regeneration. PLoS Genet 12(2):e1005831
Xu J, Li Z, Ren X, Dong M, Li J, Shi X, et al (2015) Investigation of pathogenic genes in Chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci Rep [cited 22 Sep 2016] 5:16609. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4647833/
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding
The Shomron laboratory is supported by the Israel Cancer Research Fund (ICRF), Research Career Development Award (RCDA); Wolfson Family Charitable Fund; Claire and Amedee Maratier Institute for the Study of Blindness and Visual Disorders; I-CORE Program of the Planning and Budgeting Committee, The Israel Science Foundation (Grant number 41/11); the Israeli Ministry of Defense, Office of Assistant Minister of Defense for Chemical, Biological, Radiological and Nuclear (CBRN) Defense; Foundation Fighting Blindness; Saban Family Foundation, Melanoma Research Alliance; Binational Science Foundation (BSF); Israel Cancer Research Fund (ICRF) Acceleration Grant; Israel Cancer Association (ICA); Donation from the Kateznik K. Association Holocaust; Margot Stoltz Foundation through the Faculty of Medicine grants of Tel-Aviv University; The Varda and Boaz Dotan Research Center in Hemato-Oncology, Idea Grant; ‘Lirot’ Association and the Consortium for Mapping Retinal Degeneration Disorders in Israel; Interdisciplinary grant of the Israeli Ministry of Science, Technology and Space on the Science, Technology and Innovation for the Third Age; The Edmond J. Safra Center for Ethics at Tel Aviv University; Check Point Institute for Information Security; Joint Core Program of Research on the Molecular Basis of Human Disease, Israel Science Foundation (ISF, 1852/16); The Edmond J. Safra Center for Bioinformatics at Tel Aviv University; A kind gift from Irwin Blitt; The Adelis Foundation.
Additional information
Nir Pillar, Oren Pleniceanu, and Mingyan Fang are equal first authors.
Le Cheng, Benjamin Dekel, and Noam Shomron are equal last authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pillar, N., Pleniceanu, O., Fang, M. et al. A rare variant in the FHL1 gene associated with X-linked recessive hypoparathyroidism. Hum Genet 136, 835–845 (2017). https://doi.org/10.1007/s00439-017-1804-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1804-9