
Abstract
Assigning a pathogenic role to mitochondrial DNA (mtDNA) variants and unveiling the potential involvement of the mitochondrial genome in diseases are challenging tasks in human medicine. Assuming that rare variants are more likely to be damaging, we designed a phylogeny-based prioritization workflow to obtain a reliable pool of candidate variants for further investigations. The prioritization workflow relies on an exhaustive functional annotation through the mtDNA extraction pipeline MToolBox and includes Macro Haplogroup Consensus Sequences to filter out fixed evolutionary variants and report rare or private variants, the nucleotide variability as reported in HmtDB and the disease score based on several predictors of pathogenicity for non-synonymous variants. Cutoffs for both the disease score as well as for the nucleotide variability index were established with the aim to discriminate sequence variants contributing to defective phenotypes. The workflow was validated on mitochondrial sequences from Leber’s Hereditary Optic Neuropathy affected individuals, successfully identifying 23 variants including the majority of the known causative ones. The application of the prioritization workflow to cancer datasets allowed to trim down the number of candidate for subsequent functional analyses, unveiling among these a high percentage of somatic variants. Prioritization criteria were implemented in both standalone (http://sourceforge.net/projects/mtoolbox/) and web version (https://mseqdr.org/mtoolbox.php) of MToolBox.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The exponential growth of human mitochondrial DNA (mtDNA) sequences available in public databases (Brandon et al. 2005; van Oven and Kayser 2009; Rubino et al. 2012) is probably the best current hallmark of the central role the mitochondrial genome plays in medicine, forensics and anthropology. In particular, clinicians have recently re-discovered the ‘neglected genome’ (Pesole et al. 2012) as a pivotal determinant or modifier of an increasing number of pathologies, including Alzheimer (Adeghate et al. 2013), cancer (Verschoor et al. 2013), diabetes (Patti and Corvera 2010; Mercader et al. 2012; Adeghate et al. 2013), spinocerebellar ataxias (Mancini et al. 2013) and several types of sclerosis (Patti and Corvera 2010). Because the role of mtDNA variants in most of these phenotypes is still a matter of debate, it is fundamental that either novel or previously reported mtDNA variants of interest are highlighted and brought forward to subsequent analyses, since functional studies aimed at ascertaining the potential pathogenicity often require cumbersome efforts. After sequencing and assembling an mtDNA genome, the first analytical step is usually the identification of positions differing from the chosen reference sequence. Next, with the aim of identifying variants of potential interest for a disease or a particular phenotype, such positions should be further filtered after determining the correct genetic background (haplogroup), so that fixed evolutionary allelic variants may be promptly recognized. Indeed, human mitochondrial phylogeny is described by haplogroup classification based on clusters of closely related evolutionary haplotypes, defined by the pattern of genetic markers occurring in the entire mtDNA and reflecting the migration of human populations over continents (Watson et al. 1997; Balter 2011).
Although clinicians are seldom familiar with the complexity of haplogroups, evolutionary and adaptive aspects should not be ignored in clinical studies, whereby the genetic association between haplogroup-defining variants and clinical phenotypes has been traced (Ghelli et al. 2009; Khan et al. 2013; Peng et al. 2013; Zhang et al. 2013). However, in the search for clinically relevant mtDNA variants, it is useful to rule out those evolutionary fixed, thus limiting variability analyses to few variants, annotated with estimation of conservation and prediction of pathogenicity, to obtain a shortlist of candidates that affect function of the gene/protein. Also, the heteroplasmic fraction of a sequence variant, whenever available, may not be neglected. Indeed, the advent of high-throughput sequencing technologies in mitochondrial genetics has revealed that a wide range of mtDNA variants at low heteroplasmy occurs also in healthy individuals (Payne et al. 2013; Diroma et al. 2014), and varies among tissues (He et al. 2010).
Overall, there is an urgent need for a common workflow based on stringent criteria, to be implemented by researchers who face the challenge of in-depth analysis of mtDNA sequences, which would allow them to recognize the influence of few variants on the phenotype, thus facilitating the functional assay.
In this paper, we propose and validate a workflow for prioritizing functionally important non-synonymous variants, starting from the variant annotation process already implemented in MToolBox based on the use of the Macro Haplogroup Consensus Sequences (MHCS) (Calabrese et al. 2014) and taking into account pathogenicity predictors. A nucleotide variability cutoff and a disease score threshold are established, to prioritize a pool of candidate variants affecting function, which may then be further investigated. Disease scores and prioritization criteria are now implemented in both the standalone version (http://sourceforge.net/projects/mtoolbox/) as well as in the web version of MToolBox at MSeqDR portal (https://mseqdr.org/mtoolbox.php) (Falk et al. 2014).
Materials and methods
The MToolBox variant annotation process
MtDNA variants identified by the MToolBox pipeline (Calabrese et al. 2014) are thoroughly parsed through an annotation process which is mainly based on the comparison with both the two widely used rCRS and RSRS reference sequences and the recognition of alleles that are not shared with the sample-specific MHCS. MHCSs, integrated in the MToolBox package, were generated from 32 multiple alignments of complete mitochondrial sequences from 14,144 healthy individuals available in HmtDB (November 2013 update) belonging to the 32 chosen macro-haplogroups. Each macro-haplogroup-specific multi-alignment was then subjected to nucleotide composition analysis by applying the SiteVar algorithm (Pesole and Saccone 2001) to determine the allele occurring most frequently in each position thus generating the MHCSs.
The annotations provided by MToolBox for each variant include:
-
nucleotide site-specific variability estimated on the multi-alignment of the updated healthy genomes reported in HmtDB;
-
predictions of pathogenicity for non-synonymous variants by applying MutPred (Li et al. 2009), HumDiv- and HumVar-trained PolyPhen-2 models (Adzhubei et al. 2013), SNPs&GO, PhD-SNP (Capriotti et al. 2013), and PANTHER algorithms (Thomas and Kejariwal 2004). Each predictor assigns to each sequence variant a probability score of pathogenicity as well as a qualitative prediction ‘disease’, ‘neutral’ or ‘unclassified’.
-
Mitomap (Lott et al. 2013) annotations referring to disease-associated mutations, occurring in coding and control regions and somatic mutations together with their state of homoplasmy/heteroplasmy;
-
links to OMIM (http://omim.org).
All these data are available in the ‘patho_table’, a tab delimited file provided by the MToolBox package, listing all possible 24,195 non-synonymous nucleotide substitutions which may occur within the 13 human mitochondrial protein encoding genes, as previously reported (https://sourceforge.net/projects/mtoolbox/; Pereira et al. 2011). Links to Mamit-tRNA (Pütz et al. 2007) web resources were added to provide the user with a general view of variants localization within the mitochondrial tRNA sequence structure.
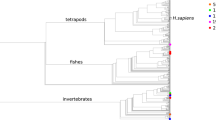
Macro-haplogroup consensus sequences (MHCS) phylogeny
The robustness of 32 MHCSs was tested by generating a phylogenetic tree including all MHCSs and two complete mitochondrial sequences, for each macro-haplogroup, derived from Phylotree (van Oven and Kayser 2009). Those sequences are also available in the Human mitochondrial Data Base (HmtDB—Rubino et al. 2012) which reports all publicly available human mitochondrial genomes. Genomes associated to population studies are stored and analyzed as ‘healthy’; genomes from subjects affected by mitochondriopathies are reported in a separate category and annotated as ‘patient’. All data required to establish if an mtDNA genome sequence belongs to the ‘healthy’ category are obtained from the GenBank entry, papers and upon request to the authors. In addition, the multi-alignment and its manual editing allow to check the quality of sequences to detect any sequencing errors.
The MHCS phylogenetic tree was produced according to the Maximum Likelihood method, based on the Jukes–Cantor substitution model (Jukes and Cantor 1969). The quality of the tree topology was assessed by bootstrap analysis of 500 replicates (Felsenstein 1985). Analyses were performed through the functions implemented in MEGA5 software (Hall 2013).
Datasets
The prioritization workflow was validated on a dataset of 125 mtDNA genomes belonging to individuals affected by Leber’s Hereditary Optic Neuropathy (LHON), sequenced by Sanger technology and stored in HmtDB (http://www.hmtdb.uniba.it) (Rubino et al. 2012) (Supplementary Table 1—SampleData). This dataset was chosen to evaluate the performance of this workflow in identifying the known LHON-causative mutations since 42 % of these genomes was expected to harbor at least one of the primary mutations included in the panel of the ‘Top 14 LHON’ annotated in Mitomap (Lott et al. 2013).
The efficiency of the workflow to prioritize and recognize tumor-specific variants with a functional impact was also tested on mtDNA sequences obtained from 20 ovarian cancer samples, collected within a concluded clinical study, at S.Orsola-Malpighi Hospital, Bologna, Italy, during the period 2012–2013. Informed consent had been obtained in compliance with the Helsinki Declaration and the study had been approved by the local ethical committee. DNA was available also from the corresponding non-tumor tissue of patients and was used in the context of this study to test the germline nature of identified variants. List of specimens and HmtDB identifiers, obtained after submission of sequences to the HmtDB, are reported in Supplementary Table 2—SampleData. Sanger sequencing of the whole mtDNA was performed as previously described (Kurelac et al. 2013), to prevent nuclear mitochondrial sequence (NumtS) (Simone et al. 2011) co-amplification. The somatic (tumor-specific) nature of mitochondrial variants was ascertained by sequencing mtDNA from matched non-tumor tissue and validated on a second PCR product.
Finally, to test our workflow also on data generated from high-throughput technologies, Whole Exome Sequencing (WXS) BAM files (Binary Alignment/Map) from 90 matched samples (primary solid tumor/peripheral blood) from Colorectal Adenocarcinoma (COAD) patients were downloaded from the online repository of the consortium dbGaP (https://cghub.ucsc.edu/). These data were generated from the Baylor College of Medicine (BMC) center of The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) on Illumina platform and mapped on GRCh37-lite reference (Genome Reference Consortium Human Build 37, accession = “GCA_000001405.1”). The samples featuring a mean read depth >10X across the mitochondrial genome after its extraction through MToolBox were brought forward in the analysis (Supplementary Table 3—BloodSamples and TumorSamples sheets).
Denaturing high-performance liquid chromatography (dHPLC) analysis on OC samples
For the variants found in Ovarian Cancer (OC) samples, PCR was performed using AmpliTaq Gold polymerase (Applied Biosystems). For m.3380G>A in MT-ND1 fw-5′-ATACCCACACCCACCCAAGA-3′ and rv-5′-AGATGTGGCGGGTTTTAGGG-3′ primers were used, for m.9837G>A in MT-CO3 fw-5′-TCAATCACCTGAGCTCACCA-3′ and rv-5′-ACCACATCTACAAAATGCCAGT-3′ primers were used, for m.14969T>A in MT-CYB fw-5′-AACTTCGGCTCACTCCTTGG-3′ and rv-5′-TCACGGGAGGACATAGCCTA-3′ primers were used. The amplification product was analyzed by WAVE Nucleic Acid Fragment Analysis System (Transgenomic, Omaha, NE, USA). Data analysis was performed as previously described (Frueh and Noyer-Weidner 2003; Kurelac et al. 2012).
Mitochondrial DNA extraction, variant detection and annotation
MToolBox pipeline (Calabrese et al. 2014), including several steps as read mapping and NumtS filtering, post-mapping processing, genome assembly, haplogroup prediction and variant annotation, was used to extract the off-target mitochondrial genomes from the WXS COAD BAM files obtained from the TCGA repository, and then to annotate each variant allele. Fasta files from LHON and ovarian cancer samples were also used as input for MToolBox to annotate mitochondrial variants and related features.
Disease Score definition for non-synonymous variants
A training dataset of 53 mtDNA non-synonymous variants (Table 1; Supplementary Table 4), previously validated as affecting function, including 28 disease-associated mutations annotated in Mitomap as ‘confirmed’ to be pathogenic by two or more independent laboratories (Lott et al. 2013), and 25 clearly pathogenic cancer-associated mutations (Gasparre et al. 2007; Porcelli et al. 2010; Pereira et al. 2012), was used to define the ‘disease score’ (as described in the “Results” section ‘Disease Score definition’) of any non-synonymous mtDNA variants, by weighting the 6 above-listed pathogenicity predictions (Thomas and Kejariwal 2004; Adzhubei et al. 2013; Capriotti et al. 2013) available in the ‘patho_table’ implemented in MToolBox (https://sourceforge.net/projects/mtoolbox/). These six methods were chosen among the most widely used pathogenicity predictors, available online for a fast evaluation of large-scale data from sequencing, although their often-contradictory predictions demand a way to weigh their reliability. More details regarding the features used by any method to predict the impact of amino acid allelic variants on protein structure/function are available in (Thomas and Kejariwal 2004; Adzhubei et al. 2013; Capriotti et al. 2013).
Disease Score threshold
To derive the Disease Score Threshold (DST) for assessing the potential functional impact of the non-synonymous variants, the mixture model of two normal distributions (McLachlan and Peel 2000) was fitted to the disease scores related to 1872 non-synonymous variants observed in 15,385 mtDNA genomes from healthy individuals, carefully selected as complete sequences (http://webservice.cloud.ba.infn.it/hmtdb/HmtDBHealthyGenomes_References.xlsx) and stored in HmtDB (May 2014, Rubino et al. 2012). It was not possible to define a DST from non-synonymous variants found in mtDNA genomes from patients, as stored in HmtDB. This dataset suffered a sampling bias, in which certain pathologies (as LHON, Alzheimer’ disease, etc.) were over-represented, while others (as MELAS, etc.) were under-represented. The analysis was performed using the normalmixEM function from the mixtools package (Benaglia et al. 2009) implemented in R version 3.1.1.



Discussion
The assessment of the role of mitochondrial variants in the onset and/or progression of human diseases and cancer is a difficult task. It requires robust and statistically reliable methods to support clinicians in the functional investigation of variants potentially affecting protein function. The question of determining the potential pathogenicity of mtDNA variants is in fact a matter of debate among clinicians (McCormick et al. 2013). Because of the peculiarities of mitochondrial polyplasmic genetics, assignment of pathogenicity should take into account the degree of heteroplasmy, the haplogroup background, and even environmental factors (Wallace et al. 2003). Several methods have been recently published to meet these goals, such as mit-o-matic (Vellarikkal et al. 2015), MitImpact (Castellana et al. 2015) and MtSNPscore (Bhardwaj et al. 2009). Here, we contribute with a robust statistical approach based on the introduction of two thresholds, NVC and DST. NVC threshold is calculated from the updated site-specific nucleotide variability values in a large dataset of complete healthy genomes available in HmtDB database, whose sequence quality was supported by a correct haplogroup prediction (see “Materials and methods”); DST is a statistically estimated robust threshold based on the pathogenicity predictions estimated by MutPred (Li et al. 2009), HumDiv- and HumVar-trained PolyPhen-2 models (Adzhubei et al. 2013), SNPs&GO, PhD-SNP (Capriotti et al. 2013), and PANTHER (Thomas and Kejariwal 2004), combined with the nucleotide variability distribution in the same HmtDB healthy dataset. To date, the information on biochemical and structural features of mitochondrial respiratory chain proteins are not so rich as for nuclear ones. Accordingly, all the in silico pathogenicity predictions may not be reliable and hence they should be considered with caution, since the determinants of pathogenicity for an mtDNA variant are unclear. The integration of pathogenicity predictions with the functional assays is an essential and strongly recommended step to consolidate the validation for pathogenicity of an mtDNA variant. The prioritization workflow is fully implemented in MToolBox (Calabrese et al. 2014) thus contributing to highlight mitochondrial DNA variants as suitable candidates for subsequent functional analyses in clinical studies—an issue which has often revealed to be problematic in analyses seeking correlations between mtDNA genotypes and disease phenotypes.
The in silico prioritization criteria here developed were established on the assumption that rare variants may more likely affect the gene/protein function than polymorphic variants (Bannwarth et al. 2013). In this context, we used the Macro Haplogroup Consensus Sequences to move the fixed evolutionary variants to the background and highlight rare ones. These rare variants may be characterized by nucleotide variability values below the established cutoff, while their potential functional impact, particularly for non-synonymous variants, may be assessed from the disease score definition. Variants mapping on the non protein-coding regions may be also prioritized by taking into account the filtering against the three references and the nucleotide variability cutoff (NVC) only. Pathogenicity data regarding tRNAs and rRNAs variants will be soon integrated in the proposed pipeline.
The application of the NVC and DST on the dataset of all possible non-synonymous variants (patho_table, https://sourceforge.net/projects/mtoolbox/) suggested that the loci MT-ATP6, MT-ATP8 and MT-CYB harbor the variants featuring the highest nucleotide variability values and number of sites. Accordingly, these genes seem to be the least conserved regions of the human mtDNA and the most prone to harbor non-synonymous variants (Supplementary Fig. 4a), as previously reported in Mitomap. On the other hand, the distribution of disease scores related to all the potentially pathogenic variants occurring in the thirteen protein-coding regions did not show any gene-specific peculiarity (Supplementary Fig. 4b).
We applied the prioritization workflow to LHON, COAD and ovarian cancer sample sets to validate the robustness of our approach.
The majority of the causative mutations expected (8 out of 11 LHON-causing mutations) on the LHON dataset was recognized as affecting function by the application of the whole stepwise prioritization workflow with the exception of the mutations m.3700G>A (MT-ND1), m.14502T>C (MT-ND6) and m.14484T>C (MT-ND6) (Supplementary Table 1). These mutations were kept out by the application of chosen variability and/or disease score thresholds but were retained by the other filtering steps. Specifically, the mutation m.14484T>C was found in healthy individuals (0.11 % of total mtDNA genomes stored in HmtDB) despite the high DS suggestive of its potential functional impact. Such mutation was formerly considered a non-pathogenic variant, resulting in a conservative change into an amino acid with similar physiochemical properties (p.M64 V) (Mackey and Howell 1992) and found in population surveys without expressing any pathological phenotype (Achilli et al. 2012). However, it was previously assumed that it may exert a pathogenic potential if found in association with the haplogroups J and I (Achilli et al. 2012). 12 % of LHON-derived sequences here analyzed harbored this mutation; among these, only two belong to haplogroup J1 whereas most of the genomes belong to the macro-haplogroup M (26 % of total). The mutation m.3700G>A, although absent from the healthy population and for this reason more likely to have a functional impact, was suggested to be benign by the disease score. It may be due to half of pathogenicity predictors used to calculate the disease score, suggesting this mutation as benign (Supplementary Table 1—AllVariants sheet). This points out that pathogenicity predictions should always be treated with caution and that functional validation of variants is of paramount importance to clarify their role in contributing to phenotype, especially in the context of specific haplogroups and upon taking into account that penetrance is a cogent clinical issue particularly in canonical mitochondriopathies such as LHON. The mutation m.14502T>C, excluded since predicted as benign and found in the healthy population, is reported in the literature in sporadic LHON cases and/or in combination with other primary LHON mutations (Zhao et al. 2009; Zhang et al. 2010). In addition, our study suggested a pool of new variants not previously associated with LHON warranting further investigation with the aim to recognize novel causative mutations which could be added in the list of “top 14” LHON-specific mutations (Brandon et al. 2005). Finally, two additional variants were prioritized removing the haplogroup-filtering step. This result does not imply that our prioritization process may give false negatives because the output of the pipeline reports the entire list of variants with the prioritized ones at the top. The user is free to filter the variants according to his preferred criteria. The system simply suggests and reports the thresholds that may facilitate the selection of functional important variants.
With respect to cancer, it needs to be underlined that our workflow may provide an additional advantage, namely the efficient selection of potentially affecting function variants that are candidates for being somatic that only transformed cells may withstand, in the context of a deregulated cell metabolism. For too long, associations between cancer types and mtDNA variants have been reported without verifying whether they were somatic or germline, in which latter case they may be speculated to be predisposing to transformation. Even further, many polymorphisms have been classified in the past years as cancer-associated mtDNA mutations (Máximo and Sobrinho-Simões 2000; Setiawan et al. 2008), a risky statement likely driven by the lack of well-curated databases and of a commonly agreed protocol to define such associations. The high frequency of mtDNA variants that is often detected in cancer samples may also be a great obstacle to select few candidates to bring forward to functional studies, with the aim to assess in which way they may impact metabolism. This is a key step that this workflow intends to simplify, as we have shown in both COAD and ovarian cancer sample sets by verifying the somatic nature of prioritized variants. However, we have here shown that our criteria are not too stringent, as they allow inclusion of a few variants that were not found to be somatic. Although this finding may somewhat decrease the performing index of our workflow in highlighting specifically cancer-associated mutation, it may on the other hand permit to select variants that may still affect the gene/protein function, even though not associated to the patient’s disease.
In this paper, we successfully demonstrate that the prioritization of human mtDNA variants based on the workflow here proposed is able to recognize potential affecting function variants. Compared to the existing pipelines, capable of annotating mitochondrial variations from next-generation datasets exclusively (mit-o-matic, Vellarikkal et al. 2015), or extract information from lists of variants or FASTA sequences exclusively (MtSNPscore, Bhardwaj et al. 2009), our prioritization workflow, currently being implemented in MToolBox, appears to be more flexible allowing a functional annotation of variants on large datasets from both next-generation (in BAM, SAM, FASTQ format) and Sanger sequencing (in FASTA format) data.
Finally, the recent publication of MitImpact (Castellana et al. 2015) reports a database of the entire dataset of non-synonymous human mtDNA variants annotated with nucleotide variability available through the HmtDB resource and pathogenicity scores estimated by applying the same predictors implemented in MToolBox, with three additional ones. The estimation of the level of concordance among predictions is provided through two alternative “uniformity scores”, similar to our percentage of concordance, previously implemented in MToolBox and now replaced by the disease scores here reported. The different type of inputs required by mit-o-matic, MitImpact and MtSNPscore did not allow to report here the results of a quantitative comparison with MToolBox. Those results may be roughly comparable according to predicted pathogenicity of the considered variants.
In conclusion, our prioritization workflow based on (1) the use of MHCSs together with RSRS and rCRS, to filter out variants fixed during evolution; (2) the availability of the NV scores obtained through the HmtDB annotated genomes and (3) the implementation of the NVC and DST, reducing the large amount of variants from both NGS and conventional Sanger technologies, offers one of the most complete tools to guide clinicians in selecting the non-synonymous mtDNA variants with a potential functional impact. However, functional assays are strongly required to confirm the pathogenicity of all mtDNA variants prioritized by this workflow, as well as by any automated method, with the aim to establish their exact role and involvement in disease phenotypes.
References
Abu-Amero KK, Bosley TM (2006) Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Invest Ophthalmol Vis Sci 47:4211–4220
Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, La Morgia C, Valentino ML, Liguori R, Pizza F, Barboni P, Sadun F et al (2012) Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber’s hereditary optic neuropathy. PLoS One 7:e42242
Adeghate E, Donáth T, Adem A (2013) Alzheimer disease and diabetes mellitus: do they have anything in common? Curr Alzheimer Res 10:609–617
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, Chapter 7:Unit7.20. doi:10.1002/0471142905.hg0720s76
Alonso A, Martin P, Albarran C, Aquilera B, Garcia O, Guzman A, Oliva H, Sancho M (1997) Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and single-strand conformation analysis. Electrophoresis 18:682–685
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147
Balter M (2011) Was North Africa the launch pad for modern human migrations? Science 331:20–23
Bannwarth S, Procaccio V, Lebre AS, Jardel C, Chaussenot A, Hoarau C, Maoulida H, Charrier N, Gai X, Xie HM, Ferre M, Fragaki K et al (2013) Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet 50:704–714
Behar DM, van Oven M, Rosset S, Metspalu M, Loogväli E-L, Silva NM, Kivisild T, Torroni A, Villems R (2012) A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet 90:675–684
Benaglia T, Chauveau D, Hunter DR, Young D (2009) Mixtools: an R package for analyzing finite mixture models. J Stat Softw 32(6):1–29
Bhardwaj A, Mukerji M, Sharma S, Paul J, Gokhale CS, Srivastava AK, Tiwari S (2009) MtSNPscore: a combined evidence approach for assessing cumulative impact of mitochondrial variations in disease. BMC Bioinform 10(Suppl 8):S7
Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC (2005) MITOMAP: a human mitochondrial genome database—2004 update. Nucleic Acids Res 33:D611–D613
Brandon M, Baldi P, Wallace DC (2006) Mitochondrial mutations in cancer. Oncogene 25:4647–4662
Brown MD, Torroni A, Reckord CL, Wallace DC (1995) Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum Mutat 6:311–325
Calabrese C, Simone D, Diroma MA, Santorsola M, Guttà C, Gasparre G, Picardi E, Pesole G, Attimonelli M (2014) MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinform Oxf Engl 30:3115–3117
Capriotti E, Calabrese R, Fariselli P, Martelli PL, Altman RB, Casadio R (2013) WS-SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics 14(Suppl 3):S6
Castellana S, Rónai J, Mazza T (2015) MitImpact: an exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum Mutat 36:E2413–E2422
Chinnery PF, Hudson G (2013) Mitochondrial genetics. Br Med Bull 106:135–159
Chinnery PF, Howell N, Lightowlers RN, Turnbull DM (1997) Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain J. Neurol 120(Pt 10):1713–1721
Chinnery PF, Brown DT, Andrews RM, Singh-Kler R, Riordan-Eva P, Lindley J, Applegarth DA, Turnbull DM, Howell N (2001) The mitochondrial ND6 gene is a hot spot for mutations that cause Leber’s hereditary optic neuropathy. Brain J Neurol. 124:209–218
Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero-Jimenez P, Thilly WG (2001) High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet 28:147–150
Diroma MA, Calabrese C, Simone D, Santorsola M, Calabrese FM, Gasparre G, Attimonelli M (2014) Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data. BMC Genomics 15(Suppl 3):S2
Falk MJ, Shen L, Gonzalez M, Leipzig J, Lott MT, Stassen APM, Diroma MA, Navarro-Gomez D, Yeske P, Bai R, Boles RG, Brilhante V et al (2014) Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol Genet, Metab
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fraser JA, Biousse V, Newman NJ (2010) The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol 55:299–334
Frueh FW, Noyer-Weidner M (2003) The use of denaturing high-performance liquid chromatography (DHPLC) for the analysis of genetic variations: impact for diagnostics and pharmacogenetics. Clin Chem Lab Med CCLM FESCC 41:452–461
Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, Iommarini L, Ghelli A, Moretti M, Betts CM, Martinelli GN, Ceroni AR, Curcio F et al (2007) Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci USA 104:9001–9006
Gasparre G, Kurelac I, Capristo M, Iommarini L, Ghelli A, Ceccarelli C, Nicoletti G, Nanni P, De Giovanni C, Scotlandi K, Betts CM, Carelli V, Lollini PL, Romeo G, Rugolo M, Porcelli AM (2011) A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res 71(19):6220–6229
Ghelli A, Porcelli AM, Zanna C, Vidoni S, Mattioli S, Barbieri A, Iommarini L, Pala M, Achilli A, Torroni A, Rugolo M, Carelli V (2009) The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS One 4:e7922
Gropman A, Chen T-J, Perng C-L, Krasnewich D, Chernoff E, Tifft C, Wong L-JC (2004) Variable clinical manifestation of homoplasmic G14459A mitochondrial DNA mutation. Am J Med Genet A 124A:377–382
Hall BG (2013) Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol 30:1229–1235
He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N (2010) Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 464:610–614
Howell N, Bindoff LA, McCullough DA, Kubacka I, Poulton J, Mackey D, Taylor L, Turnbull DM (1991) Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am J Hum Genet 49:939–950
Iommarini L, Kurelac I, Capristo M, Calvaruso MA, Giorgio V, Bergamini C, Ghelli A, Nanni P, De Giovanni C, Carelli V, Fato R, Lollini PL et al (2014) Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet 23:1453–1466
Jandova J, Shi M, Norman KG, Stricklin GP, Sligh JE (2012) Somatic alterations in mitochondrial DNA produce changes in cell growth and metabolism supporting a tumorigenic phenotype. Biochim Biophys Acta 1822:293–300
Johns DR, Neufeld MJ, Park RD (1992) An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun 187:1551–1557
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism. Academic Press, New York, pp 21–132
Keogh M, Chinnery PF (2013) Hereditary mtDNA heteroplasmy: a baseline for aging? Cell Metab 18:463–464
Khan NA, Govindaraj P, Jyothi V, Meena AK, Thangaraj K (2013) Co-occurrence of m.1555A>G and m.11778G>A mitochondrial DNA mutations in two Indian families with strikingly different clinical penetrance of Leber hereditary optic neuropathy. Mol Vis 19:1282–1289
Kim JY, Hwang J-M, Park SS (2002) Mitochondrial DNA C4171A/ND1 is a novel primary causative mutation of Leber’s hereditary optic neuropathy with a good prognosis. Ann Neurol 51:630–634
Kurelac I, Lang M, Zuntini R, Calabrese C, Simone D, Vicario S, Santamaria M, Attimonelli M, Romeo G, Gasparre G (2012) Searching for a needle in the haystack: comparing six methods to evaluate heteroplasmy in difficult sequence context. Biotechnol Adv 30:363–371
Kurelac I, MacKay A, Lambros MBK, Di Cesare E, Cenacchi G, Ceccarelli C, Morra I, Melcarne A, Morandi L, Calabrese FM, Attimonelli M, Tallini G et al (2013) Somatic complex I disruptive mitochondrial DNA mutations are modifiers of tumorigenesis that correlate with low genomic instability in pituitary adenomas. Hum Mol Genet 22:226–238
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinform Oxf Engl 25:2744–2750
Lièvre A, Chapusot C, Bouvier A-M, Zinzindohoué F, Piard F, Roignot P, Arnould L, Beaune P, Faivre J, Laurent-Puig P (2005) Clinical value of mitochondrial mutations in colorectal cancer. J Clin Oncol Off J Am Soc Clin Oncol 23:3517–3525
Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, Wallace DC (2013) mtDNA variation and analysis using MITOMAP and MITOMASTER. Curr Protoc Bioinformatics 1(123):1.23.1–1.23.26
Mackey D, Howell N (1992) A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology. Am J Hum Genet 51:1218–1228
Mancini C, Roncaglia P, Brussino A, Stevanin G, Lo Buono N, Krmac H, Maltecca F, Gazzano E, Bartoletti Stella A, Calvaruso MA, Iommarini L, Cagnoli C et al (2013) Genome-wide expression profiling and functional characterization of SCA28 lymphoblastoid cell lines reveal impairment in cell growth and activation of apoptotic pathways. BMC Med Genomics 6:22
Máximo V, Sobrinho-Simões M (2000) Hürthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Arch Int J Pathol 437:107–115
McCormick E, Place E, Falk MJ (2013) Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics 10(2):251–261. doi:10.1007/s13311-012-0174-1
McLachlan P (2000) Finite mixture models. Wiley, New York
Mercader JM, Puiggros M, Segrè AV, Planet E, Sorianello E, Sebastian D, Rodriguez-Cuenca S, Ribas V, Bonàs-Guarch S, Draghici S, Yang C, Mora S et al (2012) Identification of novel type 2 diabetes candidate genes involved in the crosstalk between the mitochondrial and the insulin signaling systems. PLoS Genet 8:e1003046
Patti M-E, Corvera S (2010) The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 31:364–395
Payne BAI, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M, Chinnery PF (2013) Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet 22:384–390
Peng GH, Zheng BJ, Fang F, Wu Y, Liang LZ, Zheng J, Nan BY, Yu X, Tang XW, Zhu Y, Lu JX, Chen BB, Guan MX (2013) Mitochondrial 12S rRNA A1555G mutation associated with nonsyndromic hearing loss in twenty-five Han Chinese pedigrees. Yi Chuan 35(1):62–72
Pereira L, Soares P, Radivojac P, Li B, Samuels DC (2011) Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am J Hum Genet 88:433–439
Pereira L, Soares P, Máximo V, Samuels DC (2012) Somatic mitochondrial DNA mutations in cancer escape purifying selection and high pathogenicity mutations lead to the oncocytic phenotype: pathogenicity analysis of reported somatic mtDNA mutations in tumors. BMC Cancer 12:53
Pesole G, Saccone C (2001) A novel method for estimating substitution rate variation among sites in a large dataset of homologous DNA sequences. Genetics 157(2):859–865
Pesole G, Allen JF, Lane N, Martin W, Rand DM, Schatz G, Saccone C (2012) The neglected genome. EMBO Rep 13:473–474
Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B (1998) Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet 20:291–293
Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, Pennisi LF, Morra I, Ciccarelli E, Melcarne A, Bartoletti-Stella A, Salfi N et al (2010) The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet 19:1019–1032
Pütz J, Dupuis B, Sissler M, Florentz C (2007) Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 13(8):1184–1190
Reynier P, Penisson-Besnier I, Moreau C, Savagner F, Vielle B, Emile J, Dubas F, Malthièry Y (1999) mtDNA haplogroup J: a contributing factor of optic neuritis. Eur J Hum Genet EJHG 7:404–406
Rubino F, Piredda R, Calabrese FM, Simone D, Lang M, Calabrese C, Petruzzella V, Tommaseo-Ponzetta M, Gasparre G, Attimonelli M (2012) HmtDB, a genomic resource for mitochondrion-based human variability studies. Nucleic Acids Res 40:D1150–D1159
Schon EA, DiMauro S, Hirano M (2012) Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 13:878–890
Setiawan VW, Chu L-H, John EM, Ding YC, Ingles SA, Bernstein L, Press MF, Ursin G, Haiman CA, Neuhausen SL (2008) Mitochondrial DNA G10398A variant is not associated with breast cancer in African-American women. Cancer Genet Cytogenet 181:16–19
Simone D, Calabrese FM, Lang M, Gasparre G, Attimonelli M (2011) The reference human nuclear mitochondrial sequences compilation validated and implemented on the UCSC genome browser. BMC Genom 12:517
Skonieczna K, Malyarchuk BA, Grzybowski T (2012) The landscape of mitochondrial DNA variation in human colorectal cancer on the background of phylogenetic knowledge. Biochim Biophys Acta 1825:153–159
Thomas PD, Kejariwal A (2004) Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci USA. 101:15398–15403
Valentino ML, Barboni P, Ghelli A, Bucchi L, Rengo C, Achilli A, Torroni A, Lugaresi A, Lodi R, Barbiroli B, Dotti M, Federico A et al (2004) The ND1 gene of complex I is a mutational hot spot for Leber’s hereditary optic neuropathy. Ann Neurol. 56:631–641
van Oven M, Kayser M (2009) Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30:E386–E394
Vellarikkal SK, Dhiman H, Joshi K, Hasija Y, Sivasubbu S, Scaria V (2015) mit-o-matic: a comprehensive computational pipeline for clinical evaluation of mitochondrial variations from next-generation sequencing datasets. Hum Mutat 36:419–424
Verschoor ML, Ungard R, Harbottle A, Jakupciak JP, Parr RL, Singh G (2013) Mitochondria and cancer: past, present, and future. BioMed Res Int 2013:612369
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242:1427–1430
Wallace DC, Ruiz-Pesini E, Mishmar D (2003) mtDNA variation, climatic adaptation, degenerative diseases, and longevity. Cold Spring Harb Symp Quant Biol 68:479–486
Wang C-Y, Li H, Hao X-D, Liu J, Wang J-X, Wang W-Z, Kong Q-P, Zhang Y-P (2011) Uncovering the profile of somatic mtDNA mutations in Chinese colorectal cancer patients. PLoS One 6:e21613
Watson E, Forster P, Richards M, Bandelt HJ (1997) Mitochondrial footprints of human expansions in Africa. Am J Hum Genet 61:691–704
White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, DiMauro S, Dahl HH, Thorburn DR (1999) Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet 65:474–482
Zhang J, Zhou X, Zhou J, Li C, Zhao F, Wang Y, Meng Y, Wang J, Yuan M, Cai W, Tong Y, Sun Y-H et al (2010) Mitochondrial ND6 T14502C variant may modulate the phenotypic expression of LHON-associated G11778A mutation in four Chinese families. Biochem Biophys Res Commun 399:647–653
Zhang J, Zhao F, Fu Q, Liang M, Tong Y, Liu X, Lin B, Mi H, Zhang M, Wei Q-P, Xue L, Jiang P et al (2013) Mitochondrial haplotypes may modulate the phenotypic manifestation of the LHON-associated m.14484T>C (MT-ND6) mutation in Chinese families. Mitochondrion 13:772–781
Zhao F, Guan M, Zhou X, Yuan M, Liang M, Liu Q, Liu Y, Zhang Y, Yang L, Tong Y, Wei Q-P, Sun Y-H et al (2009) Leber’s hereditary optic neuropathy is associated with mitochondrial ND6 T14502C mutation. Biochem Biophys Res Commun 389:466–472
Acknowledgments
The results shown in this work are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) Grant No. IG14242 and EU FP7 Marie Curie project MEET-317433 to GG. The computational work has been executed on the IT resources made available by ReCaS, a project financed by the MIUR (Italian Ministry for Education, University and Research) in the “PON Ricerca e Competitività 2007–2013—Azione I—Interventi di rafforzamento strutturale” PONa3_00052, Avviso 254/Ric. We thank Dr. I. Kurelac for technical support in DHPLC experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
G. Gasparre and M. Attimonelli: co-last authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Santorsola, M., Calabrese, C., Girolimetti, G. et al. A multi-parametric workflow for the prioritization of mitochondrial DNA variants of clinical interest. Hum Genet 135, 121–136 (2016). https://doi.org/10.1007/s00439-015-1615-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1615-9