
Abstract
Fiber quality and yield are important traits of cotton. Quantitative trait locus (QTL) mapping is a prerequisite for marker-assisted selection (MAS) in cotton breeding. To identify QTLs for fiber quality and yield traits, 4 backcross-generation populations (BC1F1, BC1S1, BC2F1, and BC3F0) were developed from an interspecific cross between CCRI36 (Gossypium hirsutum L.) and Hai1 (G. barbadense L.). A total of 153 QTLs for fiber quality and yield traits were identified based on data from the BC1F1, BC1S1, BC2F1 and BC3F0 populations in the field and from the BC2F1 population in an artificial disease nursery using a high-density genetic linkage map with 2292 marker loci covering 5115.16 centimorgans (cM) from the BC1F1 population. These QTLs were located on 24 chromosomes, and each could explain 4.98–19.80% of the observed phenotypic variations. Among the 153 QTLs, 30 were consistent with those identified previously. Specifically, 23 QTLs were stably detected in 2 or 3 environments or generations, 6 of which were consistent with those identified previously and the other 17 of which were stable and novel. Ten QTL clusters for different traits were found and 9 of them were novel, which explained the significant correlations among some phenotypic traits in the populations. The results including these stable or consensus QTLs provide valuable information for marker-assisted selection (MAS) in cotton breeding and will help better understand the genetic basis of fiber quality and yield traits, which can then be used in QTL cloning.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton is an important economic crop worldwide that produces natural fibers used as raw materials in the textile industry. With the development of spinning technologies and mechanization of cotton harvesting, the demand for higher cotton fiber quality has increased. The current goal of cotton breeding is the development of new cultivars with higher fiber yield and superb fiber quality. Gossypium barbadense and G. hirsutum are the two most economically important tetraploid cultivated species. The former (including Egyptian cotton, Pima cotton and Sea–Island cotton) has longer, stronger and finer fibers; However, its disadvantages were a low yield and narrow adaptation. The latter (Upland cotton), has a high yield and is widely grown in many countries, accounting for approximately 95% of global cotton production (Chen et al. 2007); however, it has a relatively low fiber quality. Therefore, it is of great significance to effectively transfer genes associated with high-quality traits from G. barbadense to G. hirsutum for synchronous genetic improvement of yield and fiber quality. No breakthrough has been reported in creating elite varieties with both high yield from G. hirsutum and high fiber quality from G. barbadense using traditional breeding methods (Zhang and Percy 2007). However, it was possible to transfer desirable genes from G. barbadense to G. hirsutum based on quantitative trait loci (QTL) mapping and new biotechnology.
Since the first cotton molecular genetic map was reported by Reinisch et al. (1994). More than 1000 QTLs for fiber quality and yield in cotton have been mapped using interspecific populations of G. hirsutum × G. barbadense and intraspecific populations of G. hirsutum (Fang et al. 2014; Jamshed et al. 2016; Keerio et al. 2018; Li et al. 2017a; Liu et al. 2017; Ma et al. 2017; Said et al. 2015a, b; Shang et al. 2016; Wang et al. 2015, 2016, 2017a, b; Yu et al. 2014; Zhai et al. 2016; Zhang et al. 2015b). The cotton genome draft sequences have laid a foundation for further QTL localization and molecular breeding at the genomic level (Li et al. 2014, 2015; Wang et al. 2012a; Zhang et al. 2015a). Many QTLs have been detected using natural populations, recombinant inbred or backcross inbred line populations or other populations based on single nucleotide polymorphism (SNP) markers (Ademe et al. 2017; Islam et al. 2016; Keerio et al. 2018; Li et al. 2016b; Ma et al. 2018; Sun et al. 2017; Zhang et al. 2016b). These studies provided important information for further study of QTLs/genes related to fiber quality and fiber yield and for marker-assisted selection (MAS) in breeding. However, only a few QTLs have been used in MAS (Cao et al. 2015). Most, if not all, linkage-based QTL mapping studies report results from only one mapping population.
To transfer desirable genes from G. barbadense to G. hirsutum, we developed different interspecific backcross populations and chromosome segment substitution line (CSSL) populations (Li et al. 2016a, 2017b; Lu et al. 2017; Song et al. 2017; Zhai et al. 2016; Lan et al. 2011; Liang et al. 2010; Kong et al. 2018) and constructed a high-density simple sequence repeat (SSR) genetic linkage map of the BC1F1 population, which comprised 2292 loci covering 5115.16 centimorgans (cM) of the cotton genome with an average distance of 2.23 cM between markers (Shi et al. 2015). This study reports the identification of QTLs related to fiber yield and fiber quality traits using a high-density map and 5 backcross populations from the same interspecific hybrid of the two cultivated tetraploid species. Consistent QTLs across different populations or environments were identified, which provides useful information to facilitate the understanding of the genetic basis of fiber quality and yield traits and lays a foundation for the simultaneous improvement of fiber quality and yield.
Materials and methods
Plant materials
Three backcross generations (BC1F1, BC1S1 and BC2F1) were derived from an interspecific cross between CCRI36 (G. hirsutum L.) and Hai1 (G. barbadense L.), of which CCRI36, the recipient parent, is a high yield cultivar bred by the Institute of Cotton Research (ICR), Chinese Academy of Agricultural Sciences (CAAS) and Hai1, the donor parent, is a cultivated cotton line of G. barbadense with superb fiber quality, and high resistance to Verticillium wilt (VW) and the dominant glandless trait (Shi et al. 2016).
The BC1F1 population included 135 individual plants, and the two parents were grown in the field (Anyang Experiment Farm, ICR, CAAS) in 2004. Each row was 8 m long and the rows were 0.8 m apart. The BC2F1 population (with 133 family lines), the BC1S1 population (with 120 family lines), and the two parents were planted in the field (Anyang Experiment Farm, ICR, CAAS) in 2005, with single-row plots for each family and two-row plots for the two parents. Each row was 8 m long and 0.8 m wide and included 32 plants. Additionally, 133 BC2F1 families were backcrossed with CCRI36, and the contemporary backcross bolls were named the BC3F0 population. The BC2F1 population (with 133 family lines) was planted in an artificial disease nursery (the BC2F1-DN population) in 2005 following a randomized complete block design with three replicates and one row (plot) for each family. Each row was 2.5 m long and contained 15 plants, and the rows were 0.6 m apart (Shi et al. 2016).
The glanded plants in the BC1F1, BC2F1 and BC1S1 population were removed at the seedling stage. Five populations (BC1F1, BC2F1, BC1S1, BC2F1-DN, and BC3F0) in 3 environments (the field in 2004, the artificial disease nursery in 2005, the field in 2005) were used to detect QTLs for fiber quality and yield traits in the present study.
Phenotypic evaluation
We harvested naturally opened self-pollinated bolls from each plant in the BC1F1 population in 2004, 30 naturally opened self-pollinated bolls from each plot for the BC1S1, BC2F1 and BC2F1-DN populations, and naturally opened backcrossed bolls (BC3F0) from each family in the BC2F1 population in the field in 2005 to evaluate fiber quality and yield traits. Three fiber quality traits and four yield components were analyzed, namely, boll weight (BW), lint percentage (LP), seed index (SI), lint index (LI), fiber length (i.e., mean upper-half length, FL), fiber strength (FS), and fiber micronaire (FM).
The phenotypic data for each of the 5 populations (BC1F1, BC2F1, BC1S1, BC2F1-DN, and BC3F0) were independent.
Map construction
Cotton genomic DNA was extracted from young leaves of the 135 BC1F1 plants and the two parents as described by Paterson et al. (1993). Genotyping and genetic linkage map construction for the BC1F1 population were previously described (Shi et al. 2015). Briefly, the genetic map consists of 2292 marker loci on 26 chromosomes with a total length of 5115.16 cM and an average distance of 2.23 cM between markers. This high-density genetic linkage map served as a foundation for QTL analysis of the 5 populations (BC1F1, BC1S1, BC2F1, BC2F1-DN and BC3F0).
QTL analysis
The QTLs for fiber quality and yield traits were analyzed using the composite interval mapping method (Zeng 1994) and Windows QTL Cartographer 2.5 (Wang et al. 2006) with a walk speed of 1 cM, 5 control markers, and 1000 permutation tests (Shi et al. 2015). Positive additive effects indicated that Hai1 alleles decreased the phenotypic trait values, and negative scores indicated that Hai1 alleles increased the phenotypic trait values. The CCRI36 alleles had the opposite effects. The QTLs were named as follows: (q + trait abbreviation) + chromosome/linkage group + QTL number. For the same trait, QTL across different generations or environments were considered stable when their confidence intervals overlapped (Sun et al. 2012).
Results
Performance of fiber quality and yield traits of the populations and their parents
The CCRI36 and Hai1 parents differed in yield and fiber quality traits in that the former had a greater BW and LI, lower SI and intermediate fiber quality, while the latter had longer, stronger fibers, a lower BW and LI, and a higher SI (Table 1).
The mean values of FL and FS in the 5 populations were slightly higher than those of the recurrent parent (CCRI36), and the mean values of FM in the 5 populations were slightly lower or similar to those of the parent (CCRI36). The mean values of BW and LI in 4 populations were slightly higher than those of the parent Hai1. The mean values of SI were similar to that of the parent Hai1. Large variation in all traits was observed among interspecific populations and transgressive segregation was detected in all the populations (Table 1; Fig. 1). FM exhibited more variation than the other fiber quality traits, and BW displayed more variation than SI. As expected, the earlier-generation populations exhibited more phenotypic variation than the later-generation populations. The FL, FS and FM in all 5 populations, SI and LI in 4 populations (BC1S1, BC2F1, BC3F0 and BC2F1-DN), and BW in 4 populations (BC1F1, BC1S1, BC2F1 and BC2F1-DN) exhibited a continuous and normal distribution (Fig. 1).

Frequency distribution of 6 traits of fiber quality and yield in the interspecific backcross populations of Gossypium hirsutum × Gossypium barbadense
An overall description of the QTLs detected in the 5 populations
A total of 2292 marker loci distributed on 26 chromosomes (Shi et al. 2015) were used to map QTLs in the BC1F1, BC1S1, BC2F1, BC2F1-DN and BC3F0 populations. Two yield traits (SI and LI) were not measured in the BC1F1 population due to a low seed number for individual plants, and BW was not measured for the BC3F0 population because of artificial-hybridization.
In the 2004 BC1F1 population, a total of 22 QTLs were detected, including 4 for FL, 6 for FS, 7 for FM, and 5 for BW. In the 2005 BC1S1 population, 37 QTLs were detected, including 10 for FL, 5 for FS, 6 for FM, 5 for SI, 8 for LI, and 3 for BW, which showed that the number of QTLs detected for FL was the largest and the number of QTLs detected for BW was the smallest. In the 2005 BC2F1 population, more QTLs (44) were detected, including 7 for FL, 7 for FS, 7 for FM, 10 for SI, 6 for LI, and 7 for BW. In the 2005 BC2F1-DN population, a total of 37 QTLs were detected, including 7 for FL, 6 for FS, 5 for FM, 5 for SI, 8 for LI, and 6 for BW. In the 2005 BC3F0 population, 39 QTLs were detected, including 8 for FL, 5 for FS, 9 for FM, 11 for SI and 6 for LI. Most QTLs were detected in the BC2F1 population, the fewest QTLs in the BC1F1 population, and similar numbers of QTLs were identified in the BC1S1, BC2F1-DN, and BC3F0 populations.
QTL mapping of fiber quality traits in interspecific backcross populations
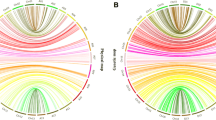
A total of 80 QTLs controlling fiber quality traits were identified in all 5 interspecific backcross populations: 33 for FL, 24 for FS, and 23 for FM. These QTLs were distributed on 19 chromosomes and explained 5.17–19.80% of the total phenotypic variation. Of these QTLs, 16 were stable, i.e., detected in 2 or 3 populations: 3 of them for FL, 5 for FS, and 8 for FM (Table 2; Fig. 2).


Chromosomal locations of QTLs for 6 traits of fiber quality and yield in the interspecific backcross populations of Gossypium hirsutum × Gossypium barbadense. Asterisk, stable QTL; the number of asterisk, the number of environments in which the QTL was detected
Fiber length
A total of 33 QTLs for FL were identified in the 5 populations (BC1F1, BC1S1, BC2F1, BC2F1-DN and BC3F0), each with a phenotypic variation explained (PVE) value of 5.67–19.80%; the QTLs were distributed on 14 chromosomes: 6 on C16, 4 each on C2, C10 and C11, 3 each on C4 and C8, 2 on C21, and only 1 on C3, C9, C12, C15, C20, C23 and C25. Thus, the QTLs for FL were clustered on C16, C2, C10 and C11.
Twenty-five of the 33 QTLs had positive additive effects, in which CCRI36 alleles increased FL by about 0.51–1.66 mm, whereas 8 QTLs had negative additive effects, in which Hai1 alleles increased FL by about 0.56–1.13 mm.
Notably, three of them could be stable: qFL-C16-4 was detected in both the BC1S1 and BC3F0 populations, with a PVE of 13.63% and 16.15%, respectively, and qFL-C16-5 was detected in both the BC2F1 and BC3F0 populations, with a PVE of 7.25% and 18.25%, respectively. The QTL qFL-C20-1 was detected in both the BC2F1-DN and BC3F0 populations, with a PVE of 5.95% and 11.80%, respectively. CCRI36 alleles increased FL by approximately 0.52–1.17 mm.
Fiber strength
In total 24 QTLs for FS were detected in the 5 populations (BC1F1, BC1S1, BC2F1, BC2F1-DN and BC3F0), each with PVE 6.13–14.05%, and located on 10 chromosomes, 5 QTLs on C21, 4 each on C11 and C16, 3 each on C5 and C10, and only 1 on C4, C9, C12, C13 and C20, respectively. It showed that C21, C11 and C16 contained more QTL for FS. Thirteen of the 24 QTLs had negative additive effects, in which Hai1 alleles increased FS about 0.85–1.99 cN/tex.
Importantly, 5 of them could be stable: 2 (qFS-C11-1 and qFS-C11-2) were detected in both the BC1S1 and BC3F0, with PVE 7.98–11.29%, and 2 (qFS-C11-3 and qFS-C11-4) were detected in both the BC2F1 and BC1F1-DN, explaining the phenotypic variations by 6.86–8.41%. Hai1 alleles increased FS by approximately 0.85–1.43 cN/tex. One of them was mapped on C16, qFS-C16-1 was detected in both the BC1S1 and BC3F0, explaining the phenotypic variations by 6.28% and 10.46%. CCRI36 alleles increased FS by approximately 0.91–1.44 cN/tex.
Fiber micronaire
In total, 23 QTLs for FM were identified in the 5 populations, each with a PVE of 5.17–13.75%; the QTLs were distributed on 8 chromosomes: 7 on C21, 4 on C15, 5 on C10, 3 on C24, and only 1 on C1, C5, C11 and C25. Therefore, C21, C15, C10 and C24 contained most QTLs for FM. Hai1 alleles decreased FM by approximately 0.14–0.40 units for 12 of the 23 QTLs, whereas CCRI36 alleles decreased FM by approximately 0.13–0.36 units for the other 11 QTLs.
Notably, 8 of the QTLs were stable: qFM-C21-1 was detected in both the BC1F1-DN and BC3F0 populations, with a PVE of 11.24% and 13.75%, respectively, qFM-C21-2 was detected in 3 populations (BC2F1, BC1F1-DN and BC3F0), with a PVE of 8.10–13.74%, and qFM-C21-3 and qFM-C21-4 were detected in both the BC2F1 and BC3F0 populations, with a PVE of 7.34–11.51%. CCRI36 alleles decreased FM by approximately 0.16–0.23 units. The QTL qFM-C15-3 and qFM-C15-4 were simultaneously detected in the BC2F1, BC2F1-DN and BC3F0 populations, with a PVE of 5.82–10.77% and 8.06–12.22%, respectively. Hai1 alleles at the 2 QTLs decreased FM by approximately 0.14–0.19 units and 0.17–0.20 units, respectively. The QTL qFM-C10-5 was detected in both the BC1S1 and BC1F1 populations, with a PVE of 6.93% and 11.34%, respectively. Hai1 alleles increased FM by approximately 0.24–0.36 units. The QTL qFM-C1-2 was detected in both the BC1F1 and BC3F0 populations, with a PVE of 7.18% and 12.73%, respectively. Hai1 alleles decreased FM by approximately 0.16–0.40 units.
QTL mapping of yield traits in interspecific backcross populations
A total of 73 QTLs for 3 fiber yield traits were detected on 20 chromosomes in 4 of the 5 interspecific backcross populations. The PVE of each QTL was 4.98–18.92%. Seven of the QTLs were stable, i.e., detected in 2 or 3 populations simultaneously: 4 for SI and 3 for LI (Table 3; Fig. 2).
Boll weight
In total, 21 QTLs for BW were identified in the 4 populations (BC1F1, BC1S1, BC2F1, and BC1F1-DN), each with a PVE of 5.87–16.58%. Among the QTLs for BW, 5, 3, 7 and 6 QTLs were detected in the BC1F1, BC1S1, BC2F1 and BC1F1-DN populations, respectively. The QTLs were found on 12 chromosomes: 4 each on C18 and C21, 2 each on C10, C14 and C17, and only 1 on C2, C3, C4, C11, C15, C19 and C20. Six QTLs (qBW-C10-1, qBW-C10-2, qBW-C11-1, qBW-C17-1, qBW-C17-2, and qBW-C21-1) for BW on C10, C11, C17 and C21 had negative additive effects, in which Hai1 alleles increased BW from 0.34 to − 0.57 g. The other 15 QTLs had positive additive effects, in which the CCRI36 alleles increased BW from 0.20 to 0.57 g.
However, no stable QTLs were detected in 2 or more populations, generations or environments.
Seed index
A total of 27 QTLs for SI were identified in the 4 populations (BC1S1, BC2F1, BC1F1-DN and BC3F0), each with a PVE of 4.98–18.92%; the QTLs were distributed on 12 chromosomes: 5 on C3, 4 each on C14, C17 and C20, 3 on C22, and only 1 on C6, C8, C9, C10, C18, C21 and C25. Hai1 alleles increased SI by approximately 0.31–1.97 g for 15 of the 27 QTLs and decreased SI by approximately 0.29–1.55 g for the other 12 QTLs.
Notably, 4 of these QTLs were stable, and 2 that were detected in both the BC2F1 and BC2F1-DN populations mapped to C14 (qSI-C14-1) and C17 (qSI-C17-2), with a PVE of 7.00–7.35% and 15.75–18.92%, respectively. Hai1 alleles at the 2 QTLs increased SI by approximately 0.47–0.49 g and 0.70–0.81 g, respectively. Two QTLs were detected in both the BC2F1 and BC3F0 populations were mapped to C20 (qSI-C20-1 and qSI-C20-2), with a PVE of 9.48–9.60% and 6.18–8.93%, respectively. Hai1 alleles at the 2 QTLs decreased SI by approximately 0.54–0.59 g and 0.47–0.52 g, respectively.
Lint index
In total, 25 QTLs for LI were identified in 4 populations (BC1S1, BC2F1, BC1F1-DN and BC3F0), each with a PVE of 6.01–14.85%; the QTLs were found on 10 chromosomes: 7 on C16, 4 on C6, 3 each on C8 and C17, 2 each on C11 and C21, and only 1 on C12, C14, C23 and C25. Hai1 alleles increased LI by approximately 0.29–1.55 g for 14 of the 25 QTLs and decreased LI by approximately 0.31–1.97 g for the other 11 QTLs.
Importantly, 3 of the QTLs were stable. One detected in both the BC2F1 and BC1S1 populations mapped to C14 (qLI-C14-1), with a PVE of 8.34–12.29%. Hai1 alleles increased LI by 0.33–0.65 g. Two QTLs (qLI-C16-1 and qLI-C16-2) detected in both the BC2F1 and BC3F0 populations were mapped to C16, with a PVE of 10.93–13.23% and 9.60–12.30%, respectively. Hai1 alleles increased LI by approximately 0.42–1.95 g and 0.40–1.83 g, respectively.
QTL clusters
Using the same linkage map from the BC1F1 population, a total of 179 QTLs for fiber quality and yield traits (7 traits: FL, FS, FM, BW, LP, SI, LI) were identified, of which 26 QTLs for LP were identified on 9 chromosomes with phenotype data related to the BC1F1 population (Shi et al. 2015).
Through comprehensive analysis, we found that 10 QTL clusters, including 71 QTLs, were related to 2 or more traits, which is a common phenomenon in cotton (Rong et al. 2007; Lacape et al. 2010; Shen et al. 2007; Said et al. 2013, 2015b; Li et al. 2016b; Keerio et al. 2018). Two QTL clusters were detected on C16, C17 and C21, and one was detected on C4, C6, C10 and C11 (Table 4; Table S1).
There were 6 QTL clusters related to both fiber quality and yield traits, namely, C16-Cluster-1, C16-Cluster-2, C21-Cluster-1, C21-Cluster-2, C10-Cluster-1 and C4-Cluster-1. Thirteen QTLs were in C16-Cluster-1 on C16 (137–161 cM), including 3 for FL (qFL-C16-1, qFL-C16-2, and qFL-C16-3) and 1 for FS (qFS-C16-1) with positive additive effects and 5 for LP (qLP-C16-1, qLP-C16-2, qLP-C16-3, qLP-C16-4, and qLP-C16-5) and 4 for LI (qLI-C16-1, qLI-C16-2, qLI-C16-3, and qLI-C16-4) with negative additive effects. The same results were observed for C16-Cluster-2 on C16 (169–193 cM), which harbored 12 QTLs, including 3 for FL (qFL-C16-4, qFL-C16-5, and qFL-C16-6) and 3 for FS (qFS-C16-2, qFS-C16-3, and qFS-C16-4) with positive additive effects and 3 for LP (qLP-C16-6, qLP-C16-7, and qLP-C16-8) and 3 for LI (qLI-C16-5, qLI-C16-6, and qLI-C16-7) with negative additive effects. C21-Cluster-1, in a region (60–76 cM) on C21, contained 4 QTLs, namely, qFL-C21-1 for FL and qFS-C21-4 for FS with positive additive effects, and qFM-C21-1 for FM and qLP-C21-1 for LP with negative additive effects. C10-Cluster-1, in a region (167–192 cM) on C10, harbored 9 QTLs, including 4 for FL (qFL-C10-1, qFL-C10-2, qFL-C10-3, and qFL-C10-4) with positive additive effects and 3 for FM (qFM-C10-3, qFM-C10-4, and qFM-C10-5) and 2 for BW (qBW-C10-1 and qBW-C10-2) with negative additive effects. C4-Cluster-1, on C4 (61–86 cM), harbored 5 QTLs, 3 (qFL-C4-1, qFL-C4-2, and qFL-C4-3) for FL, one (qFS-C4-1) for FS and one (qBW-C4-1) for BW, all with a positive additive effect. C21-Cluster-2, on C21 (83–101 cM), harbored 6 QTLs, namely, qFL-C21-2 for FL and qSI-C21-1 for SI with positive additive effects and qFM-C21-2, qFM-C21-3, qFM-C21-4, and qFM-C21-5 for FM with negative additive effects.
There were 3 QTL clusters related to fiber yield, namely, C6-Cluster-1, C17-Cluster-1 and C17-Cluster-2. Six QTLs were located in C17-Cluster-2 on C17 (111–120 cM): 2 (qBW-C17-1 and qBW-C17-2) for BW with negative additive effects and 2 (qLP-C17-4 and qLP-C17-5) for LP and 2 (qLI-C17-2 and qLI-C17-3) for LI with positive additive effects. C6-Cluster-1, in a region (36–59 cM) on C6, harbored 5 QTLs, namely, qSI-C6-1 for SI and qLI-C6-1, qLI-C6-2, qLI-C6-3, and qLI-C6-4 for LI, all with positive additive effects. C17-Cluster-1, on C17 (77–100 cM), harbored 7 QTLs, namely, qLP-C17-1, qLP-C17-2, and qLP-C17-3 for LP with positive additive effects and qSI-C17-1, qSI-C17-2, qSI-C17-3, and qSI-C17-4 for SI with negative additive effects.
C11-Cluster-1, on C11 (63–90 cM), was associated with fiber quality, harboring qFL-C11-2 and qFL-C11-3 for FL and qFS-C11-1 and qFS-C11-2 for FS, all with negative additive effects.
Discussion
The location of QTLs
The linkage map was constructed with 2292 SSR marker loci, covering 5115.16 cM of the AD genome with an average distance of 2.23 cM between markers (Shi et al. 2015). The length of the linkage map is similar to that of the AD genome in cotton, and the SSR marker loci are distributed throughout the genome. We used the high-density genetic linkage map and data from 5 backcross populations (BC1F1, BC2F1, BC1S1, BC2F1-DN, and BC3F0) in 4 different generations (BC1F1, BC2F1, BC1S1, and BC3F0) to identify the QTLs of fiber yield and fiber quality in 3 environments (the field in 2004, an artificial disease nursery in 2005, and the field in 2005). A total of 153 QTLs were identified, including 80 QTLs for 3 fiber quality traits and 73 QTLs for 3 yield traits in the present paper (Tables 2, 3; Fig. 2). Twenty-three QTLs were consistently detected in 2 or 3 populations, including 3 for FL, 5 for FS, 8 for FM, 4 for SI, and 3 for LI.
Among the 153 QTLs detected in the present study, 30 QTLs were reported in previous studies with a common marker in the confidence interval on the same chromosome. Ten QTLs for FL (qFL-C3-1, qFL-C4-2, qFL-C8-1, qFL-C8-3, qFL-C10-2, qFL-C10-4, qFL-C11-2, qFL-C16-2, qFL-C16-3, and qFL-C20-1) were reported previously (Chen et al. 2018; Kong et al. 2018; Lan et al. 2011; Said et al. 2015b; Sun et al. 2012; Wang et al. 2011, 2013, 2016, 2017a; Zhai et al. 2016; Zhang et al. 2016a). Of these QTLs, qFL-C3-1 was the same as that in three previous reports (Wang et al. 2013, 2016; Chen et al. 2018), and qFL-C16-3 was the same as that in three previous reports (Wang et al. 2011; Said et al. 2015b; Lan et al. 2011). Eight QTLs for FS (qFS-C5-1, qFS-C5-3, qFS-C10-3, qFS-C11-2, qFS-C16-1, qFS-C20-1, qFS-C21-3, and qFS-C21-5) were consistent with those reported previously (Said et al. 2015b; Shao et al. 2014; Tang et al. 2015; Wang et al. 2016; Zhai et al. 2016; Kong et al. 2018). Among these 8 common QTLs for FS, qFS-C10-3 may be the same as that in three previous reports (Shao et al. 2014; Tang et al. 2015; Kong et al. 2018). Of 7 common QTLs for FM, one QTL (qFM-C10-3) was reported by three researchers (Said et al. 2015b; Si et al. 2017; Yu et al. 2013). One QTL (qFM-C5-1) was reported by Yu et al. (2013) and Said et al. (2015b). The other 5 QTLs for FM (qFM-C15-2, qFM-C21-1, qFM-C21-2, qFM-C24-3 and qFM-C25-1) were reported in the previous studies (Said et al. 2015b; Liang et al. 2010; Song et al. 2017; Yang et al. 2015). Five QTLs for yield traits (qBW-C10-2, qBW-C21-3, qSI-C14-1, qSI-C18-1 and qLI-C17-1) were reported previously (Kong et al. 2018; Ning et al. 2014; Si et al. 2017; Wang et al. 2015; Wu et al. 2009). Nine QTLs detected in previous studies were confirmed in our advanced backcross populations, including 3 QTLs (qFL-C20-1, qFS-C11-2, and qFS-C20-1) reported by Zhai et al. (2016), 3 QTLs (qBW-C10-2, qFL-C10-4, and qFS-C10-3) reported by Kong et al. (2018), and 3 QTLs (qFL-C16-3, qFM-C21-1 and qFM-C24-3) reported by Lan et al. (2011), Liang et al. (2010), and Song et al. (2017), respectively. Therefore, 30 of the QTLs detected in this study were previously reported, and the other 123 QTLs are considered novel.
In summary, 23 QTLs (qFL-C16-4, qFL-C16-5, qFL-C20-1, qFS-C11-1, qFS-C11-2, qFS-C11-3, qFS-C11-4, qFS-C16-1, qFM-C1-1, qFM-C10-5, qFM-C15-3, qFM-C15-4, qFM-C21-1, qFM-C21-2, qFM-C21-3, qFM-C21-4, qSI-C14-1, qSI-C17-2, qSI-C20-1, qSI-C20-2, qLI-C14-1, qLI-C16-5, and qLI-C16-6) were stable and detected in 2 or more populations in this paper and 30 common QTLs (qFL-C3-1, qFL-C4-2, qFL-C8-1, qFL-C8-3, qFL-C10-2, qFL-C10-4, qFL-C11-2, qFL-C16-2, qFL-C16-3, qFL-C20-1, qFS-C5-1, qFS-C5-3, qFS-C10-3, qFS-C11-2, qFS-C16-1, qFS-C20-1, qFS-C21-3, qFS-C21-5, qFM-C5-1, qFM-C10-3, qFM-C15-2, qFM-C21-1, qFM-C21-2, qFM-C24-1, qFM-C25-1, qBW-C10-2, qBW-C21-3, qSI-C14-1, qSI-C18-1, and qLI-C17-1) were reported in the previous studies (Table S3).
There were 47 stable or common QTLs (qFL-C3-1, qFL-C4-2, qFL-C8-1, qFL-C8-3, qFL-C10-2, qFL-C10-4, qFL-C11-2, qFL-C16-2, qFL-C16-3, qFL-C16-4, qFL-C16-5, qFL-C20-1, qFS-C5-1, qFS-C5-3, qFS-C10-3, qFS-C11-1, qFS-C11-2, qFS-C11-3, qFS-C11-4, qFS-C16-1, qFS-C20-1, qFS-C21-3, qFS-C21-5, qFM-C1-1, qFM-C5-1, qFM-C10-3, qFM-C10-5, qFM-C15-2, qFM-C15-3, qFM-C15-4, qFM-C21-1, qFM-C21-2, qFM-C21-3, qFM-C21-4, qFM-C24-3, qFM-C25-1, qBW-C10-2, qBW-C21-3, qSI-C14-1, qSI-C17-2, qSI-C18-1, qSI-C20-1, qSI-C20-2, qLI-C14-1, qLI-C16-5, qLI-C16-6 and qLI-C17-1), of which 6 QTLs (qFL-C20-1, qFS-C11-2, qFS-C16-1, qFM-C21-1, qFM-C21-2, and qSI-C14-1) were both stable and common QTLs. Therefore, 17 QTLs (qFL-C16-4, qFL-C16-5, qFS-C11-1, qFS-C11-3, qFS-C11-4, qFM-C1-1, qFM-C10-5, qFM-C15-3, qFM-C15-4, qFM-C21-3, qFM-C21-4, qSI-C20-2, qSI-C17-2, qSI-C20-1, qLI-C14-1, qLI-C16-5, and qLI-C16-6) were considered novel stable QTLs.
The detection of QTLs in multiple populations or different genetic backgrounds indicated the stabilities of the genetic effects. These stable and common QTLs might increase the reliability and efficiency of selection and play important roles in the simultaneous improvement of fiber yield and quality by MAS (Shen et al. 2007; Sun et al. 2012; Tang et al. 2015; Wang et al. 2015; Zhai et al. 2016; Zhang et al. 2009).
Contributions of the A and D subgenomes to allotetraploid cottons
In the past 20 years, most of QTL mapping studies have clearly shown that more QTLs for fiber quality traits were situated on the D subgenome than on the A subgenome for fiber quality traits (Fang et al. 2014; Jiang et al. 1998; Paterson et al. 2003; Rong et al. 2007; Said et al. 2013; Shen et al. 2007; Yang et al. 2015). Some studies have shown that almost equal numbers of QTLs are on the two subgenomes (Ning et al. 2014; Wang et al. 2012b, 2015).
In the present study, more QTLs for FL and FS were located on the A subgenome than on the D subgenome (35 on the A subgenome and 22 on the D subgenome), while more QTLs or genes for FM were located on the D subgenome than on the A subgenome (8 on the A subgenome and 15 on the D subgenome). Overall, the number of QTLs for fiber quality traits on the A subgenome was larger than that on the D subgenome (43 vs. 37). However, for yield component traits, more QTLs were distributed on the D subgenome than on the A subgenome in our study. Using the same map and the same populations, 26 QTLs for LP were identified (Shi et al. 2015). A total of 99 QTLs for yield traits (BW, SI, LI, and LP) were found, 33 of which were located on the A subgenome, and 66 QTLs were located on the D subgenome (Table 5).
In summary, more QTLs for both fiber quality and fiber yield traits were detected on the D subgenome than that on the A subgenome (76 vs. 103) (Table 5).
QTL clusters for fiber yield and quality
Ten clusters were identified in this study. These clusters harbored many stable QTLs or common QTLs, of which 17 (qFL-C16-4, qFL-C16-5, qFM-C10-5, qFM-C21-1, qFM-C21-2, qFM-C21-3, qFM-C21-4, qFS-C11-1, qFS-C11-2, qFS-C16-1, qLI-C16-5, qLI-C16-6, qSI-C17-2, qLP-C16-6, qLP-C16-7, qLP-C17-1, and qLP-C17-2) were stable QTLs and 16 (qFL-C4-2, qFL-C10-2, qFL-C10-4, qFL-C11-2, qFL-C16-2, qFL-C16-3, qFM-C10-3, qFM-C21-1, qFM-C21-2, qFS-C11-2, qFS-C16-1, qBW-C10-2, qLP-C16-1, qLP-C16-6, qLP-C17-2, and qLP-C17-5) were common QTLs. Thus, 6 QTLs (qFS-C11-2, qFS-C16-1, qFM-C21-1, qFM-C21-2, qLP-C16-6, and qLP-C17-2) were both stable and common QTLs (Table 4; Table S1).
Among the ten clusters, one (C16-Cluster-1) resembled the C16-Cluster-Gh-2 cluster reported by Said et al. (2015a, b) and Wang et al. (2011). The other 9 clusters are considered novel.
All QTLs for both FL and FS showed the same additive effect direction in all 5 clusters (C4-Cluster-1, C11-Cluster-1, C16-Cluster-1, C16-Cluster-2, and C21-Cluster-1), and QTLs for FL and FM showed opposite additive effect directions in each of 3 clusters (C10-Cluster-1, C21-Cluster-1, and C21-Cluster-2). These QTL clusters revealed a strong positive genetic correlation between FL and FS and a strong negative genetic correlation between FL and FM. Some previous reports revealed similar clusters (Sun et al. 2012; Zhang et al. 2013, 2016a; Fang et al. 2014; Zhai et al. 2016; Islam et al. 2016; Diouf et al. 2018), but these correlations were not explicitly mentioned in their reports. All QTLs for both LP and LI had the same additive effect direction in 3 clusters (C16-Cluster-1, C16-Cluster-2, and C17-Cluster-2), and QTLs for LP and SI had opposite additive effect directions in C17-Cluster-1. These QTL clusters revealed a strong positive genetic correlation between LP and LI and a strong negative genetic correlation between LP and SI. To the best of our knowledge, the same or similar results have not been reported. All QTLs for both FL and FS had positive additive effects and all QTLs for both LP and LI had negative additive effects in 2 clusters (C16-Cluster-1 and C16-Cluster-2). All QTLs for both FL and FS had positive additive effects and all QTLs for both LP and FM had negative additive effects in one cluster (C21-Cluster-1). These QTL clusters revealed a strong negative genetic correlation between LP and FL and between LP and FS. Some previous reports detected similar clusters (Wang et al. 2011; Li et al. 2016b; Si et al. 2017), but these correlations were not explicitly mentioned in their reports.
These results provide an explanation for the significant phenotypic correlations between the related traits in different populations (Table S2) and indicate that these loci might function as pleiotropic genes or are closely linked to various other genes (Rong et al. 2007; Yu et al. 2013; Zhai et al. 2016).
In other words, 5 of the 10 clusters (C10-Cluster-1, C11-Cluster-1, C16-Cluster-1, C16-Cluster-2, and C17-Cluster-1), with stable or common QTLs related to 2 or more different traits, are important clusters and warrant further study for the simultaneous genetic improvement of fiber yield and quality in cotton (Table 4; Table S1). However, 3 clusters (C10-Cluster-1, C16-Cluster-1, and C16-Cluster-2) harbored negatively correlated stable QTLs or common QTLs between fiber quality and yield traits and could hinder the simultaneous improvement of these traits. Therefore, an in-depth study of this linkage mechanism and breaking the linkage drag between unfavorable QTLs/genes through gene fine mapping (Cao et al. 2015), gene-editing technology (Dahan-Meir et al. 2018; Durr et al. 2018) or other new technologies and methods would play an important role in cotton molecular breeding. In addition, with the same two parents (CCRI36 and Hai1), we have developed CSSLs that can be used for further study.
Conclusion
In conclusion, a total of 153 QTLs for fiber quality and yield traits were identified in 5 interspecific backcross populations using a high-density genetic linkage map. Of these QTLs, 30 were consistent with those identified previously. Importantly, 23 QTLs were stably detected in 2 or 3 populations or generations, 17 of which were novel stable QTLs. Ten QTL clusters for different traits were found, and 9 of them were novel.
The results provide valuable information for MAS in cotton breeding, QTL/gene cloning, understanding the genetic basis of fiber quality and yield traits in the two cultivated tetraploid species of cotton (G. hirsutum L. and G. barbadense L.) and genetically improving fiber quality and yield in cotton.
References
Ademe MS, He SP, Pan ZE, Sun JL, Wang QL, Qin HD, Liu JH, Liu H, Yang J, Xu DY, Yang JL, Ma ZY, Zhang JB, Li ZK, Cai ZM, Zhang XL, Zhang X, Huang AF, Yi XD, Zhou GY, Li L, Zhu HY, Pang BY, Wang LR, Jia YH, Du XM (2017) Association mapping analysis of fiber yield and quality traits in Upland cotton (Gossypium hirsutum L.). Mol Genet Genom 292(6):1267–1280
Cao ZB, Zhu XF, Chen H, Zhang TZ (2015) Fine mapping of clustered quantitative trait loci for fiber quality on chromosome seven using a Gossypium barbadense introgressed line. Mol Breed 35(11):215
Chen ZJ, Scheffler BE, Dennis E (2007) Toward sequencing cotton (Gossypium) genomes. Plant Physiol 145(4):1303–1310
Chen Y, Liu GD, Ma HH, Song ZQ, Zhang CY, Zhang JX, Zhang JH, Wang FR, Zhang J (2018) Identification of introgressed alleles conferring high fiber quality derived from Gossypium barbadense L. in secondary mapping populations of G. hirsutum L. Front Plant Sci 9:1023
Dahan-Meir T, Filler-Hayut S, Melamed-Bessudo C, Bocobza S, Czosnek H, Aharoni A, Levy AA (2018) Efficient in planta gene targeting in tomato using geminiviral replicons and the CRISPR/Cas9 system. Plant J 95(1):5–16
Diouf L, Magwanga RO, Gong W, He S, Pan Z, Jia YH, Kirungu JN, Du X (2018) QTL mapping of fiber quality and yield-related traits in an intra-specific Upland cotton using genotype by sequencing (GBS). Int J Mol Sci 19(2):441
Durr J, Papareddy R, Nakajima K, Gutierrez-Marcos J (2018) Highly efficient heritable targeted deletions of gene clusters and non-coding regulatory regions in Arabidopsis using CRISPR/Cas9. Sci Rep 8(1):4443
Fang DD, Jenkins JN, Deng DD, McCarty JC, Li P, Wu J (2014) Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genom 15:397
Islam MS, Zeng L, Thyssen GN, Delhom CD, Kim HJ, Li P, Fang DD (2016) Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes. Theor Appl Genet 129(6):1071–1086
Jamshed M, Jia F, Gong J, Palanga KK, Shi Y, Li J, Shang H, Liu A, Chen T, Zhang Z, Cai J, Ge Q, Liu Z, Lu Q, Deng X, Tan Y, Or Rashid H, Sarfraz Z, Hassan M, Gong W, Yuan Y (2016) Identification of stable quantitative trait loci (QTLs) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genom 17:197
Jiang C, Wright RJ, El-Zik KM, Paterson AH (1998) Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc Natl Acad Sci USA 95(8):4419–4424
Keerio AA, Shen C, Nie Y, Ahmed MM, Zhang X, Lin Z (2018) QTL mapping for fiber quality and yield traits based on introgression lines derived from Gossypium hirsutum × G. tomentosum. Int J Mol Sci 19(1):243
Kong LL, Shi YZ, Li SQ, Li BT, Li JW, Liu AY, Gong JW, Shang HH, Gong WK, Ge Q, Wang YL, Song WW, Yuan YL (2018) QTL mapping for yield and fiber quality traits in double-cross population of chromosome segment introgression lines from Gossypium hirsutum × Gossypium barbadense. Cotton Sci 30(2):119–127
Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, Al-Ghazi Y, Liu S, Palai O, Georges S, Giband M, de Assuncao H, Barroso PA, Claverie M, Gawryziak G, Jean J, Vialle M, Viot C (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol 10:132
Lan MJ, Yang ZM, Shi YZ, Ge RH, Li AG, Zhang BC, Li JW, Shang HH, Liu AY, Wang T, Yuan YL (2011) Assessment of substitution lines and identification of QTL related to fiber yield and quality traits in BC4F2 and BC4F3 populations from Gossypium hirsutum × Gossypium barbadense. Scientia Agricultura Sinica 44(15):3086–3097
Li FG, Fan GY, Wang KB, Sun FM, Yuan YL, Song GL, Li Q, Ma ZY, Lu CR, Zou CS, Chen WB, Liang XM, Shang HH, Liu WQ, Shi CC, Xiao GH, Gou CY, Ye WW, Xu X, Zhang XY, Wei HL, Li ZF, Zhang GY, Wang JY, Liu K, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu SX (2014) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46(6):567–572
Li FG, Fan GY, Lu CR, Xiao GH, Zou CS, Kohel RJ, Ma ZY, Shang HH, Ma XF, Wu JY, Liang XM, Huang G, Percy RG, Liu K, Yang WH, Chen WB, Du XM, Shi CC, Yuan YL, Ye WW, Liu X, Zhang XY, Liu WQ, Wei HL, Wei SJ, Huang GD, Zhang XL, Zhu SJ, Zhang H, Sun FM, Wang XF, Liang J, Wang JH, He Q, Huang LH, Wang J, Cui JJ, Song GL, Wang KB, Xu X, Yu JZ, Zhu YX, Yu SX (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33(5):524
Li B, Shi Y, Gong J, Li J, Liu A, Shang H, Gong W, Chen T, Ge Q, Jia C, Lei Y, Hu Y, Yuan Y (2016a) Genetic effects and heterosis of yield and yield component traits based on Gossypium Barbadense chromosome segment substitution lines in two Gossypium Hirsutum backgrounds. PLoS One 11(6):e0157978
Li C, Dong Y, Zhao T, Li L, Li C, Yu E, Mei L, Daud MK, He Q, Chen J, Zhu S (2016b) Genome-wide SNP linkage mapping and QTL analysis for fiber quality and yield traits in the upland cotton recombinant inbred lines population. Front Plant Sci 7:1356
Li L, Zhao S, Su J, Fan S, Pang C, Wei H, Wang H, Gu L, Zhang C, Liu G, Yu D, Liu Q, Zhang X, Yu S (2017a) High-density genetic linkage map construction by F2 populations and QTL analysis of early-maturity traits in upland cotton (Gossypium hirsutum L.). PloS One 12(8):e0182918
Li PT, Wang M, Lu QW, Ge Q, Rashid MHO, Liu AY, Gong JW, Shang HH, Gong WK, Li JW, Song WW, Guo LX, Su W, Li SQ, Guo XP, Shi YZ, Yuan YL (2017b) Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and chromosome segment substitution lines from G. hirsutum × G. barbadense. BMC Genom 18(1):705
Liang Y, Jia YJ, Li AG, Zhang BC, Liu GP, Li JZ, Shi YZ, Li JW, Liu AY, Gong JW, Wang T, Shang HH, Gong WK, Yuan YL (2010) Phenotyping traits related to yield and quality of BC5F2 substitution lines in cotton (Gossypium) and their QTL mapping. Mol Plant Breed 8(2):221–230
Liu X, Teng Z, Wang J, Wu T, Zhang Z, Deng X, Fang X, Tan Z, Ali I, Liu D, Zhang J, Liu D, Liu F, Zhang Z (2017) Enriching an intraspecific genetic map and identifying QTL for fiber quality and yield component traits across multiple environments in Upland cotton (Gossypium hirsutum L.). Mol Genet Genom 292(6):1281–1306
Lu Q, Shi Y, Xiao X, Li P, Gong J, Gong W, Liu A, Shang H, Li J, Ge Q, Song W, Li S, Zhang Z, Rashid MHO, Peng R, Yuan Y, Huang J (2017) Transcriptome analysis suggests that chromosome introgression fragments from Sea–Island cotton (Gossypium barbadense) increase fiber strength in Upland cotton (Gossypium hirsutum). G3 (Bethesda) 7(10):3469–3479
Ma L, Zhao Y, Wang Y, Shang L, Hua J (2017) QTLs analysis and validation for fiber quality traits using maternal backcross population in Upland cotton. Front Plant Sci 8:2168
Ma Z, He S, Wang X, Sun J, Zhang Y, Zhang G, Wu L, Li Z, Liu Z, Sun G, Yan Y, Jia Y, Yang J, Pan Z, Gu Q, Li X, Sun Z, Dai P, Liu Z, Gong W, Wu J, Wang M, Liu H, Feng K, Ke H, Wang J, Lan H, Wang G, Peng J, Wang N, Wang L, Pang B, Peng Z, Li R, Tian S, Du X (2018) Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat Genet 50(6):803–813
Ning ZY, Chen H, Mei HX, Zhang TZ (2014) Molecular tagging of QTLs for fiber quality and yield in the upland cotton cultivar Acala-Prema. Euphytica 195(1):143–156
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Bio Reporter 11:122–127
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright RJ (2003) QTL analysis of genotype × environment interactions affecting cotton fiber quality. Theor Appl Genet 106(3):384–396
Reinisch AJ, Dong JM, Brubaker CL, Stelly DM, Wendel JF, Paterson AH (1994) A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138(3):829–847
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL, Smith CW, Gannaway JR, Wendel JF, Paterson AH (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176(4):2577–2588
Said JI, Lin Z, Zhang X, Song M, Zhang J (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14:776
Said JI, Knapka JA, Song M, Zhang J (2015a) Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol Genet Genom 290(4):1615–1625
Said JI, Song M, Wang H, Lin Z, Zhang X, Fang DD, Zhang J (2015b) A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol Genet Genom 290(3):1003–1025
Shang L, Wang Y, Wang X, Liu F, Abduweli A, Cai S, Li Y, Ma L, Wang K, Hua J (2016) Genetic analysis and QTL detection on fiber traits using two recombinant inbred lines and their backcross populations in Upland cotton. G3 (Bethesda) 6(9):2717–2724
Shao QS, Zhang FJ, Tang SY, Liu Y, Fang XM, Liu DX, Liu DJ, Zhang J, Teng ZH, Paterson AH, Zhang ZS (2014) Identifying QTL for fiber quality traits with three upland cotton (Gossypium hirsutum L.) populations. Euphytica 198(1):43–58
Shen XL, Guo WZ, Lu QX, Zhu XF, Yuan YL, Zhang TZ (2007) Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in Upland cotton. Euphytica 155(3):371–380
Shi Y, Li W, Li A, Ge R, Zhang B, Li J, Liu G, Li J, Liu A, Shang H, Gong J, Gong W, Yang Z, Tang F, Liu Z, Zhu W, Jiang J, Yu X, Wang T, Wang W, Chen T, Wang K, Zhang Z, Yuan Y (2015) Constructing a high-density linkage map for Gossypium hirsutum × Gossypium barbadense and identifying QTLs for lint percentage. J Integr Plant Biol 57(5):450–467
Shi YZ, Zhang BC, Liu AY, Li WT, Li JW, Lu QW, Zhang Z, Li SQ, Gong WK, Shang HH, Gong JW, Chen TT, Ge Q, Wang T, Zhu HQ, Liu Z, Yuan YL (2016) Quantitative trait loci analysis of Verticillium wilt resistance in interspecific backcross populations of Gossypium hirsutum x Gossypium barbadense. Bmc Genomics 17:877
Si ZF, Chen H, Zhu XF, Cao ZB, Zhang TZ (2017) Genetic dissection of lint yield and fiber quality traits of G. hirsutum in G. barbadense background. Mol Breed 37(1):9
Song W, Wang M, Su W, Lu Q, Xiao X, Cai J, Zhang Z, Li S, Li P, Gong J, Gong W, Shang H, Liu A, Li J, Chen T, Ge Q, Shi Y, Yuan Y (2017) Genetic and phenotypic effects of chromosome segments introgressed from Gossypium barbadense into Gossypium hirsutum. PLoS One 12(9):e0184882
Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, Li JW, Gong JW, Shang HH, Yuan YL (2012) QTL mapping for fiber quality traits across multiple generations and environments in Upland cotton. Mol Breed 30(1):569–582
Sun Z, Wang X, Liu Z, Gu Q, Zhang Y, Li Z, Ke H, Yang J, Wu J, Wu L, Zhang G, Zhang C, Ma Z (2017) Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol J 15(8):982–996
Tang SY, Teng ZH, Zhai TF, Fang XM, Liu F, Liu DJ, Zhang J, Liu DX, Wang SF, Zhang K, Shao QS, Tan ZY, Paterson AH, Zhang ZS (2015) Construction of genetic map and QTL analysis of fiber quality traits for Upland cotton (Gossypium hirsutum L.). Euphytica 201(2):195–213
Wang S, Basten CJ, Zeng ZB (2006) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh. http://www.statgen.ncsu.edu/qtlcart/WQTLCart.htm
Wang XQ, Feng CH, Lin ZX, Zhang XL (2011) Genetic diversity of Sea–Island cotton (Gossypium barbadense) revealed by mapped SSRs. Genet Mol Res 10(4):3620–3631
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, Yin Y, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu S (2012a) The draft genome of a diploid cotton Gossypium raimondii. Nat Genet 44(10):1098–1103
Wang P, Zhu Y, Song X, Cao Z, Ding Y, Liu B, Zhu X, Wang S, Guo W, Zhang T (2012b) Inheritance of long staple fiber quality traits of Gossypium barbadense in G. hirsutum background using CSILs. Theor Appl Genet 124(8):1415–1428
Wang FR, Xu ZZ, Sun R, Gong YC, Liu GD, Zhang JX, Wang LM, Zhang CY, Fan SJ, Zhang J (2013) Genetic dissection of the introgressive genomic components from Gossypium barbadense L. that contribute to improved fiber quality in Gossypium hirsutum L.. Mol Breeding 32(3):547–562
Wang H, Huang C, Guo H, Li X, Zhao W, Dai B, Yan Z, Lin Z (2015) QTL mapping for fiber and yield traits in Upland cotton under multiple environments. PLoS One 10(6):e0130742
Wang FR, Zhang CY, Liu GD, Chen Y, Zhang JX, Qiao QH, Yuan ZC, Fan SJ, Zhang J (2016) Phenotypic variation analysis and QTL mapping for cotton (Gossypium hirsutum L.) fiber quality grown in different cotton-producing regions. Euphytica 211(2):169–183
Wang B, Draye X, Zhuang Z, Zhang Z, Liu M, Lubbers EL, Jones D, May OL, Paterson AH, Chee PW (2017a) QTL analysis of cotton fiber length in advanced backcross populations derived from a cross between Gossypium hirsutum and G. mustelinum. Theor Appl Genet 130(6):1297–1308
Wang B, Zhuang Z, Zhang Z, Draye X, Shuang LS, Shehzad T, Lubbers EL, Jones D, May OL, Paterson AH, Chee PW (2017b) Advanced backcross QTL analysis of fiber strength and fineness in a cross between Gossypium hirsutum and G. mustelinum. Front Plant Sci 8:1848
Wu JX, Gutierrez OA, Jenkins JN, McCarty JC, Zhu J (2009) Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165(2):231–245
Yang XL, Zhou XD, Wang XF, Li ZK, Zhang Y, Liu HW, Wu LQ, Zhang GY, Yan GJ, Ma ZY (2015) Mapping QTL for cotton fiber quality traits using simple sequence repeat markers, conserved intron-scanning primers, and transcript-derived fragments. Euphytica 201(2):215–230
Yu JW, Yu SX, Gore M, Wu M, Zhai HH, Li XL, Fan SL, Song MZ, Zhang JF (2013) Identification of quantitative trait loci across interspecific F2, F2:3 and testcross populations for agronomic and fiber traits in tetraploid cotton. Euphytica 191(3):375–389
Yu JZ, Ulloa M, Hoffman SM, Kohel RJ, Pepper AE, Fang DD, Percy RG, Burke JJ (2014) Mapping genomic loci for cotton plant architecture, yield components, and fiber properties in an interspecific (Gossypium hirsutum L. × G. barbadense L.) RIL population. Mol Genet Genom 289(6):1347–1367
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhai H, Gong W, Tan Y, Liu A, Song W, Li J, Deng Z, Kong L, Gong J, Shang H, Chen T, Ge Q, Shi Y, Yuan Y (2016) Identification of chromosome segment substitution lines of Gossypium barbadense introgressed in G. hirsutum and quantitative trait locus mapping for fiber quality and yield traits. PloS One 11(9):e0159101
Zhang JF, Percy RG (2007) Improving Upland cotton by introducing desirable genes from Pima cotton. World Cotton Research Conference-4, Lubbock, Texas, USA. http://wcrc.confex.com/wcrc/2007/techprogram/P1901.HTM
Zhang ZS, Hu MC, Zhang J, Liu DJ, Zheng J, Zhang K, Wang W, Wan Q (2009) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol Breed 24(1):49–61
Zhang TZ, Qian N, Zhu XF, Chen H, Wang S, Mei HX, Zhang YM (2013) Variations and transmission of QTL alleles for yield and fiber qualities in Upland cotton cultivars developed in China. PLoS One 8(2):57220
Zhang TZ, Hu Y, Jiang WK, Fang L, Guan XY, Chen JD, Zhang JB, Saski CA, Scheffler BE, Stelly DM, Hulse-Kemp AM, Wan Q, Liu BL, Liu CX, Wang S, Pan MQ, Wang YK, Wang DW, Ye WX, Chang LJ, Zhang WP, Song QX, Kirkbride RC, Chen XY, Dennis E, Llewellyn DJ, Peterson DG, Thaxton P, Jones DC, Wang Q, Xu XY, Zhang H, Wu HT, Zhou L, Mei GF, Chen SQ, Tian Y, Xiang D, Li XH, Ding J, Zuo QY, Tao LN, Liu YC, Li J, Lin Y, Hui YY, Cao ZS, Cai CP, Zhu XF, Jiang Z, Zhou BL, Guo WZ, Li RQ, Chen ZJ (2015a) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33(5):531
Zhang Z, Li J, Muhammad J, Cai J, Jia F, Shi Y, Gong J, Shang H, Liu A, Chen T, Ge Q, Palanga KK, Lu Q, Deng X, Tan Y, Li W, Sun L, Gong W, Yuan Y (2015b) High resolution consensus mapping of quantitative trait loci for fiber strength, length and micronaire on chromosome 25 of the Upland cotton (Gossypium hirsutum L.). PloS One 10(8):e0135430
Zhang SW, Zhu XF, Feng LC, Gao X, Yang B, Zhang TZ, Zhou BL (2016a) Mapping of fiber quality QTLs reveals useful variation and footprints of cotton domestication using introgression lines. Sci Rep 6:31954
Zhang Z, Shang H, Shi Y, Huang L, Li J, Ge Q, Gong J, Liu A, Chen T, Wang D, Wang Y, Palanga KK, Muhammad J, Li W, Lu Q, Deng X, Tan Y, Song W, Cai J, Li P, Rashid H, Gong W, Yuan Y (2016b) Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative trait loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum). BMC Plant Biol 16:79
Acknowledgements
This work was funded by the National Key R&D Program for Crop Breeding (2016YFD0100203) and the National Agricultural Science and Technology Innovation Project of the CAAS.
Author information
Authors and Affiliations
Contributions
YY conceived and designed the experiments. YS and BZ performed the experiments. AL and JL participated in field trials. JZ and MJ helped in data analyses. QL, JG, GQ, HS, WG SL, XX, XD and JP contributed reagents/materials/analysis tools. YS analyzed the data and drafted the manuscript. YY and JZ revised the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
The authors declare that this study complies with the current laws of the country in which the experiments were performed. This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shi, Y., Liu, A., Li, J. et al. Dissecting the genetic basis of fiber quality and yield traits in interspecific backcross populations of Gossypium hirsutum × Gossypium barbadense. Mol Genet Genomics 294, 1385–1402 (2019). https://doi.org/10.1007/s00438-019-01582-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01582-8