
Abstract
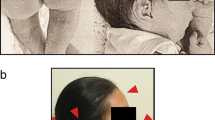
We report a nine-year-old girl (patient 1934) and a five-year-old boy (patient 2170) with small, de novo supernumerary marker chromosomes (SMCs) derived from proximal 17p. The clinical features of patient 1934 include developmental delay, triangular face, prominent forehead, low set ears, dental abnormalities, a high arched palate, long, flexible fingers, and joint laxity. Patient 2170 is affected with developmental delay, oral-motor dyspraxia/verbal apraxia, thick upper and lower lips, bilateral fifth finger clinodactyly, joint laxity and mild hypotonia. G-banded chromosome analysis of patient 1934 revealed mosaicism for a SMC in 72% of peripheral lymphocytes analyzed, whereas analysis of patient 2170 identified a smaller SMC present in 100% of cells analyzed. Fluorescence in situ hybridization (FISH) studies demonstrated that both of the SMCs derived from 17p10-p11.2. Using FISH and array-CGH analysis, the proximal breakpoints mapped within the centromere and the distal breakpoints were both located within the Smith-Magenis syndrome (SMS) common deletion region. We compare the clinical characteristics of our patients with those previously reported to have either SMC including 17p or duplications of proximal 17p in an effort to further delineate the phenotype of trisomy 17p10-p11.2 and to elucidate genotype-phenotype correlations.



Similar content being viewed by others


References
Balarin MA, da Silva Lopes VL, Varella-Garcia M (1999) A dup(17)(p11.2p11.2) detected by fluorescence in situ hybridization in a boy with Alport syndrome. Am J Med Genet 82:183–186
Bi W, Yan J, Stankiewicz P, Park SS, Walz K, Boerkoel CF, Potocki L, Shaffer LG, Devriendt K, Nowaczyk MJ, Inoue K, Lupski JR (2002) Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res 12:713–728
Brown A, Phelan MC, Patil S, Crawford E, Rogers RC, Schwartz C (1996) Two patients with duplication of 17p11.2: the reciprocal of the Smith-Magenis syndrome deletion? Am J Med Genet 63:373–377
Bruder CE, Hirvela C, Tapia-Paez I, Fransson I, Segraves R, Hamilton G, Zhang XX, Evans DG, Wallace AJ, Baser ME, Zucman-Rossi J, Hergersberg M, Boltshauser E, Papi L, Rouleau GA, Poptodorov G, Jordanova A, Rask-Andersen H, Kluwe L, Mautner V, Sainio M, Hung G, Mathiesen T, Moller C, Pulst SM, Harder H, Heiberg A, Honda M, Niimura M, Sahlen S, Blennow E, Albertson DG, Pinkel D, Dumanski JP (2001) High resolution deletion analysis of constitutional DNA from neurofibromatosis type 2 (NF2) patients using microarray-CGH. Hum Mol Genet 10:271–282
Buckton KE, Spowart G, Newton MS, Evans HJ (1985) Forty-four probands with an additional “marker” chromosome. Hum Genet 69:353–370
Chance PF, Bird TD, Matsunami N, Lensch MW, Brothman AR, Feldman GM (1992) Trisomy 17p associated with Charcot-Marie-Tooth neuropathy type 1A phenotype: evidence for gene dosage as a mechanism in CMT1A. Neurology 42:2295–2299
Crolla JA (1998) FISH and molecular studies of autosomal supernumerary marker chromosomes excluding those derived from chromosome 15: II. Review of the literature. Am J Med Genet 75:367–381
Docherty Z, Hulten MA, Honeyman MM (1983) De novo tandem duplication 17p11 leads to cen. J Med Genet 20:138–142
Dupont C, Pipiras E, Chantot-Bastaraud S, Verloes A, Baumann C, Wolf JP, Benzacken B (2003) CGH and direct diagnosis of mosaic structural chromosomal abnormalities: description of a mosaic ring chromosome 17 and review of the literature. Eur J Hum Genet 11:452–456
Fagan K, Edwards M (1997) Supernumerary ring chromosome 17 identified by fluorescent in situ hybridization. Am J Med Genet 69:352–355
Inoue K, Dewar K, Katsanis N, Reiter LT, Lander ES, Devon KL, Wyman DW, Lupski JR, Birren B (2001) The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res 11:1018–33
King PH, Waldrop R, Lupski JR, Shaffer LG (1998) Charcot-Marie-Tooth phenotype produced by a duplicated PMP22 gene as part of a 17p trisomy-translocation to the X chromosome. Clin Genet 54:413–416
Kozma C, Meck JM, Loomis KJ, Galindo HC (1991) De novo duplication of 17p [dup(17)(p12----p11.2)]: report of an additional case with confirmation of the cytogenetic, phenotypic, and developmental aspects. Am J Med Genet 41:446–450
Kozma C, Blancato J, Meck J, Jiang Y (1998) Characterization of a supernumerary marker derived from chromosome 17 by microdissection in an adult with MR/MCA. Am J Med Genet 77:19–22
Kulharya AS, Garcia-Heras J, Radtke HB, Norris KS, Keppen LD, Flannery DB (1998) Prenatal diagnosis of a trisomy 17p derived from a de novo non-mosaic satellited marker. Clin Genet 54:421–425
Lupski JR, Wise CA, Kuwano A, Pentao L, Parke JT, Glaze DG, Ledbetter DH, Greenberg F, Patel PI (1992) Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat Genet 1:29–33
Lupski JR, Garcia CA (2001) Charcot-Marie-Tooth peripheral neuropathies and related disorders. In: Scriver CR, Beaudet AL, Valle D, et al (eds) The metabolic and molecular bases of inherited diseases, vol IV. McGraw-Hill, New York, pp 5759–5788
Magenis RE, Brown MG, Allen L, Reiss J (1986) De novo partial duplication of 17p [dup(17)(p12----p11.2)]: clinical report. Am J Med Genet 24:415–420
Morrison PJ, Smith NM, Martin KE, Young ID (1997) Mosaic partial trisomy 17 due to a ring chromosome identified by fluorescence in situ hybridisation. Am J Med Genet 68:50–53
Murakami T, Lupski JR (1996) A 1.5-Mb cosmid contig of the CMT1A duplication/HNPP deletion critical region in 17p11.2-p12. Genomics 34:128–133
Potocki L, Chen KS, Koeuth T, Killian J, Iannaccone ST, Shapira SK, Kashork CD, Spikes AS, Shaffer LG, Lupski JR (1999) DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am J Hum Genet 64:471–478
Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR (2000) Molecular mechanism for duplication 17p11.2 — the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 24:84–87
Roa BB, Greenberg F, Gunaratne P, Sauer CM, Lubinsky MS, Kozma C, Meck JM, Magenis RE, Shaffer LG, Lupski JR (1996) Duplication of the PMP22 gene in 17p partial trisomy patients with Charcot-Marie-Tooth type-1 neuropathy. Hum Genet 97:642–649
Rosenberg C, Borovik CL, Canonaco RS, Sichero LC, Queiroz AP, Vianna-Morgante AM (1995) Identification of a supernumerary marker derived from chromosome 17 using FISH. Am J Med Genet 59:33–35
Sachs ES, Van Hemel JO, Den Hollander JC, Jahoda MG (1987) Marker chromosomes in a series of 10,000 prenatal diagnoses. Cytogenetic and follow-up studies. Prenat Diagn 7:81–89
Schneider MC, Hughes CR, Forrester S, Kimonis V (2000) Mild phenotype due to tandem duplication of l7p11.2. Am J Med Genet 94:296–299
Shaffer LG, Kennedy GM, Spikes AS, Lupski JR (1997) Diagnosis of CMT1A duplications and HNPP deletions by interphase FISH: implications for testing in the cytogenetics laboratory. Am J Med Genet 69:325–331
Shaw CJ, Shaw CA, Yu W, Stankiewicz P, White LD, Beaudet AL, Lupski JR (2004a) Comparative genomic hybridisation using a proximal 17p BAC/PAC array detects rearrangements responsible for four genomic disorders. J Med Genet 41:113–119
Shaw CJ, Stankiewicz P, Christodoulou J, Smith E, Jones K, Lupski JR (2004b) A girl with duplication 17p10-p12 associated with a dicentric chromosome. Am J Med Genet 124A:173–178
Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH (2003) Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 33:466–468
Stankiewicz P, Park SS, Holder SE, Waters CS, Palmer RW, Berend SA, Shaffer LG, Potocki L, Lupski JR (2001) Trisomy 17p10-p12 resulting from a supernumerary marker chromosome derived from chromosome 17: molecular analysis and delineation of the phenotype. Clin Genet 60:336–344
Stankiewicz P, Shaw CJ, Dapper JD, Wakui K, Shaffer LG, Withers M, Elizondo L, Park SS, Lupski JR (2003) Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet 72:1101–1116
Upadhyaya M, Roberts SH, Farnham J, MacMillan JC, Clarke A, Heath JP, Hodges IC, Harper PS (1993) Charcot-Marie-tooth disease 1A (CMT1A) associated with a maternal duplication of chromosome 17p11.2→12. Hum Genet 91:392–394
Veltman JA, Jonkers Y, Nuijten I, Janssen I, van der Vliet W, Huys E, Vermeesch J, Van Buggenhout G, Fryns JP, Admiraal R, Terhal P, Lacombe D, van Kessel AG, Smeets D, Schoenmakers EF, van Ravenswaaij-Arts CM (2003) Definition of a critical region on chromosome 18 for congenital aural atresia by array CGH. Am J Hum Genet 72:1578–1584
Warburton D (1991) De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 49:995–1013
Yu W, Ballif BC, Kashork CD, Heilstedt HA, Howard LA, Cai WW, White LD, Liu W, Beaudet AL, Bejjani BA, Shaw CA, Shaffer LG (2003) Development of a comparative genomic hybridization microarray and demonstration of its utility with 25 well-characterized 1p36 deletions. Hum Mol Genet 12:2145–2152
Acknowledgements
We thank the patients and their parents for participation, Dr. Lorraine Potocki for critical review of the manuscript and providing additional clinical information on patients with dup(17)(p11.2p11.2) syndrome, and Dr. Wei Yu for technical assistance. This work was supported in part by the National Institute for Neurological Disorders and Strokes (RO1 NS27042 to J.R.L.), the National Institute for Child Health and Human Development (PO1 HD39420 to J.R.L.), and FIS-ISCIII CO3/07 and GO3/184 to L.P.J. and X.E..
Author information
Authors and Affiliations
Corresponding author
Electronic database information
Electronic database information
BACPAC Resource Center, http://www.chori.org/bacpac
Rights and permissions
About this article
Cite this article
Shaw, C.J., Stankiewicz, P., Bien-Willner, G. et al. Small marker chromosomes in two patients with segmental aneusomy for proximal 17p. Hum Genet 115, 1–7 (2004). https://doi.org/10.1007/s00439-004-1119-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1119-5