
Abstract
Mutations in the gene encoding the mitochondrial protein high temperature requirement A2 (HTRA2) are inconsistently associated with a risk of Parkinson’s disease (PD). We assessed the presence of HTRA2 mutations among patients with PD and performed functional assay of identified mutations or variants. Among the total 1,373 subjects, the entire HTRA2 coding region was sequenced in 113 early-onset PD (EOPD), 20 familial PD patients and 150 control subjects. An additional 390 sporadic late-onset PD patients and 700 controls were subsequently screened to validate possible mutations found in the first set. We identified two novel heterozygous variants, c.427C > G (Pro143Ala) and c.906 +3 G > A, in 2 (1.5%) EOPD patients. The missense variant, Pro143Ala, was also observed in one late-onset PD patient but was absent in total 850 control subjects (relative risk 2.3, 95% CI 1.5–2.8, P = 0.04). Expressing Pro143Ala variant of HTRA2 in primary dopaminergic neurons causes neurite degeneration. Following exposure to rotenone, the ultra-structural mitochondrial abnormality, the percentage of mitochondrial dysfunction and apoptosis in cells carrying the HTRA2 Pro143Ala variant was significantly higher than wild-type cells. Mechanistically, protein level of phosphorylated HTRA2 was increased in cells carrying the Pro143Ala variant, suggesting Pro143Ala variant promotes HTRA2 phosphorylation with resultant mitochondrial dysfunction. Our results support a biologically relevant role of HTRA2 in PD susceptibility in Taiwanese. Further large-scale association studies are warranted to confirm the role of HTRA2 Pro143Ala variant in the risk of PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders and is characterized by intra-neuronal α-synuclein-positive aggregations (Forno 1996). Mutations in a number of pathogenic genes (e.g., SNCA, Parkin, UCHL1, DJ-1, PINK1, ATP13A2, and LRRK2) have been associated with both familial and sporadic PD (for review, see Lesage and Brice 2009). Both mitochondrial dysfunction and ubiquitin–proteasome system damage have been proposed as possible mechanisms leading to dopaminergic neuronal degeneration (Lin and Beal 2006; Rubinsztein et al. 2006; Malkus et al. 2009). The role of mitochondria in PD was first noticed with the description of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity causing PD in humans, and was later supported by the association of several PD causative genes, including PINK1 and DJ-1, with mitochondrial dysfunction (Langston and Ballard 1983; Bonifati et al. 2003; Valente et al. 2004).
Recent identification of Omi/HTRA2 (MIM# 606441) as a novel PD locus (PARK13) has further supported the role of mitochondrial dysfunction in PD pathogenesis (Strauss et al. 2005). High temperature requirement A2 (HTRA2)-mutant mice present a progressive neurodegenerative phenotype with parkinsonism features (Jones et al. 2003). HTRA2 encodes a 50-kDa serine protease that localizes to the mitochondrial intermembrane space (IMS) and protects cells from apoptotic stimuli by either preventing the accumulation of damaged protein in the mitochondria directly or exposing an inhibitor of apoptosis protein (IAP) binding motif to antagonize the interaction with cytosolic IAP-caspase (Weibezahn et al. 2004). A subsequent genetic study of PD patients found that a heterozygous mutation of HTRA2, Arg404Trp, is associated with the development of PD (Bogaerts et al. 2008). However, further study of a North American case–control series detected the same potential HTRA2 mutation in healthy control subjects (Simon-Sanchez and Singleton 2008). Another mutation screen performed in German PD patients showed an association between the Ala141Ser polymorphism near the protease domain and the risk of PD but no association for the G399S variant (Strauss et al. 2005). Nevertheless, one large-scale association study revealed no association of five known HTRA2 polymorphisms with PD in mixed populations, including a small sample size of Taiwanese patients (Krüger et al. 2009). However, one recent study conducted in the Chinese population showed an association of one intronic variant with risk of PD (Wang et al. 2011). These inconsistent findings raise a question about the role of mitochondrial HTRA2 in PD susceptibility. Furthermore, to date, the functional evidence for possible HTRA2 risk variants in neuronal toxicity is still unclear.
To elucidate the contribution of HTRA2 in PD pathogenesis, we investigated HTRA2 mutations in PD patients of Taiwanese origin. Our group has previously performed a comprehensive analysis of mutations in multiple candidate genes in a cohort of early-onset PD (EOPD) patients of Taiwanese origin (Lockhart et al. 2004; Wu et al. 2005; Lin et al. 2008a, b ; Lee et al. 2009). However, the major genetic causes in the majority of EOPD patients in our population are still unclear. The recent identification of HTRA2 polymorphisms as a risk factor in PD patients suggests that rare genetic variants may play a role in some populations. Although the disease onset age of originally reported PD patients with potential HTRA2 mutations were not totally early-onset (range 49–77 years, mean 57.3 years, Strauss et al. 2005), we sequenced the complete HTRA2 coding region in a cohort of 133 Taiwanese patients with EOPD or familial PD and 150 age- and gender-matched controls in the first study set. We then confirmed the relevance of the identified risk substitutions in a large-scale case series of late-onset PD patients. We also conducted in vitro functional assays to examine the potential toxicity of identified HTRA2 variants on mitochondria in neurons.
Materials and methods
Subjects
A total of 1,373 subjects were included in this study: 523 PD patients (113 early-onset PD (EOPD) patients, 20 familial PD (FPD) patients, and 390 sporadic late-onset PD patients and 850 control subjects who exhibited no evidence of PD. All PD patients were recruited from the Movement Disorder Clinic of the National Taiwan University Hospital, a tertiary referral center in Taiwan. Of the 133 probands enrolled in the first set of the study, 113 were sporadic EOPD patients (onset before 50 years of age) and the remaining 20 were FPD patients with a family history of the disease (at least one affected first- and/or second-degree relatives with parkinsonism). None of the subjects were from consanguineous families. The majority of the EOPD and FPD patients were ever screened for mutations of Parkin, PINK1, DJ-1 and ATP13A2 (Lockhart et al. 2004; Wu et al. 2005; Lin et al. 2008a, b; Lee et al. 2009). All patients fulfilled the diagnostic criteria for PD (Gelb et al. 1999). The patients received standard neurological examinations, including the Unified Parkinson’s Disease Rating Scale (UPDRS) and Mini-Mental Status Evaluation (MMSE) (Folstein et al. 1975). Relatives of the probands carrying the HTRA2 Pro143Ala substitution were also subjected to the University of Pennsylvania Smell Identification Test (UPSIT) (Doty et al. 1984). Among the 850 control subjects, 500 controls are healthy spouses or caregivers of PD patients without neurological signs and the other 350 control subjects were end stage renal disease patients under regular dialysis without any symptoms or signs of PD. Informed consent was obtained from all participants. The study was approved by the institutional ethics board committees.
HTRA2 screening in PD and control subjects
Complete sequencing of HTRA2 was carried out in 133 EOPD and familial PD patients and 150 age-, gender-, and ethnicity-matched controls. To further clarify the association of the novel HTRA2 substitution identified in the first part of the screening, the second part of the study was performed using an additional 390 independent sporadic OPD patients and 700 control subjects. Genomic DNA was extracted from venous blood using standard protocols. The eight exons and intron–exon boundaries of HTRA2 were amplified using polymerase chain reaction (PCR) and sequenced by ABI 3730 (Applied Biosystems). The primer sequences and PCR conditions were as previously described (Bogaerts et al. 2008). Base variants were labeled from the ATG start of protein translation (NM_013247.4).
Relative quantification of HTRA2 gene expression and RT-PCR analysis in subjects with HTRA2 c.906 +3 G > A variation
To clarify whether the potential pathogenic variation in the exon–intron boundary junction, c.906 +3 G > A, disrupts splicing efficacy of HTRA2, we performed quantitative real-time PCR (q-PCR) and reverse transcription PCR (RT-PCR) analyses to check the gene expression of HTRA2 in one of our EOPD patient, labeled as Y63, and seven age-/gender-matched control subjects. Total RNA was extracted from human whole blood using the QIAamp® RNA Blood Mini Kit (Qiagen, USA) and reverse transcription was performed using the Reverse Transcription System Kit (Promega, USA) according to the manufacturer’s instructions. For q-PCR analysis, the sequences of the primer pairs were: HTRA2, forward 5′- GTGGCAGACATCGCAACGCT -3′, reverse 5′- ACCCACTGATTCCGGAGGAG - 3′; hGAPDH, forward 5′- ATGGGTGTGAACCATGAGAAG - 3′, reverse 5′- AGTTGTCATGGATGACCTTGG - 3′. Thermal cycler conditions were 30 s at 95°C followed by 40 cycles of 5 s at 95°C to denature the DNA, 10 s at 55°C to anneal, and 15 s at 60°C to extend the template. All reactions were performed in triplicate. The values obtained for the expression of the target gene were normalized to GAPDH and quantified relative to the expression in control samples. For the relative quantification, the \( 2^{{ - \Updelta \Updelta C_{\text{T}} }} \) formula was used, where −∆∆C T = (C T, target − C T, GAPDH) experimental sample − (C T, target − C T, GAPDH) control sample. For RT-PCR analysis, the sequences of the primer pairs were: HTRA2, forward 5′-GTGGCAGACATCGCAACGCT-3′, reverse 5′-ACCCACTGATTCCGGAGGAG-3′; hGAPDH, forward 5′-ATGGGTGTGAACCATGAGAAG-3′, reverse 5′-AGTTGTCATGGATGACCTTGG-3′. A 20 μl PCR contained 50 ng cDNA, 0.25 μM of each primer and 10 μl 2× GoTaq Green Master Mix (Promega). Thermal cycler parameters were 35 cycles at 95°C for 30 s, annealing at 55°C for 30 s, and 72°C for 35 s, with preheat at 95°C for 2 min 30 s and last extension at 72°C for 7 min. The reaction was performed in triplicate.
In silico modeling of the crystal structure of HTRA2 with and without the Pro143Ala variation
The crystal structure of Omi/HTRA2 (1LCY), from which the N-terminal 133 residues were removed, was obtained from the RSCB protein data bank. The mutated model was generated using DS/Build Mutant Mutants (Discovery Studio 2.1, Accelrys Software, San Diego). The three-dimensional structural model was constructed based on the structure of 1LCY. Residue 143 was selected and assigned to be the Ala residue, and the loop region of residues 139–149 was optimized using the MODELER program. The model was calculated by default parameters, and the non-bonded Melo’s potential was adapted for loop refinement.
In vitro Functional assay of HTRA2 Pro143Ala variation
Construction of HTRA2 expression plasmids and transfection in neuronal cells
To explore the effects of potential pathogenic variation of HTRA2, Pro143Ala, in the cells, we constructed both wild-type and variant HTRA2 plasmids and performed a series of functional assay in transfected neurons. Human HTRA2 cDNA was constructed from venous blood of healthy controls and cloned into the pCR®8/GW/TOPO® vector (Invitrogen) using the primers 5′-ACTTCTGAAGGACTTCAGGTACCGGCGTGC-3′ and 5′-GAAAGGCAAGGAGGAAATCAGAGCAGGAGCC-3′. The c.427C > G (Pro143Ala) variant of HTRA2 was generated by PCR-mediated site-directed mutagenesis (Stratagene). The integrity of the construct was confirmed by direct DNA sequencing. The wild-type and variant HTRA2 were then sub-cloned into the pcDNA-DEST47 vector (Invitrogen) in-frame with the eGFP coding sequences.
The transient transfection of HTRA2 plasmid was performed in both human dopaminergic neuroblastoma cell line (SH-SY5Y) and rat primary dopaminergic neuronal culture. SH-SY5Y cell line was maintained at 37°C in a humidified 95% air/5% CO2 incubator and grown on polyornithine-precoated 6-well plates in MEM/F12 medium (Invitrogen) containing 10% fetal bovine serum and 2 mM l-glutamine. Embryonic day 14 (E14) mesencephalic cells were obtained from rat fetuses and were incubated in Ca2+, Mg2+-free HBSS containing papin at 37°C for 30 min. The dissociated cells were then fed with Neurobasal media containing B27 supplement. Both types of neurons were transfected overnight with pcDNA/GW-47/CAT vector and different HTRA2 plasmids using FuGENE HD (Roche Applied Science) according to the manufacturer’s instructions. These transfected cells were then used for the following experiments.
Localization of HTRA2 protein and quantification of neurite outgrowth in primary dopaminergic neuron culture
To elucidate whether the variant HTRA2 changes the localization of the encoded protein in mitochondria, we used immunohistochemistry assay to recognize the localization of HTRA2 protein in both SH-SY5Y and rat primary dopaminergic neurons. The transfected cells were stained with 500 nM MitoTracker Red CMXRos (Molecular Probes) according to the manufacturer’s instructions. Cells were also co-stained with nucleus marker, DAPI (0.1 μg/ml), for 30 min and then mounted with CC/Mount™ Aqueous Mounting Medium (Sigma). These cells were observed by confocal laser scanning microscopy (LSM510 Meta, Zeiss).
Given that change of neurite morphology is often the preceding feature of neuron damage (Takeuchi et al. 2005), we next examined the effect of HTRA2 Pro143Ala variant on neurite arborization in rat primary dopaminergic neurons. For the quantification of neurite outgrowth in neurons, at least 17 confocal images of tyrosine hydroxylase (TH)-positive cells per condition were taken at 40× magnification. The measurement of neurite length was carried out using the NIH freeware Image-J starting where the neurite emerges from the cell body and tracing its length out to the distal tip.
Flow cytometric analysis of mitochondrial membrane potential and apoptosis induced by rotenone
To investigate the effect of HTRA2 Pro143Ala variant on mitochondrial function, we investigate the change of mitochondrial membrane potential (MMP) in SH-SY5Y cells transfected with the wild-type or Pro143Ala variant HTRA2 by flow cytometric analysis using the probe 3,3’-dihexyloxacarbocyanine iodide (DiOC6(3) (Invitrogen). Furthermore, the transfected SH-SY5Y neurons were treated with rotenone, a mitochondria complex I inhibitor, at 50, 100, and 300 nM to test the vulnerability of cells to this specific mitochondrial neurotoxin. The percentage of cell apoptosis was assessed by flow cytometry using the annexin V (AV)-FITC/propidium iodine (PI) apoptosis detection kit (Strong Biotech Co.) as previously described (Lin et al. 2008a, b).
Transmission electron microscopy (TEM) imaging of mitochondrial morphology
The ultra-structure of SH-SY5Y cells was obtained using transmission electron microscopy. Cells were briefly rinsed in 1× phosphate-buffered saline, pH 7.4, and then fixed in 2.5% glutaraldehyde in the same buffer overnight at 4°C. They were then rinsed again and post-fixed with 1% osmium tetroxide in buffer, for 30 min at room temperature. After rinsing with buffer, slides were dehydrated in ethanol. Cells were then infiltrated with graded mixtures of propylene oxide/Spurr’s resin, and embedded in the same resin, allowing specimens to polymerize at 70°C for 8 h. Blocks were cut by a Reichert Ultracut S ultramicrotome, thus obtaining ultrathin sections (70 nm) which were collected on nickel grids. Grids were stained with uranyl acetate and then followed by staining with lead citrate. Sections were examined in a Hitachi H7100 electron microscope.
Immunoprecipitation assay of phospho-HTRA2 protein
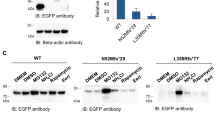
It is known that the phosphorylation status of HTRA2 protein correlates with its anti-apoptotic activity (Plun-Favreau et al. 2007). It is therefore important to examine whether Pro143Ala substitution affects the phosphorylation level of HTRA2 protein. We thus examine the phospho-HTRA2 protein level using SPM101 antibody (Gene Tex) that specifically recognizes phosphorylated serine/threonine/tyrosine sites of protein to perform immunoprecipitation (IP) assay in SH-SY5Y cell lysate transiently expressing either wild-type or Pro143Ala variant HTRA2. The ectopic HTRA2 protein in the IP effluent was analyzed by western blot using anti-eGFP antibody.
Statistical analysis
All experiments were repeated at least three times. The data were expressed as mean ± standard error (SE) in the functional assay. Categorical variables were analyzed using the Chi-square test. Numerical data were analyzed using student t test, and non-parametric Mann–Whitney test. Significance was defined as P < 0.05.
Results
For the 133 index patients in the first part of the screening study, the mean age at symptom onset of 113 EOPD patients was 40.6 ± 8.2 years (range 18–49 years) and the age at the time of sampling was 50.2 ± 7.9 years (range 20–61 years). The mean age at symptom onset of 20 FPD patients was 54.3 ± 7.8 years (range 48–66 years) and the age at the time of sampling was 59.2 ± 8.9 years (range 54–77 years). The sample consisted of 65 men and 68 women. For the 390 late-onset PD patients in the second part of the study, the mean age at onset was 56.8 ± 6.4 years, the age at the time of sampling was 61.7 ± 9.1 years, and 51.7% of the patients were men.
We identified two novel heterozygous variants among the 133 index patients in the first screening set. One variant was c.427C > G (Pro143Ala) in exon 1, which was found in an EOPD patient (labeled as Y121) with an onset age of 42 years, and the other variant was c.906 +3 G > A in the splicing junction between exon 3 and exon 4, which was found in another EOPD patient (labeled as Y63) with an onset age of 39 years (Fig. 1a). In the family of Y121, two siblings carried the same heterozygous variant (Supplementary Fig. 1, subjects II:2 and II:10). They did not show motor symptoms of parkinsonism, suggesting that HTRA2 Pro143Ala variant did not obviously segregate with PD in Y121 family. However, the UPSIT score of smelling test in one carrier who was 71 years old (Supplementary Fig. 1, subjects II:2) was markedly decreased (9/40) indicating severe anosmia, implying a premotor stage of PD. These two carriers should be closely followed up for motor symptoms of PD. Of these two substitutions, Pro143Ala was also observed in one late-onset PD patient from the second screening study of 390 sporadic PD cases, but the variant was absent in total 850 matched controls. The frequency of this novel substitution was higher in PD patients compared to controls (relative risk 2.3, 95% CI 1.5–2.8, P = 0.04). We did not screen the intronic variant, c.906 + 3G > A, in the second part of screening. Given that PINK1 has been found to regulate HTRA2 (Plun-Favreau et al. 2007), we also sequenced the entire PINK1 coding region and intron–exon boundaries in 2 probands and 2 non-affected carriers of family Y121 with HTRA2 Pro143Ala variation, and one proband with HTRA2 c.906 +3 G > A substitution. In addition, we checked the reported SNCA mutations, such as A30P and A53T, LRRK2 mutations, such as R1441C/G/H and G2019S, and Parkin mutations in the aforementioned 5 carriers with HTRA2 variants. No one has PINK1, Parkin and SNCA mutations. The clinical symptoms of the three patients were similar to typical Parkinsonism manifested as unilateral onset of tremor, rigidity, bradykinesia, and good responses to levodopa treatment. There were no atypical presentations, such as dementia, psychiatric symptoms, cerebellar ataxia or motor neuron disease, or rapid disease progression. Specifically, the mean disease duration of these three patients was 10.0 ± 5.3 years and the average Hoehn–Yahr stage was between II and III, similar to the total study cohort (mean disease duration: 9.8 ± 6.3 years, average Hoehn–Yahr stage: between II and III). Additionally, we also observed two intronic variants in our study: rs10779958:A > C and rs2241028:G > A. Both variants correspond to single nucleotide polymorphisms (SNPs) with minor allele frequencies of more than 1% in controls (Supplementary table 1).

HTRA2 substitutions identified in PD patients. a Forward sequence chromatograms obtained by direct sequencing of the HTRA2 genomic sequence. The positions of the potential variations identified in this study are indicated. Coding DNA is numbered relative to coding DNA reference sequence NM_013247.4 and starts at translation initiation site. Protein is numbered relative to largest HTRA2 isoform NP_037379.1. b The mutated residue, 143, is found between the IAP-binding motif (IBM) and the serine protease. MLS mitochondrial localizing sequence, TM predicted transmembrane domain found in unprocessed HTRA2. The C-terminal PDZ domain is also indicated. c Conservation of the HTRA2 protein residues targeted by the potential mutations identified in our patients. The closest homologues of the human HTRA2 protein were aligned using the program Vector NTI
The potential pathogenic variation in the exon–intron boundary junction, c.906 +3 G > A, seems to disrupt splicing efficacy (http://www.fruitfly.org). To test the effect of this variant allele on transcriptional activity, we performed a q-PCR analysis. We found that the HTRA2 mRNA expression level in case Y63 was significantly decreased compared to the age- and gender-matched control subjects (Fig. 2a). Specifically, HTRA2 expression was only half that of normal controls (controls: 100 ± 10.2%; PD subject Y63: 55.4 ± 9.7%; P < 0.001 by Mann–Whitney test). Additionally, the RT-PCR analysis showed that the exon 2–exon 7 transcript was significantly decreased in case Y63 as compared to the age-/gender-matched controls (Fig. 2b). This finding suggests that c.906 +3 G > A alters the transcriptional activity of HTRA2.

Quantitative real-time and reverse-transcription PCR analysis of HTRA2 c.906 +3 G > A. a Real-time PCR analysis of the c.906 +3 G > A substitution in patient Y63. HTRA2 cDNA were amplified from the whole venous blood of patient Y63 and seven age-/gender-matched control subjects. The cDNA expression of HTRA2 was shown as percentages relative to those of control subjects, averaged from three independent experiments. Statistical analysis was performed using ANOVA followed by the Mann–Whitney U test. *P < 0.01. b An agarose gel of PCR-amplified cDNA products of HTRA2 exon 2-exon 7 cDNA for proband Y63 and two age/gender matched-controls
The other novel missense substitution, c.427 C > G (Pro143Ala), targets the highly conservative region between the inhibitor of apoptosis protein (IAP)-binding motif (IBM) and the serine protease domain of the HTRA2 protein (Fig. 1b, c). In order to investigate the structural impact of the Pro143Ala variant on the HTRA2 protein, a molecular modeling analysis was performed. Residues 139–149 (PPASARSQYNF) were optimized and ten mutation Pro143Ala models generated. By visually inspecting all the models, the mutation model with the lowest energy was selected. The Pro143Ala variant induces a transformation of the shape of helix α1 compared to HTRA2 (1LCY) (Fig. 3a, red circle indicates the helix α1). Among the 11 optimized residues, the conformations of residues Ser142, Arg144, and Try147 varied significantly, whereas the other residues virtually remained at the same place (Fig. 3b).

Protein modeling of the wild-type and variant HTRA2. a Superposition of HTRA2 (purple, 1LCY) and the mutated model Pro143Ala (cyan). The optimized loop region of residues 139–149 (PPASARSQYNF) is circled in red. b A close-up view of optimized residues 139–149. Pro143 and Ala143 are shown in orange and yellow, respectively. Residues Ser142, Arg144, and Typ147 with large variations in two models are shown in stick form. The electrostatic potential surface maps for HTRA2 (1LCY) (c) and the c.427C > G (Pro143Ala) substitution (d)
To examine the effect of the potential mutant allele on the subcellular localization of HTRA2 protein, we performed immunocytochemical analysis on neuronal cells transfected with eGFP-tagged expression vectors containing either wild-type or Pro143Ala variant HTRA2. Confocal microscopy revealed that the localization of the Pro143Ala variant HTRA2 was primarily cytoplasmic, co-localizing with the mitochondrial markers (MitoTracker), similar to the wild-type protein in both SH-SY5Y cells and rat primary dopaminergic neurons (data not shown). However, expressing Pro143Ala variant HTRA2 in primary dopaminergic neurons causes defects of neurite arborization (Fig. 4a). The mean length of the neurite was significantly shorter in the Pro143Ala variant cells than in the HTRA2 wild-type cells (59.19 ± 4.28 vs. 96.01 ± 5.60, P < 0.01; Fig. 4b). Given that Lewy body deposition is the main pathological hallmark of dopaminergic neurons in PD patients. We performed immunocytochemical analysis to detect Lewy body deposition in the rat primary dopaminergic neurons expressing either wild-type or Pro143ala variant HTRA2 using anti-α-synuclein antibody. We did not detect obvious α-synuclein-positive aggregations in both wild-type and Pro143Ala variant HTRA2-expression neurons (data not shown). Whereas neurite degeneration is an early stage of cell death, we hypothesize that Pro143Ala may cause neuronal death through dysregulation of mitochondrial function.

HTRA2 Pro143Ala variant induced neurite degeneration. a Representative images of the rat primary dopaminergic neurons expressing wild-type or variant HTRA2. Scale bar, 50 μm. b Quantification of the percentage of neurite length relative to vector control in neurons expressing wild-type or variant HTRA2. Averages are the mean ± SEM of neurite length. Significance is compared by ANOVA followed by the Mann–Whitney U test. **P < 0.01
To assess the effect of Pro143Ala variant HTRA2 on the mitochondrial function, we characterized the MMP changes induced by rotenone, a mitochondria complex I inhibitor, in SH-SY5Y cell lines expressing wild-type or Pro143Ala variant HTRA2. Cells were exposed to various concentrations (50, 100 or 300 nM) of rotenone for 48 h. As shown in Fig. 5a, after 300 nM rotenone treatment, the percentage of disrupted MMP is higher in cells transfected with the Pro143Ala variant HTRA2 than in the wild-type cells (3.71 ± 0.12 vs. 1.3 ± 0.27, P < 0.05). This is confirmed by electron microscopy analysis showing that mitochondria of SH-SY5Y cells expressing wild-type HTRA2 have partially lost their cristae, become swollen, contained a dense particle and disrupted after 300 nM rotenone treatment (Fig. 5c). Mitochondria were more severely destroyed in Pro143Ala variant HTRA-transfected SH-SY5Y cells with exposure of 300 nM rotenone (Fig. 5d).

HTRA2 Pro143Ala variant impaired mitochondrial function and caused neuronal death. a Values of the DiOC6(3) ratio for different concentrations of rotenone in cell lines expressing wild-type or variant HTRA2. b Percentage of apoptotic cells (annexin V-positive) before and after treatment with different concentrations of rotenone in cell lines expressing wild-type or variant HTRA2. Values are expressed as mean ± SEM of three independent experiments. Statistical analysis was performed using ANOVA followed by the Mann–Whitney U test, *P < 0.05. TEM images showed ultrastructural mitochondrial damages in SH-SY5Y cells expressing either wild-type (c) or Pro143Ala variant HTRA2 (d) while exposing to 300 nM rotenone. Arrow double layer mitochondria membrane. Arrow head: dense particle. Asterisk: cristae. Magnification, ×50,000
Moreover, the apoptosis was significantly increased in the HTRA2 Pro143Ala variant transfected cells comparing to wild-type transfected cells after 300 nM rotenone treatment (Fig 5b, 42.4 ± 3.9 vs. 23.6 ± 3.8%, P = 0.03).
Given that Pro143Ala is the only residue from the potential phosphorylation site of HTRA2 protein for p38 stress kinase, the upstream kinase for HTRA2, and the phosphorylation status of HTRA2 protein correlates with its anti-apoptotic activity (Plun-Favreau et al. 2007), it is therefore important to examine whether Pro143Ala substitution affects the phosphorylation status of HTRA2 protein. We found that the protein level of phospho-HTRA2 was elevated in Pro143Ala variant-expressing cells, as compared to wild-type HTRA2 or vector control (Fig. 6a, b). These results suggest that Pro143Ala variant, through up-regulating phosphorylation level of HTRA2 protein, may directly or indirectly impair mitochondrial integrity with resulting neuronal death.

HTRA2 Pro143Ala substitution increased protein level of phospho-HTRA2. a Western blot analysis of SH-SY5Y cell lysates expressing empty vector, wild-type or variant HTRA2 to detect expressions of HTRA2 protein (GFP antibody) and actin (upper and lower panels). Immunoprecipitated phosphorylated proteins are shown by blotting with the anti-phosphoserine/threonine/tyrosine antibody, and the coprecipitated HTRA2 is revealed by anti-GFP antibody (middle panel). b Percentages of GFP immuno-intensity of the IP complex of cell lysates expressing wild-type or variant HTRA2 to vector control are averaged from three independent experiments. Values are expressed as mean ± SEM. Statistical analysis was performed using ANOVA followed by the Mann–Whitney U test, *P < 0.05
Discussion
We evaluated the HTRA2 gene in a relatively large series of Taiwanese patients with PD. In the first screening set, we did not find a positive association between previously reported SNPs and PD risk, which is consistent with a recent report of large-scale mixed populations (Krüger et al. 2009). In addition, we did not detect the reported intronic risk variant, IVS5 + 29T > A, in our study population (Wang et al. 2011). Notably, we identified two novel substitutions (Pro143Ala and c.906 +3 G > A) in a single heterozygous state in three PD patients, and these substitutions were absent from all 850 controls. To assess the genetic evidence for the pathogenicity of our identified variations, we examined whether HTRA2 c.427 C > G (Pro143Ala) segregates with disease in our index family Y121. The clinical phenotypes of the proband included typical PD features and a good response to levodopa treatment. Two carriers did not exhibit parkinsonism features at the time of the study. However, although the elder carrier did not have parkinsonism features yet at the age of 71, the smelling test showed severe anosmia, suggesting a pre-motor state of dopaminergic neuronal degeneration. Both carriers need to be closely followed for both motor and non-motor features of parkinsonism. Subsequently, we genotyped c.427 C > G (Pro143Ala) in a large set of total 850 control subjects and identified no additional genetic substitution. However, it is not easy to prove the pathogenicity of our identified substitutions, especially as the number of approached family members in our index family was limited. Given the result that this substitution was not observed in a total of 1,700 control chromosomes, we suggest that either c.427 C > G (Pro143Ala) or c.906 +3 G > A is a rare risk polymorphism in our population. Although we did not perform cDNA or gene dosage analysis to detect large duplications or deletions, given that the HTRA2 mutations reported to date are missense mutations, the chance that our study underestimated the mutational frequency is relatively small. Therefore, either point mutations or small deletions could be detected by the method used in our current study.
Among these two novel substitutions we identified, c.906 +3 G > A, which is located in the exon–intron boundary junction, had significantly decreased mRNA expression compared to the age- and gender-matched control subjects in the q-PCR analysis. This variant also caused decreased exon 2-exon 7 transcript in the RT-PCR analysis. This variation could dramatically change the transcriptional activity or splicing ability of HTRA2 (Fig. 2a, b). However, because this variant is in the intron, rather than in the exon, it is technically difficult to construct the expression plasmid for this HTRA2 intronic variant. Therefore, we did not perform similar functional studies as done in the Pro143Ala variant in the neuronal cell lines. Further functional studies are warranted in cellular or animal models to clarify the pathogenicity of this intronic variation.
HTRA2 Pro143Ala is an evolutionarily highly conserved amino acid residue (Fig. 1c) and is only one residue from the potential phosphorylation site for p38 stress kinase, the upstream kinase for HTRA2 (Plun-Favreau et al. 2007). Using an in silico phosphorylation site prediction tool, recent studies identified Ser142, near the protease domain, and Ser400, in the PDZ domain, of HTRA2 as two potential phosphorylation sites for proline-directed serine/threonine kinases (Plun-Favreau et al. 2007). Among these two sites, protein levels of phosphorylated Ser142 HTRA2 is increased in brain samples from patients with idiopathic PD and it is found to be specifically phosphorylated in response to activation of the p38 stress kinase. Ambiguously, the level of phopho-HTRA2 at Ser142 site is decreased in familial PD patient with PINK1 mutations, the upstream regulator of P38 and HTRA2 (Plun-Favreau et al. 2007). The reason for this condition is unclear. One of the possible mechanisms is that, although the phosphorylation of HTRA2 is mediated by the p38 pathway it is also dependent on the interaction between HTRA2 and PINK1, possibly through a trimeric protein complex. These observations suggest that the dysregulation of phospho-HTRA2 at Ser142 site contributes to the development of PD. Our molecular modeling data showed that the amino acid residue 143, the residue next to Ser142, is situated at the beginning of helix α1 of the HTRA2 protein, and the Pro143Ala substitution results in a marked helix transformation (Fig. 3a) and affects the orientation of Ser142 (Fig. 3b). Furthermore, electrostatic potential surface maps show that the surface of this region differs between wild-type and mutant HTRA2 (Fig. 3c, d). This structural variation would impact the phosphorylation of Ser142 by p38 kinase and may potentially decrease cellular protection from oxidative stress. We therefore propose that Pro143Ala HTRA2 may act through up-regulating the phosphorylation level of HTRA2 protein with resulting mitochondrial dysfunction. Our in vitro findings are in accordance with our hypothesis as evidenced by the level of phosphorylated HTRA2, which is significantly increased in cells expressing Pro143Ala variant HTRA2 than wild-type HTRA2 (Fig. 6a, b). We further observed a higher percentage of cells with disrupted MMP and a higher rate of apoptosis in the cells expressing the Pro143Ala variant compared to wild-type protein. Using a cellular stress model, we revealed that the Pro143Ala variant is associated with a greater rate of mitochondrial dysfunction, mitochondrial morphology disruption and apoptosis under conditions of increased oxidative stress (300 nM rotenone) (Fig. 5a–d). Overall, our model provides compelling evidence to support our finding that the Pro143Ala variant is biologically relevant to the pro-apoptotic mechanism by disturbing mitochondrial function.
In conclusion, our data support a role for HTRA2 in PD susceptibility in the Taiwanese population. The pathogenicity of Pro143Ala variant is needed to be confirmed by further large-scale replication studies involving more patients and controls in Asian populations. In addition, our in vitro work showed that the Pro143Ala variant is biologically relevant and may act through pro-apoptotic mechanisms. Further in vivo work to unravel the pathophysiological significance of Pro143Ala substitution and its relationship with mitochondrial dysfunction is warranted.
References
Bogaerts V, Nuytemans K, Reumers J, Pals P, Engelborghs S, Pickut B, Corsmit E, Peeters K, Schymkowitz J, De Deyn PP, Cras P, Rousseau F, Theuns J, Van Broeckhoven C (2008) Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat 29:832–840
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Doty RL, Shaman P, Dann M (1984) Development of the University of Pennsylvania Smell Identification Test: a standardized microencapsulated test of olfactory function. Physiol Behav 32:489–502
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55:259–272
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56:33–39
Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnóczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES (2003) Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425:721–727
Krüger R, Sharma M, Riess O, Gasser T, Van Broeckhoven C, Theuns J, Aasly J, Annesi G, Bentivoglio AR, Brice A, Djarmati A, Elbaz A, Farrer M, Ferrarese C, Gibson JM, Hadjigeorgiou GM, Hattori N, Ioannidis JP, Jasinska-Myga B, Klein C, Lambert JC, Lesage S, Lin JJ, Lynch T, Mellick GD, de Nigris F, Opala G, Prigione A, Quattrone A, Ross OA, Satake W, Silburn PA, Tan EK, Toda T, Tomiyama H, Wirdefeldt K, Wszolek Z, Xiromerisiou G, Maraganore DM, for the Genetic Epidemiology of Parkinson’s disease consortium (2009) A large-scale genetic association study to evaluate the contribution of Omi/HtrA2 (PARK13) to Parkinson’s disease. Neurobiol Aging (Epub ahead of print)
Langston JW, Ballard PA Jr (1983) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine. N Engl J Med 309:310
Lee MJ, Mata IF, Lin CH, Tzen KY, Lincoln SJ, Bounds R, Lockhart PJ, Hulihan MM, Farrer MJ, Wu RM (2009) Genotype-phenotype correlates in Taiwanese patients with early-onset recessive Parkinsonism. Mov Disord 24:104–108
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18:R48–R59
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, Wu RM (2008a) Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 71:1727–1732
Lin CH, Tzen KY, Yu CY, Tai CH, Farrer MJ, Wu RM (2008b) LRRK2 mutation in familial Parkinson’s disease in a Taiwanese population: clinical, PET, and functional studies. J Biomed Sci 15:661–667
Lockhart PJ, Bounds R, Hulihan M, Kachergus J, Lincoln S, Lin CH, Wu RM, Farrer MJ (2004) Lack of mutations in DJ-1 in a cohort of Taiwanese ethnic Chinese with early-onset parkinsonism. Mov Disord 19:1065–1069
Malkus KA, Tsika E, Ischiropoulos H (2009) Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: how neurons are lost in the Bermuda triangle. Mol Neurodegener 4:24
Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J (2007) The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol 9:1243–1252
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Simon-Sanchez J, Singleton AB (2008) Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet 17:1988–1993
Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Müller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Krüger R (2005) Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet 14:2099–2111
Takeuchi H, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuno R, Suzumura A (2005) Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem 280:10444–10454
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Wang CY, Xu Q, Weng L, Zhang Q, Zhang HN, Guo JF, Tan LM, Tang JG, Yan XX, Tang BS (2011) Genetic variations of Omi/HTRA2 in Chinese patients with Parkinson’s disease. Brain Res (Epub ahead of print)
Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119:653–665
Wu RM, Bounds R, Lincoln S, Hulihan M, Lin CH, Hwu WL, Chen J, Gwinn-Hardy K, Farrer M (2005) Parkin mutations and early-onset parkinsonism in a Taiwanese cohort. Arch Neurol 62:82–87
Acknowledgments
We would like to thank all of the patients who participated in this study. We also thank the staff of the Second Core Lab, Department of Medical Research, National Taiwan University Hospital for their technical support during the study. The grant is supported from the National Taiwan University Hospital (aNTUH 098-001196) and National Science Council (NSC96-2628-B-002-102-MY3, NSC98-2628-B-002-072-MY3, NSC 99-2314-B-002-078-MY3).
Conflict of interest
The authors report no conflicts of interest and have no financial disclosures to make.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
C.-H. Lin and M.-L. Chen contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Pedigree of the Y121 family with the HTRA2 Pro143Ala variant. Black symbols denote family members affected by parkinsonism. The proband is marked with an arrowhead. The genders of some asymptomatic subjects were concealed to maintain confidentiality. The patients’ ages at the time of the study are indicated above their symbols. + implies that DNA was available; m, mutated alleles; wt, normal alleles. Supplementary material 2 (PPT 81 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lin, CH., Chen, ML., Chen, G.S. et al. Novel variant Pro143Ala in HTRA2 contributes to Parkinson’s disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum Genet 130, 817–827 (2011). https://doi.org/10.1007/s00439-011-1041-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1041-6