
Abstract
X chromosome inactivation (XCI) is a process in mammals that ensures equal transcript levels between males and females by genetic inactivation of one of the two X chromosomes in females. Central to XCI is the long non-coding RNA Xist, which is highly and specifically expressed from the inactive X chromosome. Xist covers the X chromosome in cis and triggers genetic silencing, but its working mechanism remains elusive. Here, we review current knowledge about Xist regulation, structure, function and conservation and speculate on possible mechanisms by which its action is restricted in cis. We also discuss dosage compensation mechanisms other than XCI and how knowledge from invertebrate species may help to provide a better understanding of the mechanisms of mammalian XCI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In placental mammals, individuals carrying an X and a Y chromosome develop as males, whereas XX animals develop as females. The Y chromosome contains only a small number of genes, most of them male-specific. The X chromosome, in contrast, contains around 1,000 genes, posing an enormous copy number imbalance between the sexes. The potentially detrimental effects of copy number imbalances are evidenced by autosomal copy number changes, which invariably result in embryonic lethality or severe developmental defects. Nevertheless, the twofold difference in X chromosome number between males and females is part of normal development, and inheritance of aberrant X copy numbers (as in e.g. XXX or XO females, or XXY males) results in phenotypes that are relatively mild compared with autosomal aneuploidy. The reason for this exceptional behavior of X chromosomes is that, while an X- and Y-chromosomal system evolved to discriminate between the sexes, dosage compensation mechanisms co-evolved to counteract the detrimental effects of the associated copy number variations in hundreds of X chromosomal genes.
Dosage compensation is a mechanism that corrects for the sex-chromosomal dosage differences between the sexes. In 1961, Mary Lyon was one of the first to suggest that dosage compensation in mice occurs by genetic inactivation of one of the two X chromosomes in female cells (Lyon 1961). Earlier studies had reported that nerve cell nuclei from female cats have one chromosome that is structurally distinct and characterized by distinct nuclear morphology visible as a dense heterochromatic region, also known as the Barr body (Barr and Bertram 1949). Other experiments revealed that in female rat liver cells, the Barr body represents one X chromosome whereas the other appears euchromatic like the autosomes (Ohno et al. 1959). Furthermore, mice with a single X chromosome (XO) were found to be phenotypically normal, suggesting that one X is sufficient for normal viability (Welshons and Russell 1959). Lyon suggested that one of the two X chromosomes in female mice is subject to genetically programmed, random inactivation. This theory was supported by the mosaic appearance of female mice that are heterozygous for an X-linked fur color gene: random inactivation of one X chromosome in each cell in the early embryo, followed by clonal expansion accounts for this observation (Lyon 1961).
X chromosome inactivation (XCI) in females thus leads to similar transcription levels of X-chromosomal genes between males and females, who now both express genes from a single X chromosome. However, not all genes on the X are inactivated: genes in the pseudo-autosomal region (PAR), the region of the X homologous to the Y and responsible for XY-pairing during meiosis, as well as a fair number of individual genes on the X are not inactivated. The latter genes are called escapers and it has been estimated that 15–20% of human X-linked genes completely escape inactivation, and another 10% escape partially (Carrel and Willard 2005). PAR genes are expressed from two copies in both males and females, whereas escapers that lack a functional Y homolog are differentially expressed between the sexes. The PAR genes together with the escapers likely account for the phenotypes observed in for example XO and XXX females.
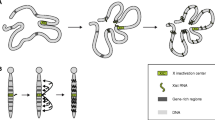
XCI occurs in all marsupials and placental mammals during early development. Interestingly, XCI is not the only solution to compensate for sex chromosome dosage differences; other species have developed completely different approaches to solve the same problem (Fig. 1a–d). In the fruit fly Drosophila melanogaster, male (XY) individuals increase expression of their single X chromosome twofold to meet expression levels of their female (XX) counterparts (Gelbart and Kuroda 2009). In the nematode Caenorhabditis elegans, males have a single X chromosome (XO) and XX individuals are hermaphrodites. Here, XX hermaphrodites reduce transcription levels from both X chromosomes by half to achieve similar transcription levels as in XO males (Meyer et al. 2004). Despite the different approaches, mice, worms and flies have in common that in one sex, specialized, X-specific complexes (dosage compensation complex, DCC) composed of RNA and/or protein target an entire chromosome for stable and inheritable changes in transcription levels through epigenetic modifications (Fig. 1a–d).

Mammalian, Caenorhabditis elegans and Drosophila melanogaster dosage compensation. a Table comparing features of dosage compensation between the three species. Proteins indicated with an asterisk are not specific for dosage compensation and are also a part of other complexes or cellular processes. b–d Images showing X chromosome-wide localization of dosage compensation components. b Mouse differentiating ES cell; the inactive X chromosome is coated by Xist RNA (green; RNA FISH for Xist). c Both X chromosomes in a C. elegans embryonic hermaphrodite nucleus are bound by the dosage compensation complex (DCC) (green; DCC component DPY-27, courtesy of Te Wen Lo and Barbara J. Meyer). d Male Drosophila cell with polytene chromosomes, showing the DCC targeting the X chromosome in green (green; DCC component MSL2, courtesy of Ina Dahlsveen and Peter Becker)
In this review, we discuss what is currently known about the initiation and establishment of XCI in placental mammals. We focus on the role of Xist, XCI’s central player, and briefly discuss how knowledge from invertebrate species may help to gain new insight in mammalian dosage compensation.
XCI initiation
In mice, XCI is initiated in the early embryo in two rounds. At an early developmental stage, around the 4- to 8-cell stage, the paternal X chromosome is inactivated in all cells of the developing embryo (imprinted XCI) (Huynh and Lee 2003; Okamoto et al. 2004). Later in development, this chromosome becomes reactivated in the inner cell mass, but remains inactive in the extra-embryonic tissues. A second round of XCI then occurs in the developing embryo proper around embryonic day 5.5. In inbred mouse strains, the choice of the X to be inactivated is random this time; the paternal and maternal X chromosomes now have equal chances of becoming inactivated (random XCI). In interspecies crosses, preferred inactivation of either the paternal or maternal X chromosome (skewing) may occur. Once the choice of the X chromosome to inactivate has been made, the inactive X is propagated clonally to daughter cells.
Xist and Tsix, the master regulators of X inactivation
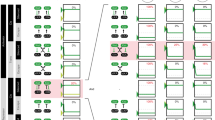
Central to XCI in mammals is the long, non-coding RNA Xist (X-inactive specific transcript). It is transcribed from the Xist gene, which lies in a region on the X chromosome called the X inactivation center (Xic), containing clustered genes and regulatory sequences involved in the X inactivation process (Fig. 2a). Xist is spliced and polyadenylated and, during XCI onset, becomes transcribed only from the future inactive X chromosome (Xi) (Borsani et al. 1991; Brockdorff et al. 1991, 1992; Brown 1991). The processed Xist transcript coats the Xi in cis (Brown et al. 1992) and recruits chromatin remodeling complexes including PRC2, which trimethylates lysine 27 on histone H3 (H3K27me3), a hallmark of facultative heterochromatin (Chadwick and Willard 2004; Mak et al. 2002; Plath et al. 2003; Silva et al. 2003; Zhao et al. 2008). Xist is absolutely essential for initiation of XCI and covers the Xi in all differentiated somatic cells, resulting in Xist RNA associating with the Xi, forming typical “clouds” when visualized by RNA FISH (Fig. 1b) (Brown et al. 1992). Once the Xi has been completely silenced, the silent state is stably inherited and can not be reversed. Tight regulation of Xist transcription to ensure inactivation of a single X chromosome only in females is, therefore, essential.

Features of the X-inactivation center (Xic) and Xist in mouse. a Schematic overview of the location of the X inactivation center (Xic) on X (top panel) and the genes contained in this region (second panel). The third panel shows the overlapping transcripts Xist and Tsix. Tsix has two annotated promoters, the one downstream being the major promoter. The last panel is a schematic overview of the Xist transcript with repetitive domains indicated in yellow (b). Overview of the repetitive regions in Xist indicated in a
In mice, antagonizing Xist function is Tsix RNA, which is transcribed in the antisense orientation from Xist and fully overlaps with the Xist gene (Fig. 2a) (Lee et al. 1999). Tsix is also a non-coding RNA, is transcribed from the active X (Xa) before and during XCI onset (Lee et al. 1999), and inhibits Xist expression in cis by several mechanisms. First, inhibition may occur by transcriptional interference (Luikenhuis et al. 2001; Sado et al. 2006; Shibata and Lee 2004). Second, Xist/Tsix duplex RNA formation and processing by the RNA interference pathway may play a role by siRNA-mediated deposition of chromatin remodeling complexes (Ogawa et al. 2008). Also, recruitment of chromatin remodeling complexes by the Tsix RNA to the Xist promoter has been postulated as a possible mechanism for Tsix-mediated repression of Xist (Sun et al. 2006). Finally, Tsix is involved in pairing of the two X chromosomes, a process which has been implicated in initiation of XCI (Bacher et al. 2006; Xu et al. 2006). Deletion or truncation of Tsix leads to up-regulation of Xist in cis and skewed XCI with preferential inactivation of the mutated allele (Lee and Lu 1999). Impaired transcription of Tsix has also been reported to lead to ectopic XCI in male cells (Luikenhuis et al. 2001; Sado et al. 2002; Vigneau et al. 2006), although one study indicated absence of XCI in Tsix mutant male ES cells (Lee and Lu 1999). The discrepancy between these studies is most likely caused by differences in differentiation protocols which has recently been shown to lead to altered expression levels of key XCI regulators, including OCT4 (Ahn and Lee 2010).
Xist and Tsix are the master regulatory switch genes in XCI. Interestingly, in female cells with a heterozygous deletion encompassing both genes that includes Xite, a positive regulator of Tsix located upstream of Tsix, XCI is still initiated on the wild type X chromosome (Monkhorst et al. 2008). This suggests that activation of Xist is regulated by other factors, but how? In C. elegans, autosomal and X-linked regulators play a key role in the counting process, by determining the relative number of X chromosomes. Here, initiation of dosage compensation is determined by the balance between autosomal and X-chromosomal signal elements (Powell et al. 2005). X-linked activators thus counteract the effect of autosomal inhibitors of dosage compensation. When the ratio of X-linked versus autosomal signal elements is 1, as in XX hermaphrodites, the dosage compensation machinery is turned on. However, if the ratio is lower than 1, as in XO males (X:A ratio 0.5), the concentration of X-linked activators is not sufficient to overcome repression of the dosage compensation machinery by autosomal inhibitors (Meyer 2000).
Activators of X chromosome inactivation
Several recent findings support a role for X-linked activators and autosomally encoded inhibitors in the regulation of mammalian XCI. The first indications came from studies with triploid and tetraploid mouse ES cell lines generated by cell fusion experiments. Analysis of XXXX, XXXY and XXYY tetraploid ES cells after differentiation showed that a single X chromosome remains active for each diploid autosome set (Monkhorst et al. 2008; Takagi 1983, 1993), as was found for mouse tetraploid embryos (Webb et al. 1992). Comparison of XCI kinetics in these different tetraploid, and also XXY triploid ES cells, indicated an important role for the X:A ratio in the probability to initiate XCI, suggesting the presence of an X-encoded activator of XCI (Monkhorst et al. 2009). The first activator, the E3 ubiquitin ligase RNF12/RLIM, was recently discovered and is one of the few known protein-coding—rather than RNA—regulators of Xist (Jonkers et al. 2009). The Rnf12 gene is located approximately 500 kb upstream of Xist (Fig. 2a) and the encoded protein stimulates Xist expression in a dose-dependent manner. RNF12 expression from a single X chromosome in males is insufficient to activate Xist, whereas the double dose in females is sufficient to initiate XCI. In contrast to Xist and Tsix, RNF12 acts in trans and activates Xist on both X chromosomes. Once the inactivation process is started on one X and silencing spreads over the chromosome, Rnf12 will also become silenced in cis. Given a relatively short half-life for RNF12, this results in an RNF12 expression level that equals that in male cells, and this is too low to activate Xist on the other X. Because initiation of XCI is driven by stochastic processes, and the feedback after XCI initiation is rapid, most XX female cells will initiate XCI on a single X chromosome only (Monkhorst et al. 2008). As would be expected for a trans-acting activator, overexpression of Rnf12 triggers XCI in male cells and leads to inactivation of both X chromosomes in a high percentage of female cells (Jonkers et al. 2009). Also, only very few Rnf12−/− cells initiate XCI (Barakat et al. 2011). A different study also reported impaired imprinted XCI in cells carrying a maternally inherited Rnf12 deletion, but observed a milder effect on random XCI (Shin et al. 2010), possibly as a consequence of differences in expression of other XCI-activators and -inhibitors. Although the target of the E3 ubiquitin ligase RNF12 remains elusive, transgenic studies indicate that Xist is the major downstream target of RNF12 (Barakat et al. 2011). Unexpectedly, XCI is skewed toward the mutated X chromosome in Rnf12+/− female ES cells (Barakat et al. 2011), despite the absence of a phenotype for this mutation in male Rnf12−/Y mice (Shin et al. 2010). This suggests that RNF12 is required for persistent Xist expression, at least during the window when XCI is established (Wutz and Jaenisch 2000). A continuous requirement for RNF12 for the activation of Xist and for maintenance of XCI may also explain why Rnf12+/− female embryos which maternally inherit the mutated allele display severe growth defects: maternal inheritance of the mutated allele would result in an Rnf12 null embryo because the wild type paternal X becomes inactivated during imprinted XCI. Due to the complete absence of Rnf12 expression that follows, these embryos may then be unable to maintain Xist expression and imprinted XCI, explaining the reported early lethality of these mice (Shin et al. 2010).
Many other positive regulators of Xist can be found in the Xic region and include the non-coding RNAs Ftx and Jpx, and the pairing element Xpr (Augui et al. 2007; Chureau et al. 2011; Sun et al. 2006; Tian et al. 2010). The region of the Xic upstream of Xist—including Ftx, Jpx and Xpr—has been shown to be enriched for H3K9 and H3K27 di- and trimethylation, respectively (Heard et al. 2001; Rougeulle et al. 2004). These epigenetic marks may contribute to transcriptional regulation of one or more genes in the region, including Xist itself, and may result from ongoing bidirectional transcription in this region as well as from immediate recruitment of PRC2 by nascent Xist transcripts. The Xpr region is involved in pairing of the two X chromosomes at the onset of XCI (Augui et al. 2007). Pairing of the Xist/Tsix region has been implicated to play an important role in the initiation of XCI (Bacher et al. 2006; Xu et al. 2006), although a regulatory role for direct interaction of two X chromosomes in XCI remains to be determined.
Deletion of Ftx in male mouse ES cells is associated with reduced transcription of Xist, Tsix and Jpx (Chureau et al. 2011). These findings could indicate a direct role for Ftx in Xist activation, but may also be explained by Ftx-mediated global activation of the Xic region. A similar role may be attributed to Jpx/Enox, which encodes another non-coding long RNA, and is located just upstream of Xist. Jpx/Enox has been shown to activate Xist in trans, possibly by interfering with Tsix (Tian et al. 2010). However, unlike Rnf12 transgenic lines, male cell lines with Ftx or Jpx/Enox transgenes did not show induction of XCI on the endogenous X chromosome (Jonkers et al. 2009), arguing against a role in trans for these genes. Similar results were obtained with Xpr transgenic male ES cell lines (Jonkers et al. 2009). Nevertheless, studies with Xist YAC transgenic male ES cell lines covering both Ftx and Jpx/Enox showed induction of XCI on the endogenous X chromosome in a small percentage of cells, but only in multicopy ES cell lines (Heard et al. 1999). These findings suggest that Ftx and Jpx/Enox may require additional factors for their trans-activating properties, and also indicate that RNF12 is a more potent activator of the XCI process.
Inhibitors of X chromosome inactivation
Other important regulators of XCI are the key pluripotency factors NANOG, OCT4, KLF4, REX1 and SOX2, and the reprogramming factor cMYC, acting as autosomally encoded inhibitors of XCI (Donohoe et al. 2009; Navarro et al. 2008, 2010). Binding of different combinations of these pluripotency factors at different locations in the locus can either result in repression of Xist or in activation of Tsix or Xite. NANOG, OCT4 and SOX2 bind the intron 1 region of Xist and binding of these factors has been implicated in direct repression of Xist (Navarro et al. 2008). OCT4 is also recruited to the Tsix regulatory region, and binds the Xite promoter region together with SOX2 (Donohoe et al. 2009). Recruitment of these factors has been implicated in X chromosome pairing and in activation of Tsix, although binding of OCT4 and SOX2 to these specific regions has been disputed by others (Navarro et al. 2010). REX1, KLF4, and cMYC are recruited to the DXPas34 region, a regulatory region in Tsix, and are involved in Tsix activation (Navarro et al. 2010), together with YY1 and CTCF (Donohoe et al. 2007). The repression of Xist is released upon differentiation, as the concentration of pluripotency factors drops, linking XCI to the pluripotent state and differentiation (Navarro et al. 2008). Interestingly, a deletion encompassing the Xist intron 1 region that recruits OCT4, SOX2 and NANOG, only has a mild effect on XCI, leading to skewed XCI at later stages of ES cell differentiation (Barakat et al. 2011). Xist expression in undifferentiated heterozygous intron 1+/− female ES cells was not affected, indicating that ES cell specific transcription factors act in concert to inhibit Xist expression by binding to various sites throughout the Xist, Tsix and Xite genes. Finally, NANOG, OCT4 and SOX2 also have a repressive effect on XCI by binding and inhibiting Rnf12 (Navarro et al. 2011). Altogether, the inhibitors are involved in setting a threshold that has to be overcome by the XCI-activators to induce XCI (Barakat et al. 2010). Indeed, gene ablation experiments of these different factors resulted in ectopic activation of XCI in mutated male cells, supporting a crucial role for these factors in maintaining the threshold for XCI.
Despite this wide plethora of known XCI regulators (see Fig. 3 for an overview), it is likely that more remain to be identified. Evidence for undiscovered activators of XCI comes from the observation that Rnf12+/− heterozygous female mouse ES cells still initiate XCI, albeit at much lower levels (Jonkers et al. 2009), and even Rnf12 null cells still display occasional XCI (Barakat et al. 2011).

Overview of regulators of Xist and XCI. Left activators and inhibitors act through the Xist/Tsix switch region to regulate initiation of XCI. Right XCI is established and maintained by a plethora of histone modifications, bound protein and protein complexes, and RNA specific for the Xi
XCI establishment
As discussed above, the end result of Xist regulation is expression from a single X chromosome in females, which is then to become the Xi. The first observation following Xist expression is coating of the Xi by Xist RNA, exclusion of RNA polymerase II (polII) from the Xist compartment (see below), and gradual accumulation of Xi-specific epigenetic marks.
X chromosome coating by Xist and formation of a nuclear compartment
Spreading of Xist must be restricted to prevent the aberrant inactivation of another X chromosome or possibly even an autosome. Thus, Xist must act in cis and should not spread onto other chromosomes. Xist-tagging experiments have shown that Xist RNA never leaves the territory of the X chromosome from which it is transcribed (Jonkers et al. 2008). How diffusion of Xist is restricted is not known, but the Xist domain forms a distinct nuclear compartment from which RNA polII is excluded. X chromosomal genes are recruited into this domain and subsequently become silenced. The formation of a compartment suggests that structural nuclear factors play a role (Chaumeil et al. 2006; Nakagawa and Prasanth 2011), and at least two proteins that are thought to be components of the nuclear matrix, SAF-A and SATB1, are involved in establishing this compartment (Nakagawa and Prasanth 2011). The nuclear matrix is referred to as the biochemical nuclear structure that is resistant to detergent and high salt treatment and that remains after treatment with nucleases (Berezney 1991). Its composition and even its mere existence are heavily debated. Heterogeneous nuclear proteins (hnRNPs) are important nuclear structural proteins and possible constituents of a nuclear matrix, and are thought to organize the genome by binding to putative matrix-associated regions (MARs) in the DNA. One such hnRNP, SAF-A, colocalizes with Xist and is important for the formation of the Xist nuclear compartment (Fackelmayer 2005; Hasegawa et al. 2010; Helbig and Fackelmayer 2003; Pullirsch et al. 2010). Cells depleted of SAF-A by RNAi fail to form Xist clouds and show lower levels of H3K27me3 (Hasegawa et al. 2010). Another nuclear protein, SATB1—a DNA binding protein involved in nuclear architecture and chromatin looping (Cai et al. 2003, 2006)—also contributes to the framework of the Xist compartment (Agrelo et al. 2009). Although Xist and SatB1 do not colocalize or interact directly, SATB1 forms a ring-like structure that contains Xist RNA in thymic cells. SATB1 is required for correct localization of Xist because SATB1 knockdown leads to impaired formation of Xist clouds in these cells (Agrelo et al. 2009). The expression pattern of this protein overlaps with the permissive window of XCI initiation, and overexpression of SatB1 together with Xist in differentiated cells allows gene silencing to occur (Agrelo et al. 2009). The structural function of SAF-A and SATB1 suggests that they contribute to XCI through the formation of the silent nuclear compartment. A role for nuclear structural proteins is further supported by the finding that the compartment is formed independently of DNA, since DNase treatment does not disrupt the Xist localization pattern (Clemson et al. 1996). However, the nature of the molecular interactions between SAF-A, SATB1 and Xist remain to be determined.
The Xist nuclear compartment is depleted of RNA polII, and initial gene silencing is established by relocalization of active genes into this RNA polII-depleted area, followed by the epigenetic modifications that contribute to long-term silencing (Chaumeil et al. 2006). Thus, genes cease to be transcribed before the appearance of silent chromatin marks such as H3K27me3. Interestingly, it was recently shown that lack of transcription is sufficient to trigger chromatin modifications. Treatment of cells with actinomycin D, which binds the transcription initiation complex and inhibits elongation, markedly reduces the size of the nuclear territory occupied by Xa, approaching the size of the Xi. Nevertheless, the basal packaging of chromatin in 30 nm fibers remains unaffected. This shows that transcription inhibition leads to chromatin compaction and that this occurs at a higher level of compaction than the 30 nm fiber (Naughton et al. 2010). Relocalization into an RNA polII-depleted area may thus be sufficient to establish some of the chromatin marks associated with the Xi, although clearly factors and events are required for stable maintenance of the Xi.
Epigenetic marks associated with XCI
During the establishment of the nuclear compartment, PRC2 (polycomb repressive complex 2) is recruited to the Xi by Xist. PRC2 is composed of the protein subunits SUZ12, EED and EZH2, and it trimethylates lysine 27 of histone H3 (H3K27me3), a hallmark of inactive chromatin. This inactivating mark first appears on active genes (Marks et al. 2009). Active genes are generally characterized by H3K4me3 enrichment in their promoters, and the deposition of H3K27me3 here may thus result in the simultaneous occurrence of opposing (active vs. inactive) chromatin marks, on the Xa and Xi, or may even be present transiently on promoters of active genes on the future Xi. Interestingly, such a bivalent state is typical for many developmental genes in ES cells (Azuara et al. 2006; Bernstein et al. 2006; Pan et al. 2007; Pasini et al. 2008). Next, the H3K4me3 mark is gradually lost (Marks et al. 2009), followed by incorporation of histone macroH2A (Costanzi and Pehrson 1998; Mermoud et al. 1999), enrichment for H3K9me2 (Heard et al. 2001; Mermoud et al. 2002; Peters et al. 2002), ubiquitylation of H2AK119 (de Napoles et al. 2007; Smith et al. 2004), H4K20 methylation (Kohlmaier et al. 2004), DNA methylation (Norris et al. 1991) and hypoacetylation of histone H4 (Jeppesen and Turner 1993) (Fig. 3). The Xi also becomes enriched for PRC1, which is associated with PRC2 and H3K27me3 (Plath et al. 2004). Surprisingly, Xist is not required for the maintenance of the Xi once the inactive state is established. Conditional deletion of Xist in differentiated cells leads to loss of macroH2A incorporation and loss of H3K27me3, but the Xi is not reactivated (Csankovszki et al. 1999; Kohlmaier et al. 2004). In fact, no conditions, except reprogramming, have been found to date that can completely reactivate the Xi. Only harsh and highly artificial conditions involving conditional knockout of Xist, combined with chemical treatments that remove DNA methylation and inhibit hypoacetylation, have been found to lead to limited reactivation of a GFP transgene on the Xi (Csankovszki et al. 2001). Thus, some epigenetic modifications persist in the absence of Xist and are sufficient to maintain the inactive state.
The differential regulation of XCI initiation, establishment and maintenance is further supported by the finding that overexpression of Xist cannot induce XCI in cells once they have differentiated (Kohlmaier et al. 2004; Wutz and Jaenisch 2000). This led to the suggestion that stem cells and early embryos go through an “XCI permissive state” during early differentiation. Only in this time window, Xist is required for and capable of XCI. Proteins that are key to stem cell identity, such as OCT4, REX1, SOX2 and NANOG, may play a role in defining this window, as these also regulate Xist and Tsix expression directly and indirectly (Navarro et al. 2008, 2010).
Conservation of Xist RNA structure and functional elements
Besides a role for nuclear structural proteins in preventing diffusion of Xist throughout the nucleus, self-aggregation properties may also be a feature of Xist. Although predictions have been made regarding the secondary structure of repetitive sequences in Xist, no models currently exist for folding of the complete Xist RNA. Whether Xist’s overall structure is conserved thus remains unknown, but conserved sequence elements may aid in understanding the intriguing characteristics of Xist. Poor overall sequence conservation of Xist between mice, humans and other mammals provides little information on functionality, and suggests that secondary structure is more important for Xist function than the primary sequence. In Drosophila dosage compensation, two non-coding RNAs—roX1 and roX2—play a central role in targeting the DCC to the X chromosome (Fig. 1). Also here, secondary structure seems important, despite these two RNAs being fully redundant; they share almost no sequence similarity (Meller and Rattner 2002). Nevertheless, some parts of Xist are conserved at the sequence level, suggestive of functional elements. Some of these are highly repetitive and are referred to as repeat A–E (Fig. 2b). In addition, the fourth exon of Xist is well conserved.
The conserved repeat A is composed of nine A-rich repeats and is the best characterized region of Xist (Fig. 2a, b) (Nesterova et al. 2001). Several predictions have been made regarding the secondary structure of this repeat, all involving the formation of hairpin structures. It is required for the silencing function of Xist by serving as a recognition and binding site for PRC2 (Maenner et al. 2010; Zhao et al. 2008). Also, the region may dimerize with other A-repeats (Duszczyk et al. 2008). It is unlikely that repeat A contributes to aggregation and localization of Xist, since studies using constructs expressing a mutant form of Xist have shown that Xist localizes normally in the absence of the A-repeat, but lacks silencing activity (Royce-Tolland et al. 2010; Wutz et al. 2002). Others have targeted the endogenous Xist locus for deletion of the A-repeat and showed that this sequence is also required for Xist expression, and deletion leads to ectopic Tsix expression in the pre-implantation embryo (Hoki et al. 2009). Interestingly, a 1.6 kb-long non-coding RNA called RepA is transcribed from the A-repeat region. This RNA recruits the PRC2 complex through interaction with the EZH2 subunit, and may itself be involved in the initiation of XCI (Zhao et al. 2008).
Seemingly contradicting data have been published regarding the role of repeat C (Fig. 2b). Wutz et al. (2002) showed that an Xist transgene with a deletion of repeat C localizes normally to DNA, and is also still capable of silencing. However, another study recently reported that blocking the same repeat C with an LNA probe (locked nucleic acid—an antisense probe with high melting temperature that stably binds the target DNA) completely disrupts Xist localization (Sarma et al. 2010). Since deletion of the same repeat does not affect localization, the mislocalization of LNA-targeted Xist may represent an indirect effect: the LNA may interfere with secondary structure formation and affect the global folding of the molecule, leading to impaired localization, whereas the C-repeat itself is not required for localization.
The other conserved sequences in Xist are less extensively characterized and seem to serve redundant functions (Fig. 2b). Deletion of repeats B, C, D and E showed unaffected localization patterns. Even combined deletion of several repeats simultaneously barely affects the localization pattern (Fig. 2b) (Wutz et al. 2002). Only deletion products lacking repeat A in combination with one or more of the other repeats show impaired localization. For repeat B, an effect on promoter activity has been reported (Hendrich et al. 1997). An Xist allele containing an inversion that includes repeat D (Xist INV) showed compromised mutant Xist localization and reduced silencing efficiency. Although random XCI initially occurs in heterozygous Xist INV/WT cells, the mutant Xist cannot sustain XCI and cells that initially inactivated this allele are gradually selected against and lost from the cell population (Senner et al. 2011). Similar to what was discussed for repeat C, deletion of repeat D was found to lead to less severe phenotypes (Wutz et al. 2002), suggesting that the overall structure of Xist is affected by the inversion. Another highly conserved region in Xist is exon IV. Deletion of this exon does not lead to detectable XCI phenotypes, except for a slight reduction in expression of the mutant Xist transcript (Caparros et al. 2002). However, this does not result in skewing such as observed for mutations in Tsix (Lee and Lu 1999).
Another conserved feature of Xist RNA is its length. A fair number of non-coding RNAs have been identified, many of them functioning in the establishment of epigenetic modifications, imprinting of genes and allele-specific expression. However, measuring 17 kb in mouse and over 19 kb in humans, Xist is the longest functional non-coding RNA described, making the poor sequence conservation even more mysterious. Secondary structure formation and size may both contribute to Xist function by unknown mechanisms. Speculatively, these mechanisms include aggregation and intermolecular interactions, or Xist might even function as a ribozyme. Finally, the size of Xist combined with a complicated secondary structure could act to limit its diffusion through the nucleus and restrict its localization to the chromosome from which it is transcribed—the nuclear matrix possibly serving as a physical barrier.
Spreading of Xist
How does Xist recognize and bind to the X chromosome? In mammals, no specific sequences that designate the X chromosome for dosage compensation are known. In Drosophila and C. elegans, however, sequences important for X chromosome identity have been defined that are involved in targeting the DCC to the X chromosomes. The mechanisms of X-recognition, binding and spreading show striking overlap in these species despite the opposing effects of dosage compensation (activation vs. silencing) (Fig. 1a–d). Sites that specify the X chromosome are necessary in both species because dosage compensation is regulated by trans-acting factors. For example, although the roX genes are X-linked in Drosophila, they act in trans since roX gene expression from autosomal transgenes drives correct assembly and spreading of the DCC on X, although autosomal spreading is also observed (Meller and Rattner 2002). Targeting to the X chromosome is driven by high affinity DCC binding sites called chromatin entry sites (CES), which share a 150 bp motif (Oh et al. 2003, 2004). Although the motif itself is only slightly enriched on the X chromosome compared with autosomes, its positioning downstream of active genes is highly specific for the X chromosome. Higher concentrations of the DCC have been demonstrated to enable occupation of lower-affinity binding sites (Fagegaltier and Baker 2004; Park et al. 2002). Strikingly, the highest affinity binding sites are the roX genes themselves (Kelley et al. 1999). This suggests that complexes are assembled at the site of roX RNA transcription, after which mature DCCs can spread to other sites on the X chromosome (Smith et al. 2001). Interestingly, spreading of the DCC onto autosomal sequences is highly correlated with gene activity, suggesting a role for active chromatin modifications, RNA polII or open chromatin in DCC spreading (Larschan et al. 2007) (Fig. 1a). Sequence motifs for DCC binding have also been identified in C. elegans (McDonel et al. 2006). The sites were mapped by extensive analysis of extra-chromosomal arrays carrying X-sequences for their ability to recruit the DCC (Csankovszki et al. 2004). Termed rex (recruiting element on X) sites, they are capable of DCC recruitment when integrated on autosomes. Their specificity was later confirmed and extended by ChIP-chip analysis, which showed preferred binding of the DCC to promoter regions (Ercan et al. 2007; Jans et al. 2009). The rex sites cooperate with so-called dox sites (dependent on X), which cannot recruit the DCC when detached from X, but are essential for spreading of the DCC (Jans et al. 2009).
The use of trans-acting initiation factors and lack of a cis-acting factor like Xist in flies and worms results in the need to target dosage compensation to X chromosomes by X-specific sequences. Importantly, flies and worms lack an equivalent of mammalian choice: once the decision to initiate dosage compensation has been made, all X chromosomes in the nucleus are subjected to dosage compensation. Studies of hermaphrodite worms with aberrant X chromosome numbers show that, as in mammals, the X:A ratio determines whether dosage compensation is on or off (Meyer 2000). However, no choice has to be made since all worm X chromosomes are subjected to dosage compensation. A similar situation exists in male flies. In contrast, mammals inactivate a single X and the other X chromosome remains active. The cis-specificity of Xist in mammals may be sufficient to recognize which chromosome is to be inactivated and prevents inactivation in trans. Mono-allelic expression might be the mechanism that omits the need for X-specific sequence elements that recruit dosage compensation elements as in worms and flies. In mammals, specification of the X chromosome for targeting dosage compensation may thus not be required. Nevertheless, sequence elements that promote spreading are potentially important.
LINE-1 or LINE elements (long interspersed elements) are retrotransposons that constitute a large part of mammalian genomes, e.g., 17% in humans (Cordaux and Batzer 2009). Because of their relatively high density on human X chromosomes, they have been suggested to play a role in promoting spreading of Xist (Lyon 1998). Several findings further support a role for LINEs in XCI. First, Xist does not spread efficiently when expressed from autosomes, which are relatively LINE-poor, and spreading is especially inhibited into LINE-poor autosomal areas (Popova et al. 2006; Tang et al. 2010). Furthermore, LINEs seem to be expressed specifically from the Xi (Chow et al. 2010), and Xist interacts with LINE elements directly (Murakami et al. 2009). Furthermore, a computational approach on human X chromosomes found that LINE elements are particularly enriched in the 5′-region of genes that are silenced during XCI (Wang et al. 2006).
The LINE hypothesis remains heavily disputed as many findings counteract the above. First, LINE enrichment could merely be a passive and non-functional consequence of the reduced meiotic recombination rate of the X chromosome compared with autosomes, in view of the lack of X recombination in the male germline. Whereas LINE elements are enriched on human X chromosomes, especially around the Xic region (Bailey et al. 2000), no such enrichment is observed for mouse X chromosomes (Chureau et al. 2002). Also, a South American rodent, Oryzomys palustris, has been reported that appears to lack LINE elements in its genome, and this is not associated with more rapid mutations in Xist (Cantrell et al. 2009). Furthermore, LINE-poor areas are generally gene-rich, and gene-poor areas tend to be LINE-rich. Impaired spreading on autosomes at LINE-poor areas is now attributed to the low LINE-density, but could result from selection against cells that efficiently silence these regions; since these same regions are also gene-rich, their silencing may severely affect cell viability. Cells that fail to silence gene-rich autosomal areas may thus be more efficiently propagated due to selection effects. Finally, Xist spreading at gene-poor LINE elements seem to contradict the observation that hallmarks of XCI are first found on active genes (Marks et al. 2009). Finally, as discussed above, the cis-acting properties of Xist may make the need for spread elements redundant.
Chromatin changes and transcription effects
In mammals, many histone modifications and other chromatin changes cooperate to establish and maintain the silent state. These are well characterized and as mentioned above, include amongst others H3K27me3, macroH2A incorporation, H2Aub and DNA methylation. How these modifications are targeted to specific genes and how others escape silencing is unknown. In flies and worms, epigenetic modifications seem to be less extensive, possibly because dosage compensation here involves fine-tuning of expression levels to a twofold change instead of global silencing, therefore, requiring different mechanisms and different chromatin modifications. In flies, the dosage compensated X chromosome in males is characterized by increased acetylation of lysine 16 on histone 4 (H4K16ac), a hallmark of active chromatin (Turner et al. 1992). Notably, this enrichment is biased toward 3′-ends of genes, whereas H4K16ac normally accumulates at 5′-gene promoters (Gelbart et al. 2009; Kind et al. 2008; Smith et al. 2001). It was recently shown that RNA polII density is also enhanced at 3′-gene ends (Larschan et al. 2011) and the current model thus proposes that the Drosophila DCC acts to enhance transcription elongation, rather than initiation. In addition to the bias toward 3′-gene ends, the DCC also preferably targets active genes for H4K16ac modification on the X chromosome, even when they are from autosomal origin and inserted as transgenes on X (Gorchakov et al. 2009). This bias may be responsible for fine-tuning to twofold increase in expression for differentially expressed genes.
In C. elegans, where transcription of both hermaphrodite X chromosomes is reduced by half, no chromatin modifications associated with the dosage compensated X chromosomes have yet been found (Fig. 1). Depletion of the histone variant H2A.Z (HTZ-1 in C. elegans) from the X chromosome is the only nucleosomal change identified so far. This seems to contribute indirectly to dosage compensation because the relative enrichment on autosomes prevents spreading of the DCC onto autosomes (Petty et al. 2009). One subunit of the DCC contains SMC (structural maintenance of chromosomes) family proteins, and thereby resembles condensin, a protein complex that condenses chromatin in preparation for cell division. This suggests that changes in higher order chromatin structure likely play a role in transcriptional silencing in C. elegans (Csankovszki 2009). Despite the highly different systems, recent findings show some parallels between mammalian and C. elegans dosage compensation. First, an SMC protein was recently found to colocalize with the Xi in mice and was implicated in DNA methylation (Blewitt et al. 2008). Furthermore, a recent study showed that one DCC component, DPY-30, is also part of the gene activating MLL/COMPASS complex (Pferdehirt et al. 2011), which stimulates H3K4me3 in mammals (Jiang et al. 2011). Both complexes bind the same genes in C. elegans despite their opposing effects on transcription (Pferdehirt et al. 2011). Speculatively, DPY-30 may be involved in targeting the DCC to active genes by switching between the two complexes.
Despite the different mechanisms of dosage compensation, the preference for targeting active genes seems to be shared by all three species. In C. elegans, DCC affinity for genes depends on transcriptional activity (Ercan et al. 2009). The DCC is targeted to active genes in Drosophila by the chromodomain subunit of MSL3, which recognizes the active H3K36 trimethyl mark (Larschan et al. 2007; Sural et al. 2008). Chromatin immunoprecipitation (ChIP) against H3K27me3 in differentiating mouse ES cells followed by high-throughput parallel sequencing revealed that the H3K27me3 mark appears first at active promoters, indicating that, also in mammals, active genes are preferred targets of dosage compensation (Marks et al. 2009). Hence, some properties of active genes might serve as a basis for targeting dosage compensation. These properties may involve active chromatin marks, ongoing transcription by RNA polII, or a combination of events.
Perspectives
Many questions about X chromosome inactivation in mammalian species remain to be solved, and knowledge about dosage compensation in worms and flies may be helpful to address different points. In mammalian XCI, unsolved issues include, for example, the working mechanism that underlies the cis-acting specificity of Xist. Also, it is likely that more activators and inhibitors of dosage compensation remain to be identified. Furthermore, the role of LINE elements as cis-acting booster elements for spreading of Xist needs to be confirmed. Finally, the key factors that contribute to set the XCI window and to achieve stable and irreversible silencing need to be elucidated. Identifying these factors would not only provide more insight in X chromosome dosage compensation, but potentially also in gene regulation mechanisms in general. Clearly, X-inactivation research is likely to uncover many more interesting insights in the future that may well extend beyond the field of dosage compensation.
References
Agrelo R, Souabni A, Novatchkova M, Haslinger C, Leeb M, Komnenovic V, Kishimoto H, Gresh L, Kohwi-Shigematsu T, Kenner L et al (2009) SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells. Dev Cell 16:507–516
Ahn JY, Lee JT (2010) Retinoic acid accelerates downregulation of the Xist repressor, Oct4, and increases the likelihood of Xist activation when Tsix is deficient. BMC Dev Biol 10:90
Augui S, Filion GJ, Huart S, Nora E, Guggiari M, Maresca M, Stewart AF, Heard E (2007) Sensing X chromosome pairs before X inactivation via a novel X-pairing region of the Xic. Science 318:1632–1636
Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M et al (2006) Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8:532–538
Bacher CP, Guggiari M, Brors B, Augui S, Clerc P, Avner P, Eils R, Heard E (2006) Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol 8:293–299
Bailey JA, Carrel L, Chakravarti A, Eichler EE (2000) Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA 97:6634–6639
Barakat TS, Jonkers I, Monkhorst K, Gribnau J (2010) X-changing information on X inactivation. Exp Cell Res 316:679–687
Barakat TS, Gunhanlar N, Gontan Pardo C, Achame EM, Ghazvini M, Boers R, Kenter A, Rentmeester E, Grootegoed JA, Gribnau J (2011) RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet 7:e1002001
Barr ML, Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163:676
Berezney R (1991) The nuclear matrix: a heuristic model for investigating genomic organization and function in the cell nucleus. J Cell Biochem 47:109–123
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326
Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N et al (2008) SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet 40:663–669
Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C et al (1991) Characterization of a murine gene expressed from the inactive X chromosome. Nature 351:325–329
Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S (1991) Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351:329–331
Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S (1992) The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71:515–526
Brown SD (1991) XIST and the mapping of the X chromosome inactivation centre. Bioessays 13:607–612
Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF (1992) The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71:527–542
Cai S, Han HJ, Kohwi-Shigematsu T (2003) Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat Genet 34:42–51
Cai S, Lee CC, Kohwi-Shigematsu T (2006) SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet 38:1278–1288
Cantrell MA, Carstens BC, Wichman HA (2009) X chromosome inactivation and Xist evolution in a rodent lacking LINE-1 activity. PLoS One 4:e6252
Caparros ML, Alexiou M, Webster Z, Brockdorff N (2002) Functional analysis of the highly conserved exon IV of XIST RNA. Cytogenet Genome Res 99:99–105
Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434:400–404
Chadwick BP, Willard HF (2004) Multiple spatially distinct types of facultative heterochromatin on the human inactive X chromosome. Proc Natl Acad Sci USA 101:17450–17455
Chaumeil J, Le Baccon P, Wutz A, Heard E (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20:2223–2237
Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M, Avner P, Wutz A, Barillot E et al (2010) LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141:956–969
Chureau C, Prissette M, Bourdet A, Barbe V, Cattolico L, Jones L, Eggen A, Avner P, Duret L (2002) Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res 12:894–908
Chureau C, Chantalat S, Romito A, Galvani A, Duret L, Avner P, Rougeulle C (2011) Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum Mol Genet 20:705–718
Clemson CM, McNeil JA, Willard HF, Lawrence JB (1996) XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol 132:259–275
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703
Costanzi C, Pehrson JR (1998) Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393:599–601
Csankovszki G (2009) Condensin function in dosage compensation. Epigenetics 4:212–215
Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R (1999) Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet 22:323–324
Csankovszki G, Nagy A, Jaenisch R (2001) Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 153:773–784
Csankovszki G, McDonel P, Meyer BJ (2004) Recruitment and spreading of the C elegans dosage compensation complex along X chromosomes. Science 303:1182–1185
de Napoles M, Nesterova T, Brockdorff N (2007) Early loss of Xist RNA expression and inactive X chromosome associated chromatin modification in developing primordial germ cells. PLoS One 2:e860
Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT (2007) Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell 25:43–56
Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT (2009) The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature 460:128–132
Duszczyk MM, Zanier K, Sattler M (2008) A NMR strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a Xist RNA A-repeat. Nucleic Acids Res 36:7068–7077
Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD (2007) X chromosome repression by localization of the C elegans dosage compensation machinery to sites of transcription initiation. Nat Genet 39:403–408
Ercan S, Dick LL, Lieb JD (2009) The C elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Curr Biol 19:1777–1787
Fackelmayer FO (2005) A stable proteinaceous structure in the territory of inactive X chromosomes. J Biol Chem 280:1720–1723
Fagegaltier D, Baker BS (2004) X chromosome sites autonomously recruit the dosage compensation complex in Drosophila males. PLoS Biol 2:e341
Gelbart ME, Kuroda MI (2009) Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136:1399–1410
Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI (2009) Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol 16:825–832
Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI (2009) Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev 23:2266–2271
Hasegawa Y, Brockdorff N, Kawano S, Tsutui K, Nakagawa S (2010) The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell 19:469–476
Heard E, Mongelard F, Arnaud D, Avner P (1999) Xist yeast artificial chromosome transgenes function as X-inactivation centers only in multicopy arrays and not as single copies. Mol Cell Biol 19:3156–3166
Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL (2001) Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107:727–738
Helbig R, Fackelmayer FO (2003) Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain. Chromosoma 112:173–182
Hendrich BD, Plenge RM, Willard HF (1997) Identification and characterization of the human XIST gene promoter: implications for models of X chromosome inactivation. Nucleic Acids Res 25:2661–2671
Hoki Y, Kimura N, Kanbayashi M, Amakawa Y, Ohhata T, Sasaki H, Sado T (2009) A proximal conserved repeat in the Xist gene is essential as a genomic element for X-inactivation in mouse. Development 136:139–146
Huynh KD, Lee JT (2003) Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 426:857–862
Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt RR, Eisen MB, Meyer BJ (2009) A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev 23:602–618
Jeppesen P, Turner BM (1993) The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell 74:281–289
Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, Roeder RG (2011) Role for Dpy-30 in ES cell-fate specification by regulation of h3k4 methylation within bivalent domains. Cell 144:513–525
Jonkers I, Monkhorst K, Rentmeester E, Grootegoed JA, Grosveld F, Gribnau J (2008) Xist RNA is confined to the nuclear territory of the silenced X chromosome throughout the cell cycle. Mol Cell Biol 28:5583–5594
Jonkers I, Barakat TS, Achame EM, Monkhorst K, Kenter A, Rentmeester E, Grosveld F, Grootegoed JA, Gribnau J (2009) RNF12 is an X-encoded dose-dependent activator of X chromosome inactivation. Cell 139:999–1011
Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI (1999) Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98:513–522
Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, Bertone P, Akhtar A (2008) Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 133:813–828
Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2:E171
Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI (2007) MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28:121–133
Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI (2011) X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471:115–118
Lee JT, Lu N (1999) Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99:47–57
Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21:400–404
Luikenhuis S, Wutz A, Jaenisch R (2001) Antisense transcription through the Xist locus mediates Tsix function in embryonic stem cells. Mol Cell Biol 21:8512–8520
Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190:372–373
Lyon MF (1998) X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet 80:133–137
Maenner S, Blaud M, Fouillen L, Savoye A, Marchand V, Dubois A, Sanglier-Cianferani S, Van Dorsselaer A, Clerc P, Avner P et al (2010) 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol 8:e1000276
Mak W, Baxter J, Silva J, Newall AE, Otte AP, Brockdorff N (2002) Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr Biol 12:1016–1020
Marks H, Chow JC, Denissov S, Francoijs KJ, Brockdorff N, Heard E, Stunnenberg HG (2009) High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res 19:1361–1373
McDonel P, Jans J, Peterson BK, Meyer BJ (2006) Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature 444:614–618
Meller VH, Rattner BP (2002) The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21:1084–1091
Mermoud JE, Costanzi C, Pehrson JR, Brockdorff N (1999) Histone macroH2A1.2 relocates to the inactive X chromosome after initiation and propagation of X-inactivation. J Cell Biol 147:1399–1408
Mermoud JE, Popova B, Peters AH, Jenuwein T, Brockdorff N (2002) Histone H3 lysine 9 methylation occurs rapidly at the onset of random X chromosome inactivation. Curr Biol 12:247–251
Meyer BJ (2000) Sex in the wormcounting and compensating X-chromosome dose. Trends Genet 16:247–253
Meyer BJ, McDonel P, Csankovszki G, Ralston E (2004) Sex and X-chromosome-wide repression in Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 69:71–79
Monkhorst K, Jonkers I, Rentmeester E, Grosveld F, Gribnau J (2008) X inactivation counting and choice is a stochastic process: evidence for involvement of an X-linked activator. Cell 132:410–421
Monkhorst K, de Hoon B, Jonkers I, Mulugeta Achame E, Monkhorst W, Hoogerbrugge J, Rentmeester E, Westerhoff HV, Grosveld F, Grootegoed JA et al (2009) The probability to initiate X chromosome inactivation is determined by the X to autosomal ratio and X chromosome specific allelic properties. PLoS One 4:e5616
Murakami K, Ohhira T, Oshiro E, Qi D, Oshimura M, Kugoh H (2009) Identification of the chromatin regions coated by non-coding Xist RNA. Cytogenet Genome Res 125:19–25
Nakagawa S, Prasanth KV (2011) eXIST with matrix-associated proteins. Trends Cell Biol
Naughton C, Sproul D, Hamilton C, Gilbert N (2010) Analysis of active and inactive X chromosome architecture reveals the independent organization of 30 nm and large-scale chromatin structures. Mol Cell 40:397–409
Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P (2008) Molecular coupling of Xist regulation and pluripotency. Science 321:1693–1695
Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, Schoorlemmer J, Rougeulle C, Chambers I, Avner P (2010) Molecular coupling of Tsix regulation and pluripotency. Nature 468:457–460
Navarro P, Moffat M, Mullin NP, and Chambers I (2011) The X-inactivation trans-activator Rnf12 is negatively regulated by pluripotency factors in embryonic stem cells. Hum Genet [Epub ahead of print]
Nesterova TB, Slobodyanyuk SY, Elisaphenko EA, Shevchenko AI, Johnston C, Pavlova ME, Rogozin IB, Kolesnikov NN, Brockdorff N, Zakian SM (2001) Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res 11:833–849
Norris DP, Brockdorff N, Rastan S (1991) Methylation status of CpG-rich islands on active and inactive mouse X chromosomes. Mamm Genome 1:78–83
Ogawa Y, Sun BK, Lee JT (2008) Intersection of the RNA interference and X-inactivation pathways. Science 320:1336–1341
Oh H, Park Y, Kuroda MI (2003) Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17:1334–1339
Oh H, Bone JR, Kuroda MI (2004) Multiple classes of MSL binding sites target dosage compensation to the X chromosome of Drosophila. Curr Biol 14:481–487
Ohno S, Kaplan WD, Kinosita R (1959) Formation of the sex chromatin by a single X-chromosome in liver cells of Rattus norvegicus. Exp Cell Res 18:415–418
Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E (2004) Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303:644–649
Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA (2007) Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1:299–312
Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH (2002) Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science 298:1620–1623
Pasini D, Hansen KH, Christensen J, Agger K, Cloos PA, Helin K (2008) Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev 22:1345–1355
Peters AH, Mermoud JE, O’Carroll D, Pagani M, Schweizer D, Brockdorff N, Jenuwein T (2002) Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 30:77–80
Petty EL, Collette KS, Cohen AJ, Snyder MJ, Csankovszki G (2009) Restricting dosage compensation complex binding to the X chromosomes by H2A.Z/HTZ-1. PLoS Genet 5:e1000699
Pferdehirt RR, Kruesi WS, Meyer BJ (2011) An MLL/COMPASS subunit functions in the C elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes Dev 25:499–515
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300:131–135
Plath K, Talbot D, Hamer KM, Otte AP, Yang TP, Jaenisch R, Panning B (2004) Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J Cell Biol 167:1025–1035
Popova BC, Tada T, Takagi N, Brockdorff N, Nesterova TB (2006) Attenuated spread of X-inactivation in an X; autosome translocation. Proc Natl Acad Sci USA 103:7706–7711
Powell JR, Jow MM, Meyer BJ (2005) The T-box transcription factor SEA-1 is an autosomal element of the X:A signal that determines C elegans sex. Dev Cell 9:339–349
Pullirsch D, Hartel R, Kishimoto H, Leeb M, Steiner G, Wutz A (2010) The Trithorax group protein Ash2 l and Saf-A are recruited to the inactive X chromosome at the onset of stable X inactivation. Development 137:935–943
Rougeulle C, Chaumeil J, Sarma K, Allis CD, Reinberg D, Avner P, Heard E (2004) Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol Cell Biol 24:5475–5484
Royce-Tolland ME, Andersen AA, Koyfman HR, Talbot DJ, Wutz A, Tonks ID, Kay GF, Panning B (2010) The A-repeat links ASF/SF2-dependent Xist RNA processing with random choice during X inactivation. Nat Struct Mol Biol 17:948–954
Sado T, Li E, Sasaki H (2002) Effect of TSIX disruption on XIST expression in male ES cells. Cytogenet Genome Res 99:115–118
Sado T, Hoki Y, Sasaki H (2006) Tsix defective in splicing is competent to establish Xist silencing. Development 133:4925–4931
Sarma K, Levasseur P, Aristarkhov A, Lee JT (2010) Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc Natl Acad Sci USA 107:22196–22201
Senner CE, Nesterova TB, Norton S, Dewchand H, Godwin J, Mak W, Brockdorff N (2011) Disruption of a conserved region of Xist exon 1 impairs Xist RNA localisation and X-linked gene silencing during random and imprinted X chromosome inactivation. Development 138(8):1541–1550
Shibata S, Lee JT (2004) Tsix transcription- versus RNA-based mechanisms in Xist repression and epigenetic choice. Curr Biol 14:1747–1754
Shin J, Bossenz M, Chung Y, Ma H, Byron M, Taniguchi-Ishigaki N, Zhu X, Jiao B, Hall LL, Green MR et al (2010) Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 467:977–981
Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4:481–495
Smith ER, Allis CD, Lucchesi JC (2001) Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem 276:31483–31486
Smith KP, Byron M, Clemson CM, Lawrence JB (2004) Ubiquitinated proteins including uH2A on the human and mouse inactive X chromosome: enrichment in gene rich bands. Chromosoma 113:324–335
Sun BK, Deaton AM, Lee JT (2006) A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell 21:617–628
Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI (2008) The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol 15:1318–1325
Takagi N (1983) De novo X-chromosome inactivation in somatic hybrid cells between the XO mouse embryonal carcinoma cell and XY rat lymphocyte. Exp Cell Res 145:397–404
Takagi N (1993) Variable X chromosome inactivation patterns in near-tetraploid murine EC x somatic cell hybrid cells differentiated in vitro. Genetica 88:107–117
Tang YA, Huntley D, Montana G, Cerase A, Nesterova TB, Brockdorff N (2010) Efficiency of Xist-mediated silencing on autosomes is linked to chromosomal domain organisation. Epigenetics Chromatin 3:10
Tian D, Sun S, Lee JT (2010) The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143:390–403
Turner BM, Birley AJ, Lavender J (1992) Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell 69:375–384
Vigneau S, Augui S, Navarro P, Avner P, Clerc P (2006) An essential role for the DXPas34 tandem repeat and Tsix transcription in the counting process of X chromosome inactivation. Proc Natl Acad Sci USA 103:7390–7395
Wang Z, Willard HF, Mukherjee S, Furey TS (2006) Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comput Biol 2:e113
Webb S, de Vries TJ, Kaufman MH (1992) The differential staining pattern of the X chromosome in the embryonic and extraembryonic tissues of postimplantation homozygous tetraploid mouse embryos. Genet Res 59:205–214
Welshons WJ, Russell LB (1959) The Y-Chromosome as the Bearer of Male Determining Factors in the Mouse. Proc Natl Acad Sci USA 45:560–566
Wutz A, Jaenisch R (2000) A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell 5:695–705
Wutz A, Rasmussen TP, Jaenisch R (2002) Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30:167–174
Xu N, Tsai CL, Lee JT (2006) Transient homologous chromosome pairing marks the onset of X inactivation. Science 311:1149–1152
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322:750–756
Acknowledgments
We gratefully acknowledge Ina Dahlsveen and Peter Becker for providing images of the Drosophila DCC, and Te Wen Lo and Barbara J. Meyer for C. elegans images and sharing of unpublished data. We thank all members of the Gribnau lab for discussions and J. Anton Grootegoed, Tahsin Stefan Barakat and Cristina Gontan Pardo for helpful comments on this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pontier, D.B., Gribnau, J. Xist regulation and function eXplored. Hum Genet 130, 223–236 (2011). https://doi.org/10.1007/s00439-011-1008-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1008-7