
Abstract
A salmon louse (Lepeophtheirus salmonis salmonis) genetic linkage map was constructed to serve as a genomic resource for future investigations into the biology of this important marine parasitic copepod species, and to provide insights into the inheritance patterns of genetic markers in this species. SNP genotyping of 8 families confirmed the presence of 15 linkage groups based upon the assignment of 93,773 markers. Progeny sample size weight adjusted map sizes in males (with the exception of SL12 and SL15) ranged in size from 96.50 cM (SL11) to 134.61 cM (SL06), and total combined map steps or bins ranged from 143 (SL09) to 203 (SL13). The SL12 male map was the smallest linkage group with a weight-averaged size of 3.05 cM with 6 recombination bins. Male:female specific recombination rate differences are 10.49:1 and represent one of the largest reported sex-specific differences for any animal species. Recombination ratio differences (M:F) ranged from 1.0 (SL12) to 29:1 (SL15). The number of markers exhibiting normal Mendelian segregation within the sex linkage group SL15 was extremely low (N = 80) in comparison to other linkage groups genotyped [range: 1459 (SL12)—10206 markers (SL05)]. Re-evaluation of Mendelian inheritance patterns of markers unassigned to any mapping parent according to hemizygous segregation patterns (models presented) identified matches for many of these markers to hemizygous patterns. The greatest proportion of these markers assigned to SL15 (N increased to 574). Inclusion of the hemizygous markers revised SL15 sex-specific recombination rate differences to 28:1. Recombination hot- and coldspots were identified across all linkage groups with all linkage groups possessing multiple peaks. Nine of 13 linkage groups evaluated possessed adjacent domains with hot-coldspot transitional zones. The most common pattern was for one end of the linkage to show elevated recombination in addition to internal regions. For SL01 and SL06, however, a terminal region with high recombination was not evident while a central domain possessing extremely high-recombination levels was present. High levels of recombination were weakly coupled to higher levels of SNP variation within domains, but this association was very strong for the central domains of SL01 and SL06. From the pooled paternal half-sib lots (several virgin females placed with 1 male), only 1 or two surviving family lots were obtained. Surviving families possessed parents where both the male and female possessed either inherently low or high recombination rates. This study provides insight into the organization of the sea louse genome, and describes large differences in recombination rate that exist among individuals of the same sex, and between the sexes. These differences in recombination rate may be coupled to the capabilities of this species to adapt to environmental and pharmaceutical treatments, given that family survivorship appears to be enhanced when parents have similar recombination levels.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The sea louse (Lepeophtheirus salmonis) is an ectoparasitic copepod that infests many marine fish species and is of particular concern to the Atlantic salmon (Salmo salar) aquaculture industry. Infestations of the sea louse on Atlantic salmon cost the industry millions of dollars of lost revenue annually (Costello 2009). Because of the economic importance of this parasite, there is considerable interest in developing genomic resources to better understand the biology of this species. This includes understanding the physiological processes and genes involved in conferring adaptive resistance to therapeutics used to kill the species and span to understanding the genes involved in regulating behaviours such as mate choice and host selection. A fundamental step in this process is the construction of a high-density genetic marker map that can be used to track allelic variants of genes which may be involved in sea louse adaptation and selection. Multiple sea louse families were also used, which facilitated an investigation into the nature of differences in recombination rates between the sexes in this species, as well as examining differences in recombination rates among individuals within the same sex.
Due to the concentrated nature of net pen reared Atlantic salmon, infestations of sea lice can multiply and spread rapidly within a single net pen in a very short period of time. Adult females can typically survive for several months and produce on average 10 or more egg strings during that time period. Although egg numbers depend upon environmental conditions and female age, hundreds of eggs are typically produced in an egg string, and maturity may be reached in 3 weeks to 2.5 months depending upon temperature and salinity (Boxaspen 2006; Mustafa et al. 2001; Pike and Wadsworth 2000). Upon hatch, sea lice undergo two moults through planktonic stages before they reach their first parasitic infectious levels during the copepodid stages of their life cycle.
Currently, one of the most widespread means of controlling sea lice prevalence is through the use of pharmaceuticals in treatment baths or administered in feed. Compounds such as pyrethroids that include cypermethrin (Besnier et al. 2014; Poley et al. 2016a), organophosphates such as azamethiphos, or avermectins such as emamectin benzoate (Skilbrei et al. 2015) have been shown to effectively treat sea lice infestations. However, all of the therapeutics used to combat salmon louse parasitism are known to show declining efficacy with continued use (Aaen et al. 2015) as the population undergoes selective resistance to treatment. Therefore, to fully understand the physiological responses of sea louse resistance to therapeutics it is essential to gain an understanding of the sea louse genome.
Genomic resources developed for copepod species and knowledge of the genetic variability present in natural populations of copepods worldwide is limited. One of the most extensively studied copepods in this regard are harpacticoid copepods of the genus Tigriopus for which there are genomic sequencing builds available as well as knowledge of their population genetic structures and hybrid compatibilities among differentiated populations (Kang et al. 2016, 2017; Li 2018; Harrison and Edmands 2006; Pritchard et al. 2011; Willett et al. 2016). The marine copepod Tigriopus californicus also represents the first copepod species for which a genetic linkage map has been constructed (Foley et al. 2011; Pritchard et al. 2011). Due to the economic importance of sea lice considerable genetic resources for this species have also been developed (Yasuike et al. 2012; Salmon louse project; Salmon louse genome).
Copepods appear to have relatively low numbers of chromosomes as diploid numbers in the range of 10–24 chromosomes have been reported for cyclopoid copepods, but up to a 2n = 34 karyotype has been reported in a calanoid copepod species (Yang et al. 2008). Tigriopus californicus has a diploid chromosome of 2n = 24 suggesting similarity between harpacticoid and cyclopoid copepods. An initial linkage map compiled for the salmon louse indicates that this species has 15 linkage groups (2n = 30) (Besnier et al. 2014) suggesting that this ectoparasitic group (Siphonostomatoida) may have a broader range of diploid chromosome numbers similar to the calanoid copepods. However, a recent phylogeny of copepods based upon 24 nuclear protein-coding genes suggests that calanoid copepods are more distantly related to harpacticoid, cyclopoid and siphonostomatoida copepods, with cyclopoids and siphonostomatoids being the most closely related (Eyun 2017). The current paper describes a detailed genetic map for L. salmonis which supports the previously described genetic map for the species (n = 15 linkage groups) and provides a basis for more detailed genomic resource comparisons among the copepod groups. The map will also increase our knowledge of how genomic rearrangements and diversity within this species are coupled to varying degrees of adaptive resistance to therapeutics.
Materials and methods
Genotyping
A custom SNP genotyping array containing 201,279 assays for L. salmonis was developed for use on the Affymetrix Axiom platform. Purified DNA extracted from 1629 individuals was quantified, normalized and genotyped according to the manufacturer’s protocol. Individual sample performance was assessed using the Affymetrix Best Practice workflow, which lead to the exclusion of 42 samples, leaving 1587. Each SNP assay was subsequently classified into one of six categories reflecting assay performance; only the genotypes from “PolyHighResolution” SNP assays were used for mapping (n = 101,622 SNPs). Information on the polymorphic SNP markers scored in this study can be found in Supplementary File 2 of Messmer et al. (2018).
Crosses
For the production of families, several 10 gallon aquaria were established and seeded using a single adult male and up to 10 virgin females. Lice were originally sourced from Bay Management Areas 1, 2A and 2B from the Bay of Fundy, Canada. Females were first harvested from the previous generation wherein a single female carrying an egg mass was sequestered to a single aquarium. Copepodids developing from these egg masses were sexed and the female progeny were placed into separate aquaria before coming in contact with any mature males. Up to 10 unrelated females were then placed into the mating aquarium with the male, and resulting copepodids produced from these half-sib crosses were harvested and had their DNA extracted for genotyping. Progeny were assigned to their respective female parent following a pedigree analysis with the software program PROBMAX (Danzmann 1997) using the first 400 SNP markers in the array.
Error filtering
Prior to map construction, genotypes were filtered for errors in genotyping calls and reconstruction of missing parental genotypes dependent upon whether the progeny SNP genotypes conformed to Mendelian expectations for aa × ab or ab × aa cross structures. Data were initially arrayed in a 2 allele column format for each locus, where rows represented the parents followed by the individual progeny assigned to each half-sib family. Crosses of type ab × ab were removed from the analysis since 50% of the progeny genotypes resulting from such crosses (i.e., ab genotypes) cannot be unambiguously assigned a correct parental phase in all instances. Markers were also removed in cases where an unexpected genotype was detected in the progeny. For example, if bb genotypes were observed in 5% or more of the progeny when the parental genotypes were only aa × ab. In instances where bb genotypes accounted for less than 5% of the genotypes, they were recoded as missing under the assumption that they were due to genotyping errors. Genotypes that were also completely missing in the progeny, based upon parental genotypes were removed from the analysis. Several programs within the LINKMFEX compendium of programs (Danzmann 2018) were used to initially filter the data according to the criteria just outlined, and the subsequent remaining SNP markers were converted to OneMap genotype format (Margarido et al. 2007). Further filtering at this stage was performed to exclude markers that deviated greatly from Mendelian expectations (P < 0.01) using the program OneMap_Segregation_Distortion_Check (Danzmann 2018).
Map construction (single family)
Initial map orders were established using the record algorithm (van Os et al. 2005) within the OneMap (Margarido et al. 2007) software program. These orders were based solely upon markers that produced zero recombination clusters or ZRC (= 2 or more markers sharing identical genotypes across all the mapping progeny within a family). Prior to identifying ZRC within each linage group, the markers were first sorted into sex-specific separate linkage group genotype files at a LOD (Logarithm of Odds) = 5 threshold using Lep-MAP (Rastas et al. 2013) due to the increased computational speed obtained with this program when sorting large genotype files. Pairwise recombination distances were generated between all markers within the linkage group, and ZRC clusters were generated using the following command in OneMap:
Only markers without missing genotypes were included in the first phase of map construction. Singleton markers were identified as all unlinked markers following this initial round of marker clustering. Singletons and markers with missing genotypes were excluded from the first round of map order construction. Reliance on ZRC for initial map construction is due to the fact that ZRC are unlikely to represent genotyping errors as they represent identical phase genotype positions among multiple markers.
To establish map orders 500 iterations of the ZRC marker clusters were evaluated for consensus in the uniformity of marker position orderings. This was done using only a single reference marker for each ZRC given that all other markers within the cluster had identical genotypes. This approach greatly reduces computational time in map ordering. If a consensus order was not obtained following the initial orderings than a further 500–1000 iterations were performed until a majority-rule consensus order was achieved. Majority-rule ZRC map orders were established separately for each family and parent resulting in the production of 16 L. salmonis salmonis genetic maps. Since recombination distances were found to differ greatly between males and females (see "Results"), only sex-specific linkage maps were finally produced.
Singleton markers and markers with missing genotypes were added to the male-specific maps during a second round of map building. All markers genotyped had greater than 90% complete genotypic vectors across all the progeny within a family and hence all markers with missing genotypes were evaluated. In many instances, multiple singleton and missing genotype markers were placed between two ZRC clusters, but their linear map order could not be unambiguously determined as the markers all differed by 1 recombination step from one another. Their linear placement would generate multiple adjacent double crossover steps and they were, therefore, considered to arise from genotyping errors. Such assemblages were all assigned an identical map position and placed within a single map step bin. Placement of singleton and missing marker genotypes into the existing framework ZRC map was performed using the program Min_Rec_Link_Assign (Danzmann 2018). This program places a query marker into an interval between two ZRC positions based upon the minimum recombination distances detected between the query marker and all interval groupings. In instances where a query marker was equally likely to be placed into the upstream versus the downstream interval surrounding a ZRC, the query marker was retained, but assigned the map step position of the ZRC. These markers were considered to arise from genotyping errors.
Markers with missing genotypes may also occasionally join two adjacent ZRC positions if a missing genotype call occurs in a progeny where a phase shift crossover occurred between the upstream and downstream ZRC locations. The missing marker would, therefore, join both the upstream and downstream ZRC with zero recombination. OneMap ordering recognizes such inconsistencies and places the marker with missing genotypes into the ZRC with the highest number of markers. This is considered to be a potentially erroneous placement and all such missing markers were detected using the program Marker_List_Link (Danzmann 2018) and removed from the analysis.
Map construction (multiple families)
Maps were constructed and are depicted in two ways in this study. The first method regards recombination crossover points in the map ordering as analogous to map steps or map bins. Hence each ZRC position in an initial map order would be considered a sequential map step for that parent. Additional map steps, as mentioned above, were added in a second round of map ordering wherein singletons and missing genotype markers were added to the framework ZRC map within each linkage group. In addition, there were a few intervals established that contained multiple singleton markers or even new ZRC composed of two or more missing marker genotypes and singletons, but whose relative ordering could not be resolved. All these ambiguous marker groupings were arbitrarily assigned the same map step number at that position. Once the initial map placements were completed all map positions were renumbered sequentially from head–tail within each linkage group. Assessment of the overlaps among all recombination bins among parents was performed using the program Recom_Block_Map_Ord (Danzmann 2018). This program uses a single source parent in turn as a reference parent and depicts the exact overlaps in the recombination bins detected in the comparison parents to those present in the reference parent.
To portray linkage maps in a more conventional manner, a single parent was chosen for each linkage group that best represented the map ordering for that linkage group. This choice was based upon four criteria. The map for the parent should (i) represent the maximum number, or close to the maximum number of map steps detected for that linkage group; (ii) should possess a large number of progeny genotyped; (iii) should possess a high number of markers genotyped; and (iv) should also possess inter-individual validation. What is meant by this latter term is that map orders depicted in the chosen parent should reflect inter-individual consistency in ordering among the largest subset of parents. The male siring families 4a and 4b was excluded from this grouping even though the map steps produced from this individual produced some of the largest maps. The reason for this is that several inconsistencies in map step orders were observed between this male and other male parents, and in the case of linkage group 4, several central small ZRC within family 4a were reassigned to adjoining ZRC as they could not be reliably ordered. Therefore, this individual did not possess inter-individual validity.
An initial ordering of assembled map for all parents was made using Combination_Map (Danzmann 2018) which uses 3 other programs in the LINKMFEX compendium to prepare the data. The advantage of using a standard reference map is that newly genotyped parents that are added to the study can be updated easily into the reference framework. The map represents a true portrayal of exact recombination breakpoints in the map for the species, and this is benchmarked against an individual that usually displays the most reliable and maximum map step order for the species. It also highlights regions where an exact cM position cannot be assigned to a marker in relation to the reference map due to the fact that all markers in overlapping recombination bins among parents are not shared and, therefore, their exact cM location cannot be ascertained. Since Combination_Map only depicts a single crossover region for all markers falling within any given map step with the reference parent, the dataset was further explored using the Combination_Map_Ref_Nodes_Adjust (Danzmann 2018) program. This program delineates the ordering of markers from all the crossover bins of comparison parents that share markers with the reference parent and that overlap the reference parent map step. The output results from both preceding programs was combined using the program Combination_Map_Integrate (Danzmann 2018) which lists all the sequential crossover regions of comparison parents in relation to the reference parent and assigns each position a sequential map step or bin number, in addition to depicting a cM distance.
To produce a final estimate of the map lengths (cM distances) for each sex, the map lengths of all individual parents were weighted by the sample sizes of progeny genotyped within each family used to produce the map. Thus, the reference map positions shown for each linkage map may in some instances be larger or smaller than the best estimate for the map length of that linkage group. An estimate of the species-adjusted length for each linkage group position can be obtained by
where pos = the cM position shown in the reference map; avglng = the estimate for averaged map length across all parents; obslng = map length observed for the reference parent.
Sex-specific recombination ratios
Adjusted map lengths for each linkage group (described above) were used to obtain a recombination ratio given as large map length/small map length. Since the heterogametic sex generally produces smaller map lengths (Trivers 1988), the sex producing the majority of smaller map lengths was used as the denominator sex throughout. To obtain an estimate of the species-specific recombination rates, the recombination ratios obtained for each linkage group were weighted by the total number of markers used within each sex to produce the map lengths.
Segregation distortion
All polymorphic markers genotyped in the study and which were not excluded because they were derived from an ab × ab cross type, were assessed for deviations from 1:1 Mendelian segregation using the program OneMap_Segregation_Distortion_Check (Danzmann 2018). This program uses a G test with 1 df to detect markers exhibiting distortion. Markers with a P < 0.01 following the test were excluded from the analysis. Tests results are reported separately for both male and female-derived markers.
Assessment of crossover distributions
To evaluate the number of crossover steps occurring throughout the length of a given linkage group, the reference map steps within each composite map were divided into intervals of three consecutive reference map steps. Each of these regions was defined as a domain. If an additional map step was present it was added to the terminal grouping, but if two additional steps were present they were used to represent a new interval. To evaluate the total number of crossovers occurring within these intervals or domains, the results matrix produced by the Recom_Block_Map_Ord program was queried using the reference parent alignments with the program Recom_Domain_X-over_Counts (Danzmann 2018) which counts the total number of crossovers recorded within a domain among all mapping parents according to the formula:
where counts within each interval or domain are recorded as the summation over all map steps (s) among all (t) possible map steps among mapping parents, where map steps are completely contained within the domain (N) span, or overlap (O) either the upstream or downstream domain, or bridge an interval (I) between two domains where no overlapping map step assignment is recorded. In the latter two instances a complete map step is shared between the adjacent domains. Counts were obtained independently for each domain evaluated according to the reference map.
Recombination hotspots and coldspots were defined as domains possessing counts that were higher or lower, respectively, than 95% confidence interval of the median. The 95% CI was calculated using the AsBio program in R (Aho 2014). Averages of counts across all domains were then obtained, and any domains exceeding 2, as well as 1.9 standard deviations of the count mean were considered ‘very high’ recombination ‘hotspots’. Since the current linkage map has not been arrayed against the genomic builds for the species it was not possible to assess cM/Mb intervals and thus obtain estimates of genome-wide averages. Such averages can then be used to identify regions that may exceed the average recombination levels by 10 × or more which is the classical definition of a recombination hotspot. Nonetheless, this larger scale overview of recombination counts over all mapping parents used to construct linkage maps can still have merit and identify broader regions of high versus low recombination activity. Relative levels within each domain were depicted in plots of the X-over counts along each domain relative to the median crossover count level.
Results
Crosses
Interestingly, very few, or no progeny were detected among most of the potential half-sib crosses. Only 6 out of the 10 experimental tanks produced enough progeny within any given half-sib family to warrant further study. Eight experimental families were obtained with sample sizes of N > 20 for genetic mapping. In two lots (3 and 4) two paternal half-sib families were identified. Family progeny sizes were as follows: Family 3a = 44; 3b = 38; 4a = 36; 4b = 33; 5a = 49; 6 = 24; 7 = 31; 9 = 41, yielding 296 progeny and 14 parents in total for analysis.
Linkage maps
Of the 101,622 markers that provided genotyping information, a total of 93,773 markers were assigned to 15 linkage groups in sea lice (Table 1), which corresponds to previously reported linkage group numbers for the species (Besnier et al. 2014). Linkage groups were named in a similar fashion to Besnier et al. (2014) using overlapping subsets of SNP markers. SL05 had the highest number of markers assigned (10,206), while SL15 had the lowest number (n = 80). Total unique marker numbers assigned across both male and female parents were as follows: SL01 = 8859; SL02 = 6863; SL03 = 8254; SL04 = 8410; SL05 = 10,206; SL06 = 8939; SL07 = 6764; S08 = 6366; SL09 = 5143; SL10 = 4058; SL11 = 3711; SL12 = 1459; SL13 = 7593, SL14 = 7109; and SL15 = 80. Total number counts in these listings exceed the number indicated in Table 1 due to the fact that 39 markers had assignments to 2 linkage groups, while one marker (#39234) was assigned to 3 linkage groups (Online Resource 1). Interestingly, a marker from SL13 was involved in 56.4% of the two linkage group pairing assignments. All the SL13 assignments only occurred in the female parent, whereas the same marker assigned to pairs of male linkage groups involved 11 different linkage groups.
A large number of markers (8953) were observed to have apparent genotyping errors in at least 1 of the 14 parents surveyed. These errors ranged from the detection of additional Non-Mendelian genotypes in the progeny that were unexpected according to the parental genotypes, as well as the complete lack of certain genotypic classes in the progeny according to the parental genotypes. For most markers, normal Mendelian expectations were observed across the majority of the parents genotyped. However, 1316 markers remained that could not be assigned due to the genotype discrepancies.
Map lengths were much greater in the male parents compared to females and ranged from 3.05 cM (SL12) to 134.61 cM (SL06) in males, and from 3.04 cM (SL12) to 15.93 cM (SL05) in females (Fig. 1). The male linkage maps and map steps (bins) assigned to the composite linkage maps are provided in Online Resource 2. The individual marker overlap map steps used to construct these maps are provided in Online Resource 3, which shows all the individual map step locations within each male linkage map arrayed in a similar ‘head–tail’ orientation across all markers. The number of composite recombination bins detected within the different male linkage groups ranged from 6 (SL12) to 203 (SL13) (Fig. 2). Marker assignments to individual female linkage map ZRC are provided in Online Resource 4, and overlap matrices for these assignments can be found in Online Resource 5. Generally, it was not possible to accurately array female ZRC within linkage groups due to the extreme lack of recombination observed in females. Generally, only one large ZRC was detected within each linkage group that contained several hundred markers (Table 2), with additional smaller ZRC within the linkage group only differing by one recombination step from the main cluster. Given that these smaller ZRC and singletons could all equally be arrayed in a unit step ordering in either an upstream or downstream direction, it was essentially impossible to obtain a supported consensus ordering for these markers. An example of this is shown in Fig. 3, which depicts the phase map for SL05. The main ZRC contains 1865 markers, but the additional 5 marker positions all differ by the same recombination distance. Their ordering is thus ambiguous. Given the general lack of consensus in female ZRC ordering across the different female maps, the ZRC clusters numbered in Supplementary File 5 are not ordered in a uniform ‘head–tail’ orientation.

Average sizes (weight adjusted by family sample size) of male (M) and female (F) linkage groups

Integrated number of recombination bins or map steps among 14 male linkage groups. Total number of map steps among all maps shown at the top of each histogram bar. Markers assigned to male and female recombination bins are listed in Online Resource 2 and Online Resource 4, respectively

Linkage phase map for SL05 in Dame 5a. The two ZRC for this map are indicated as C1 and C2 with C1 possessing 1865 markers. The phase alignments of 4 additional singleton markers are shown below. C2 and all singleton markers differ by 1 recombination step (red arrows) from the main central C1 ZRC. (Color figure online)
The greater number of recombination steps detected in the male maps facilitated a more in-depth analysis of the recombination bin overlaps among males. The different map-specific recombination steps identified in each male facilitated the assessment of comparison recombination map steps that were present in each comparison male, and that were either completely aligned to a source parent map bin, or spanned either upstream or downstream, or both upstream and downstream recombination positions within the reference parent. The matrix showing all pairwise comparisons or map steps among all the mapping parents is provided in Online Resource 6.
Collinearity of markers between established male and female map orders was very poor and is not shown. In summary, many adjacent ZRC within the female maps were interspersed throughout several male ZRC along the length of the linkage group. Ordering within male maps (with the exception of S4) was quite robust; however, (see Online Resource 6) suggesting problems with the lack of recombination in female genomes confounds the map arrangements in this sex. Genotyping errors may also have contributed to these errors as several of the female ZRC were only composed of 2 markers. Therefore, it is possible that several of these smaller female ZRC are erroneous assemblies, which would suggest that female map lengths may be even smaller than depicted.
Recombination ratios
Males had substantially greater map lengths than did females (Fig. 4), with some of the smallest linkage groups (SL09, SL10, and SL11) possessing some of the largest differences. In contrast, however, the smallest linkage group (SL12) had the most uniform recombination ratios between the sexes based upon the ordering of the ZRC. Sample size weight adjusted values used to determine linkage group sizes indicate a species-specific difference in male:female map lengths of 10.49:1 which is one of the largest values reported for any given animal species.

Male versus female (M:F) recombination ratios across 14 salmon louse linkage groups (excluding the sex linkage group SL15). M:F ratio in SL15 was 28:1 when hemizygous markers were added (see text for explanation)
Segregation distortion
It was expected that fairly uniform levels of markers exhibiting segregation distortion (SD) would be detected among linkage groups with SD numbers proportional to the number of SNPs genotyped per linkage group. While generally true for most linkage groups, there were notable exceptions. One female linkage group (SL04) and four male linkage groups (SL04, SL06, SL07, and SL11) had very high levels (Table 3). The distribution of markers exhibiting distortion was also not uniform among mapping parents, with both parents from family 5a possessing relatively high levels of distortion, along with the sire from family 9 (Table 4).
To determine if the markers with high segregation distortion were aligned to specific chromosomal regions, the proportion of markers with distortion (P < 0.01) within each recombination bin in the male map builds were assessed. Since not all female markers were genotyped in the male parents, the number overlapping each bin position was first corrected for only female-specific markers. The female markers with SD were observed to be relatively uniformly distributed throughout the length of SL04 (Fig. 5). Markers aligning to bins shown in red or blue, need to be interpreted with caution, however, as each of these bins would have contained only 6 or fewer total markers within adjacent node intervals, or directly within nodes, respectively. Alignments of the SD markers from the 3 male linkage groups with the highest levels of SD presented a different finding. Here, markers with SD were clustered within a restricted region of the linkage group. In each linkage group, one major cluster was observed. For SL06, the cluster spanning bins 59–99 also overlapped the domain containing a large recombination hotspot in this linkage group (see below).

Regions of extreme segregation distortion in female linkage group SL04, and male linkage groups SL06, 07, and 11. Recombination bins are indicated along the abscissa, and reference bins with N ≤ 6 markers in reference and comparison map steps are indicated in blue and red, respectively. Bins with N > 6 markers in reference and comparison map steps are indicated in green or empty blocks, respectively. Overlapping steps are shown in purple. (Color figure online)
Crossover counts
All of the linkage groups examined had regions characterized as hot- and coldspots of recombination (exceeding the 95% CI). The average number of recombination hotspots across 13 linkage groups was 2.23 out of an average 13.3 domains surveyed per linkage group, or roughly ~ 1/5 of all recombination domains may have elevated levels. The average number of coldspots detected was 1.92. The most variable in this regard were SL01 and SL06, each possessing 3 hot- and coldspot regions. Five of the 13 male linkage groups (excluding SL12 and SL15) that were analyzed for recombination differences along their length possessed a single domain with very high levels of recombination (> 2 std. dev) and included SL01, SL05, SL06, SL08, and SL10. In addition, two linkage groups (SL09, S14) contained a domain with high crossover rates (> 1.9 std. dev.), while SL04 contained two such domains (Online Resource 7).
Discussion
Sex-specific markers and meiosis
Extremely large male:female recombination rates were observed among all the linkage groups with the exception of SL12, making this one of the largest differences in sex-specific recombination rate reported for any animal species. The sex linkage group (SL15) was not included in the main analysis due to lack of markers genotyped in both sexes (N = 80). However, the data indicate that overall SL15 may have the highest sex-specific differences in recombination rate (29.2:1). Among all the other linkage groups the average male:female ratios were 10.49:1.
SL15 was also identified as the sex linkage group. A SNP marker localized to scaffold LSalAtl2s6658 associated with SNP AX-98427605 (#43100) has been identified as being close to the sex-specific marker in the salmon louse (Messmer et al. 2018), but differs from the exact SNP location of the RAD-seq sex marker Lsa101901 identified by Carmichael et al. (2013). This SNP which overlaps the prohibitin-2 gene is associated with segregation polymorphisms in the female parent indicating that sea lice have a ZW sex-determining system (Carmichael et al. 2013). Marker 43100 displayed unusual progeny genotype patterns that did not match Mendelian expectations across most of the families surveyed, with the exception of Dame 7, where it was observed that the marker localized to SL15. Dame 6 was also heterozygous at marker 43100 and expected genotypic patterns were observed in the progeny, but the marker was removed from the analysis due to segregation distortion. All males at marker 43100 were T/T homozygotes, whereas females were either G/G homozygotes or G/T heterozygotes. Marker 28506, along with marker 43100 were located within scaffold LSalAtl2s6658 and marker 28506 was localized to 53.35 cM on the composite male sire 5a SL15 map (overall composite map length = 106.61 cM Online Resources 2, 8). The weight adjusted map position would be 63.23 cM given that the overall map length among all male parents was 126.36 cM for SL15. One additional marker (49103) could have localized to this scaffold as well, but this marker although showing variation in all the mapping families was excluded from analysis as the progeny genotypes did not conform to Mendelian expectations. Given the large number of markers (1316) that showed unexpected genotypes in one or more of the mapping panels, and that were not observed to conform to Mendelian expectations in any of these families, we examined the possibility that some of these markers may be inherited in a hemizygous fashion. Hemizygous inheritance has been identified in invertebrate species (Kaiser and Bachtrog 2010; Lima 2014), and this mode of transmission is most often associated with markers on the sex chromosomes (Qvanström and Bailey 2009), especially highly evolved sex chromosomes that display heteromorphic differences. The 1316 markers that were refractory to demonstrating any type of normal inheritance pattern were queried across all 8 mapping families for conformation to possible hemizygous models of inheritance (Table 5) using the program Hemizygous_Recode (Danzmann 2018). It was observed that from 230 to 270 markers in the male parents, and from 113 to 155 markers in the female parents could result from resolved hemizygous inheritance in the alternate parent (Table 6). These values are actually conservative, as in many instances, one of the two parents, or both parents had overlapping genotype patterns that could potentially indicate hemizygosity, but due to confounding overlapping patterns of expressed alleles versus null alleles, the genotypes could not be resolved.
The new hemizygous marker set was first assembled into possible linkage groups using a LOD = 3.0 clustering in OneMap. In all parents examined this resulted in one large assemblage and several smaller clusters that ranged from 2 to 10 markers along with a few unlinked markers. The large cluster of markers constituted between 72.7–80.2% of all markers in females, and from 74.9 to 82.6% of the markers in males, if sire 9 is excluded. In the progeny from sire 9 it was observed that only 63.9% of the markers fell into 1 large cluster. A large number of markers were, however, excluded in sire 9 prior to analysis due to the detection of segregation distortion (P < 0.01)(Table 6). Subsequent assemblage of these markers with pre-existing markers on the map revealed that in all parents, the single large cluster of hemizygous markers was part of the sex linkage group SL15 (Online Resource 8). In total, 574 markers were placed onto SL15 in both sexes, with 494 markers having hemizygous inheritance patterns. By comparison, 419 hemizygous markers were localized to all the other linkage groups in the sea louse genome combined, with 27 hemizygous markers remaining unlinked to any linkage group at a LOD = 4.0 threshold (Online Resource 9). Two linkage groups (SL04, SL05) had more than 40 hemizygous markers localized, while only 1 hemizygous marker was detected within SL12. Other linkage groups had between 16 and 37 hemizygous markers assigned. Another feature of this analysis was the observation that the numerous markers (N = 147 within SL01-14 and N = 460 within SL15) were consistently identified as possessing hemizygous inheritance patterns across multiple parents. This attests to the widespread regulation of allelic heterogeneity at these genomic locations, and most notably within SL15. Another interesting feature of the hemizygous markers was that the SL15 markers exhibited weak linkage or co-segregation with some of the markers on SL01 and SL05 in sires 3a and 5a, respectively (data not shown).
The genetic map for SL15 was reconstructed with the inclusion of the hemizygous markers to assess whether the general 10-fold increase in marker numbers influenced map length estimates and recombination ratios. Similar to the methods described above, ZRCs were first established within SL15, and their relative ordering established using the record option in OneMap. However, given the smaller number of markers present in SL15 compared to the other linkage groups, singleton markers were also included in this build. With the inclusion of the hemizygous markers into the SL15 map, the sample size weight-averaged male linkage group size was 126.36 cM, while the estimate for the female map was 4.51 cM. As expected, average map length estimates increased by 29.79 cM in males, but only 1.17 cM in females, highlighting the extreme conservation of recombination in the heterogametic sex. Family specific male and female genetic maps and the overlap matrix of map positions for SL15 are given in Online Resource 8. The male map positions are also shown in a uniform ‘head–tail’ ordering among all the male maps. The revised estimate for male:female recombination differences (28.02:1) was the largest detected for any linkage group and was very similar to the original estimate based upon a much smaller number of markers.
The detection of such a large number of putative hemizygous markers that localize to both male and female maps is unusual. When sex chromosomes evolve, the homolog that does not contain the sex-determining region generally exhibits hemizygous segregation due to the loss of complementary alleles on the sex-bearing chromosome (Qvanström and Bailey 2009). For Z-W species, this type of segregation pattern may be expected for the heteromorphic or heterogametic sex, and, therefore, we would expect the prevalence of such markers to be much greater for female segregation patterns compared to males. Although the direction of this pattern was supported with the current dataset (i.e., a greater number of male markers were scored indicating hemizygous segregation in the female parent), there were also a very large number of female-scored markers indicating male hemizygosity. This suggests that sex chromosome diversification may be incipient in L. salmonis.
A survey of chromosome numbers in the orders Harpacticoida and Cyclopoida, which are more closely related to the Siphonostomatoida (Eyun 2017), to which L. salmonis belongs, reveals that the maximum haploid chromosome number is n = 12 (Yang et al. 2008). In contrast the higher chromosome number detected in L. salmonis, suggest that some of the smaller chromosomes within Siphonostomatoida may be derived from chromosomal rearrangements, similar to the evolution of sex chromosomes in some other invertebrates (Blackmon et al. 2017), involving fusions, fissions, small inversions, and translocations. Such rearrangements may lead to unequal and differential degrees of pairing during meiosis. During transitional states of sex chromosome evolution, varying states of hemizygosity may be detected due to varying and occasional rounds of recombination between the sex chromosomes (Qvarnström and Bailey 2009; Bachtrog et al. 2014). Recombination levels are higher between incompletely diverged sex-determining homologs (Lima 2014), and this retards the rate at which full sex determination becomes established following meiotic segregation. Incomplete sex-specific differentiation, may, however, be augmented via sex-specific differentiation in gene expression levels (Bachtrog et al. 2014; Jorden and Charlesworth 2012). Sex-specific differences in L. salmonis gene expression levels exist (Poley et al. 2016b), with higher levels detected in males when expression is detected in both sexes for certain genes, while for other transcripts male-only expression is evident. It will be of interest to couple the location of these differentially expressed transcripts to the genomic builds for sea lice and ascertain if a substantial portion align to the hemizygous markers on SL15.
Chromatin diminution has also been observed within copepod species (Wyngaard and Gregory 2001; McKinnon and Drouin 2013) which involves the complete deletion of portions of the genome during early mitotic divisions following pronuclei fusions post-fertilization. The deletion of genomic DNA is reportedly only expected to occur in somatic cell lines with gonadal cell lines retaining a complete complement of nuclear DNA (Standiford 1989). Deletion of entire segments of DNA could result in a mosaic pattern of DNA retention whereby an entire locus could be lost in one parent and wholly or partially retained in another parent at the DNA breakpoint. To investigate this possibility, we re-examined the segregation patterns of loci initially excluded due to non-conformance to Mendelian segregation patterns for the presence of null locus inheritance in one of the parents using the program Homozygous_Null_Recode (Danzmann 2018). This program identifies normal Mendelian or hemizygous inheritance modes in one parent, coupled with the complete loss of allelic segregation at the same locus in the alternate parent (see Table 7). Such patterns could result from the process of chromatin diminution. A total of 176 markers were observed to comply with these models of inheritance, and 129 of these markers were localized to the existing salmon louse linkage groups (Online Resource 10). Interestingly, only a single marker matching these models was located within SL15. While the higher proclivity to possess hemizygous markers on SL15 compared to homozygous null markers on the sex linkage group is not entirely clear at present, it may suggest that there is a need to retain at least one copy of the sex chromosome complement in all cells within the species. It will be of interest to couple the location of these differentially expressed transcripts to the genomic builds for sea lice and ascertain if a substantial portion align to the hemizygous markers on SL15. Future research should also determine the genomic location of these markers, and whether genes closely linked to genomic regions with a potential for complete or partial loss display differential sex gene expression.
Mate Selection, differential family survivorship, and recombination
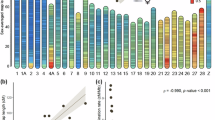
An unexpected finding from the current study was the lack of detection of multiple half-sib families despite having mating arenas established with 10 or more virgin females in each arena. In five lots, all copepodids genotyped were derived from a single family, while in 3 lots, only two paternal half-sib families were detected. In one of these lots, the number of surviving progeny was very low in one of the half-sib families (5b) and was not included in the initial map builds. When final average map lengths were calculated within each parent (which reflect recombination levels), it was evident that bi-parental averages in linkage group lengths were very similar to one another across the families (Fig. 6). This indicates that males possessing either very low, intermediate, or high recombination rates also mated with females possessing these intrinsic recombination rates. This suggests that some type of assortative mating may be occurring in the salmon louse, or that survivorship of the progeny is favoured if the parents have similar intrinsic recombination rates. Although the current dataset is small it does indicate reduced progeny viability if the parental genomic backgrounds differ too greatly in their intrinsic recombination levels. In fact, from a preliminary assessment of the viability of surviving copepodids sampled among families, it appears that an inverse relationship may exist between the number of surviving progeny produced and the level of recombination in their parents (Fig. 7). Families with higher levels of recombination in both the male and female parent produce lower numbers of surviving offspring while the opposite is true for families where both parents have lower recombination rate. This latter observation must be treated with caution, however, as too few families and populations have been tested to assess the general validity of the association.

Significant relationship between average male linkage group size (abscissa) and female linkage group size (ordinate). Family IDs are indicated on the plot

Significant relationship between combined average male and female linkage group sizes (abscissa) and surviving progeny numbers within each family (ordinate)
Assortative mating has been reported to occur in the purple sea urchin (Strongylocentrotus purpuratus) to account for the differences observed in sperm and egg binding protein patterns that exist in this species (Stapper et al. 2015), and mosquitos (Anopheles gambiae) due to X-chromosomal genomic islands of selection (Aboagye-Antwi et al. 2015). Further testing in the sea louse would be needed to confirm assortative mating using pedigreed lines of low (L) and high (H) recombination rate families. If four types of mating chambers were established (i.e., L × L, L × H, H × L, and H × H) and the highest production of egg strings with viable progeny among multiple females occurred within L × L chambers it would confirm that assortative mating does occur based upon background genomic structure within L. salmonis, and that viability may be inversely related to recombination differences between the parents. Conversely, If mating is at random, but survivorship is poorer among crosses with parents possessing higher or more divergent recombination rates, then a census of the proportion of females in mating arenas with developing egg strings would need to be performed. In other words, if initial fertilization levels were random among test arenas, with subsequent lower rates of offspring viability among H × L, L × H and H × H groups it would suggest some form of selection against genomes where recombination levels differed greatly. Attempted mating could first be assessed by examining the virgin females placed into a mating arena for the presence of spermatophore plugs, after a set time period. If all females have been potentially inseminated (i.e., all possess a spermatophore plug) but only copepodids from a small number of females are censused then it would imply that some type of differential selection has influenced family survival. Conversely, the presence of spermatophores in only a subset of the females present would indicate a degree of assortative mating by males.
If the lowest viability occurred in the female (H) × male (L) crosses, and possibly H × H backgrounds, it would support the hypothesis of accelerated evolutionary change in ZW species (Ritchie 2007; Saether et al. 2007). Recombination among mate choice loci is generally accepted as being an impediment to population diversification (Nei 1969; Felsenstein 1981) due to the fact that if many such loci are involved and they are randomly scattered among chromosomes, then no clear assemblage of distinct ‘trait choice’ loci are expected to arise within populations. Clustering of such loci into a region of linkage disequilibrium or their concentration within sex chromosomes has been postulated to be a way of accelerating evolutionary rates based upon mate choice; as such regions would be largely non-recombining. Evidence is accumulating that in ZW determining species evolutionary rates may be elevated due to tracking of male-specific traits through the female-derived Z-chromosome, upon which females subsequently make mate choice decisions (Ritchie 2007). Females of a specific Z-type prefer to mate with males of a similar Z-type. Recombination rates will be crucial in determining the extent of differentiation among family lines. These models predict that in species with higher rates of recombination, the Z-chromosome will change at a faster rate compared to ancestral populations and female mate choice patterns will seek to match males with the highest concordance to their own intrinsic Z-chromosome configuration. Otherwise, chromosomal rearrangements between fast and slow-evolving Z chromosomes will cause incompatibilities leading to lower ZZ male viability. Therefore, another caveat that arises from the proposed experiment described above is that skewed sex ratios should be observed in the progeny from hybrid recombination-type tanks, with a lower percentage of males being produced. Therefore, in H × H background crosses a greater range of variation may be observed. In certain families the Z/W allelic combination may be largely incompatible, while in others they may be highly compatible leading to a greater array of surviving progeny levels derived from H x H parental backgrounds. Variable and significant sex ratio differences have been observed among family lines of T. californicus (Voordouw et al. 2005), suggesting that skewed family sex ratios can occur in copepods which may relate to the differences in genomic compatibility conditioned upon recombination ancestry among parental pairs.
In contrast to birds, male mate choice appears to be prevalent across copepod species (Burton 1985; Heuch and Schram 1996) including L. salmonis (Ritchie et al. 1996a; Hull et al. 1998; Birkett et al. 2002; Todd et al. 2005). Male preferences are highest for unfertilized virgin females followed by later advanced copepodid stages (Hull et al. 1998), which is similar to findings in other copepods (Burton 1985). How assortative mating based upon recombination differences within individuals operates remains unclear, but may involve chemosensory cues. Indeed, L. salmonis males react, and are directionally motivated towards female extracts conditional upon the female’s sexual stage (Birkett et al. 2002) and these responses are removed by ablation of male antennae (Todd et al. 2005). Therefore, smaller demic groups of sea lice possessing intrinsically different basal levels of recombination may differ sufficiently in their chemical composition due to shared past ancestry, that mate selection may be ‘fine tuned’ among these demes. Since mate choice appears predominantly male-driven in copepods (Hull et al. 1998; Ritchie et al. 1996a), chemosensory matching across the sexes is suggested to play a role. Here, lack of recombination within chromosomes may drive “species” or even “demi-specific” traits for selection. The current study has shown that the largest male:female recombination rate differences occur with the sex chromosome SL15 making it tempting to speculate that the Z-chromosome or even the undifferentiated W-chromosome may preferentially house such “mate choice specific” genes. It will be of interest in the future, once the current linkage map is aligned to genomic scaffolds, to ascertain if the sex chromosomes possess higher complements of chemosensory genes such as olfactory G-protein coupled receptors.
Although monogamy was once thought to be prevalent across copepods, because it was thought that sperm plugs would prevent successful fertilizations by successive males (Ritchie et al. 1996b), the report of polyandry occurring in L. salmonis (Todd et al. 2005) has cast doubt upon this supposition. If polyandry does occur to a wide degree among L. salmonis populations, it could suggest some degree of female mate choice also occurs. It will be of interest to assess if females prone to polyandry are a result of more divergent recombination background in initial pairings and if subsequent pairings more closely match their own ‘background recombination level’. Future studies could include mating arena trials where both males and females of known recombination background are placed together after the females have been inseminated by another male. Higher levels of polyandrous spermatophores detected within females introduced to males of similar recombination background would suggest some degree of female mate choice. In addition, higher copepodid survival derived from ‘similar’ sperm, versus ‘dissimilar sperm’ in such crosses could also support the concept of adaptive selection occurring in such crosses.
Sex-specific sex chromosome recombination rates, in general, tend to be higher within the sex chromosome itself, than found within other autosomal pairs (Burt et al. 1991), and this was also observed in this study where the female-specific SL15 W-chromosome had extremely low levels resulting in the highest linkage group specific male:female recombination ratio. This difference was 29:1 when only the Mendelian compliant markers were assessed and was only slightly adjusted to a ratio of 28:1 (male:female) when the addition of hemizygous markers was considered. Biological reasons for the suppression of recombination in the heterogametic sex are still unclear, but the suggestion that a suppression of recombination maintains linked loci critical to sex-determination is the most powerful (Nei 1969; Burt et al. 1991). The extension of suppressed recombination levels to the autosomal pairs is, however, more of an enigma. Researchers have suggested that gametic selection in plant species differs between the sexes and that this drives recombination levels in general (Lenormand and Dutheil 2005), but empirical evidence for this hypothesis is still generally lacking in animal species.
If gametic selection occurs in animals, evolutionary rates may be accelerated in ZW species due to the fact that gametic selection is likely to be more accelerated in females compared to males. Only 1 of 4 meiosis products survives in females, whereas all 4 are potentially viable in males. If some degree of autosomal meiotic drive accompanies the segregation of the Z and W chromosomes at meiosis I, then quite significant differences (i.e., non-random) should exist in the phase configuration of gametes across linkage groups.
The large sex-specific recombination levels detected in this study appear to be unusual among crustacean species studied to date, with recombination ratio differences approximating 1:1 ratios between the sexes (Perez et al. 2004; You et al. 2010; Cui et al. 2015). Admittedly, the number of species with available sex-specific maps is limited, and, therefore, general assessments of these differences are unwarranted at the present time. However, the Chinese mitten crab (Eriocheir sinensis) has also been reported to have a ZW sex-determining mechanism and in this species, the female sex linkage ratio was actually larger than observed in males (Cui et al. 2015). The most recent genetic map for this species estimates female:male recombination rates at 1.13:1 (Qiu et al. 2017). A genetic map is currently available for the copepod species Tigriopus californicus but this map is entirely male-based (Foley et al. 2011; Pritchard et al. 2011), as female meiosis have been reported to be achiasmate in this species (Ar-Rushdi 1963; Burton et al. 1981). Environmental sex determination is also presumed to occur in this species (Burton et al. 1981; Foley et al. 2011). Our findings confirm that although female recombination levels are extremely low, they do not appear to be achiasmatic in L. salmonis. Some evidence for low levels of female recombination were also incidentally reported in Tigriopus (Foley et al. 2011) but were excluded as genotyping errors. We cannot entirely exclude the possibility of some unusual mode of female-specific gamete transmission influencing recombination events. Although most meioses generated in female copepods appear to be achiasmate, recombination may be possible through extended multiple chromosomal ring formations similar to those that have been observed during female meioses in copepod species such as Mesocyclops edax (Chinnappa and Victor 1979). Such formations could lead to ectopic recombination within homologs which may account for the poor congruence in male and female map orders observed in the current study. Current cytological studies are lacking in L. salmonis such that the extent to which bivalents or different types of meiosis I configurations exist in this species is unknown.
Crossover rates; segregation distortion; and heterozygosity
Although the current estimates of recombination differences in L. salmonis are not aligned with estimates of genomic build distances, the inferences made on the distribution of recombination events is still insightful. Plots showing the total recombination counts per recombination domain (which roughly span 10 cM intervals on the references maps) (e.g., Fig. 8 and Online Resource 7) indicate that crossover hotspots are fairly randomly distributed along the lengths of all male chromosomes. However, for all linkage groups with the exceptions of SL01, SL02 and SL06, a recombination hotspot was localized to one end of the chromosome (occurring in either the ultimate or penultimate domain). For SL01/06 a region of very high recombination was only detected in the centre of the linkage group, while for SL02, both ends of the linkage group possessed terminal domains with higher recombination levels. The predominant pattern is more consistent with the pattern of recombination observed in another crustacean Daphnia magna, where recombination rates were all reported to be elevated towards the terminal ends of the linkage groups (Dukic et al. 2016). This suggests that the ends of linkage groups surveyed in L. salmonis with elevated recombination rates may be the telomeric ends of the chromosomes.

Plot of the number of recombination events occurring in the recombination domains of linkage group SL01. Domains encompass 3 linear recombination positions in the reference map and their associated crossover positions in comparison maps. The median level of crossovers is indicated by the blue line, and recombination hot- and coldspots are indicated with hat symbols above and below the line, respectively. Extreme hotspots (> 2 sd of the mean) are indicated with an asterisk. See Online Resource 7 for additional linkage group plots. (Color figure online)
More than half of the 13 linkage groups surveyed (SL01, 03, 05, 06, 08, 09, 10, 11, 14) had a recombination hotspot immediately adjacent to a recombination coldspot, and within SL06, the region of very high recombination (domain 7) was flanked both up- and downstream by recombination coldspots. Regions of low recombination often flank hotspots in mammalian genomes (Lichten and Goldman 1995; Hey 2004) and often coincide with regions of lower gene density. In humans, approximately 25,000 hotspots have been identified (Myers et al. 2005) through linkage disequilibrium mapping, and display a great deal of heterogeneity on a fine scale, that is not readily evident from broad scale surveys such as the ones depicted in this study. Future alignments of the current genetic map to genomic scaffolds in the salmon louse will be able ascertain if crossover frequency is related to higher gene density.
Levels of overall nucleotide variability have been reported to be directly related to recombination rate differences in the genomes of animals (Begun and Aquadro 1992; Nachman 2002). Increased genetic variability is found in regions of higher recombination. This suggests that overall levels of genetic variability detected among individuals (i.e., total number of heterozygous individuals) should also be coupled to higher regions of recombination. We investigated this relationship in L. salmonis by documenting the total number of genetically variable SNPs found within each recombination domain within a linkage group, using the program Polymorph_Count_X_Recom_Domains (Danzmann 2018). This census included not only the polymorphic male markers that were used to construct the map, but also all of the polymorphic female markers occurring within a domain as well as doubly heterozygous markers of the type ab x ab. Such markers were initially excluded from the analysis since the phase of doubly heterozygous progeny for these markers cannot be ordered within phase maps. For this analysis, heterozygous markers that occurred wholly within a recombination domain were scored a value of 1.0 while markers that occurred in comparison parent recombination bins that overlapped either an upstream or downstream domain were scored a value of 0.5 within each overlapping domain (Online Resource 11). Results from this analysis weakly supported the association between increased recombination levels and genetic variability within a linkage group domain (Fig. 9; Online Resource 7; Online Resource 12). An increased level of genetic variability was significantly coupled to higher crossover levels in six (SL01, 06, 09, 10, 13, and 14) of 13 linkage groups examined, while this was marginally true (P < 0.1) for another two linkage groups (SL02 and SL08). Overall, however, in all linkage groups with the exception of SL04, the overall relationship between these two variables was positive, albeit extremely weak for SL05 and SL07.

Regression plot of relationship between the level of genetic variability detected within recombination domains versus number of crossover events within domains within SL01. Variability was recorded as total number of progeny with heterozygous genotypes. See Online Resource 12 for additional linkage group plots
The high levels of segregation distortion detected in linkage groups SL04, SL07, and SL11 (Fig. 5) may have contributed to the lack of a strong coupling between observed genetic variability and recombination rate. Markers exhibiting distortion would not have been included in map ordering and this could slightly bias the observed associations given that they were included in the polymorphism counts. Similarly, for SL06, the strong association between genetic variability and recombination level is due to the extremely high levels of variation observed within domain 7, which also possesses very high crossing-over levels. If this domain is removed from the analysis no association between the two variables is detected. SL01 also possessed an extreme polymorphism peak (> 15,000 polymorphic SNP markers in domain 9) that was coupled to extremely high crossover rates. When this point was removed, however, the association between variability and crossover rate was still significant (data not shown). Interestingly, SL06 also possessed regions of extreme segregation distortion which could have influenced map order structure (Fig. 5).
Recombination coldspots have been empirically and theoretically linked to regions that may contain higher numbers of coadapted genes related to species survival. These so-called “genomic islands of selection” have been postulated to arise through various evolutionary forces, but are recognized as regions more refractory to change that may result from recombination events that disrupt favourable allelic combinations (Yeaman 2013). Genomic rearrangements have been predicted to be important in localizing coadapted genes into smaller genomic regions that more readily ameliorate the disruptive effects of recombination. Researchers have reported that gene densities are reduced in regions of lower recombination, and that these regions may also accumulate higher densities of transposable elements (TEs) (Fontinallas et al. 2007; Dolgin and Charlesworth 2008). In contrast, other studies have reported elevated levels of recombination in regions of high TE density (Everitt et al. 2014), or have found no association between recombination and TE density (Adrion et al. 2017). The current study provides a framework map that has identified recombination hot- and coldspots within linkage groups. Future coupling of these map regions to genomic scaffolds will facilitate an in-depth analysis of the genomic regions for differences in transposable element and gene abundance in relation to differences in recombination levels within the chromosomes. Alignment of markers to scaffold builds will also help to resolve discrepancies in the ordering of markers among multiple mapping parents (see Lien et al. 2016).
Conclusions
Our findings have confirmed previous studies that identify the presence of 15 linkage groups in the sea louse (Messmer et al. 2018). Large differences in recombination rates were detected between the sexes, with the largest male:female recombination difference present within the sex linkage group SL15. Unusual segregation patterns were observed in both male and female parents that could be due to hemizygous inheritance patterns. The largest number of hemizygous inherited markers was located to SL15, with the highest proportion of these markers arising from hemizygous segregation of female alleles. However, the observation that a substantial portion is also derived from male meioses suggests the process of sex chromosome differentiation is at an incipient level in this ZW-based species. We surveyed recombination rate differences along the length of each male-specific linkage group. Recombination rate differences were coupled to overall levels of heterozygosity, but only weakly so for about half the linkage groups. It is unclear at present whether these differences may arise from selection differentials among genes housed within these chromosomes. Whether the recombination-heterozygosity associations identified in this study generally hold for other populations of sea lice will need further investigation. Of interest, was the observation that families producing surviving copepodids, were derived from parents with intrinsically similar recombination rates. Future studies will need to investigate whether such differences may arise from some type of assortative mating within this species, or may be due to differential selection against the progeny derived from parents that differ too greatly in their background recombination levels.
References
Aaen SM, Helgesen KO, Bakke MJ, Kaur BK, Horsberg TE (2015) Drug resistance in sea lice: a threat to salmonid aquaculture. Trends Parasitol 31(2):72–81
Aboagye-Antwi F, Alhafez N, Weedall GD, Brothwood J, Kandola S, Paton D, Fofana A, Olohan L, Betancourth MP, Ekechukwu NE, Baeshen R, Traore SF, Diabate A, Tripet F (2015) Experimental swap of Anopheles gambiae’s assortative mating preferences demonstrates key role of X-chromosome divergence island in incipient sympatric speciation. PLoS Genet 11(4):e1005141
Adrion JR, Song MJ, Schrider DR, Hahn MW, Schaack S (2017) Genome-wide estimates of transposable element insertion and deletion rates in Drosophila melanogaster. Genome Biol Evol 9(5):1329–1340
Aho K (2014) Foundational and applied statistics for biologists using R. CRC/Taylor and Francis, Boca Raton, FL. https://cran.r-project.org/web/packages/asbio/index.html
Ar-Rushdi AH (1963) The cytology of achiasmatic meiosis in the female Tigriopus (Copepoda). Chromosoma 13:526–539
Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman T-L, Hahn MW, Kitano J, Mayrose I, Ming R, Perrin N, Ross L, Valenzuela N, Vamose JC, The Tree of Sex Consortium (2014) Sex determination: Why so many ways of doing it? PLoS Biol 12(7):e1001899
Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356:519–520
Besnier F, Kent M, Skern-Mauritzen R, Lien S, Malde K, Edvardsen RB, Taylor S, Ljungfeldt LER, Nilsen F, Glover KA (2014) Human-induced evolution caught in action: SNP-array reveals rapid amphi-atlantic spread of pesticide resistance in the salmon ectoparasite Lepeophtheirus salmonis. BMC Genom 15:937
Birkett MA, Duce I, Mordue W, Pickett JA (2002) Role of semiochemicals in mate location by parasitic sea louse Lepeophtheirus salmonis. J Chem Ecol 28(10):2107–2117
Blackmon H, Ross L, Bachtrog D (2017) Sex determination, sex chromosomes, and karyotype evolution in insects. J Hered 108(1):78–93
Boxaspen K (2006) A review of the biology and genetics of sea lice. ICES J Mar Sci 63:1304–1316
Burt A, Bell G, Harvey PH (1991) Sex differences in recombination. J Evol Biol 4:259–277
Burton RS (1985) Mating system of the intertidal copepod Tigriopus californicus. Marine Biol 86:247–252
Burton RS, Feldman MW, Swisher SG (1981) Linkage relationships among five enzyme-coding gene loci in the copepod Tigriopus californicus: a genetic confirmation of achiasmatic meiosis. Biochem Genet 19:1237–1245
Carmichael SN, Bekaert M, Taggart JB, Christie HRL, Bassett DI, Bron JE, Skuce PJ, Gharbi K, Skern-Mauritzen R, Sturm A (2013) Identification of a sex-linked SNP marker in the salmon louse (Lepeophtheirus salmonis) using RAD sequencing. PLoS ONE 8(10):e77832
Chinnappa CC, Victor R (1979) Achiasmatic meiosis and complex heterozygosity in female cyclopoid copepods (Copepoda, Crustacea). Chromosoma 71:227–236
Costello MJ (2009) The global economic cost of sea lice to the salmonid farming industry. J Fish Dis 32:115–118
Cui Z, Hui M, Liu Y, Song C, Li X, Li Y, Liu L, Shi G, Wang S, Li F, Zhang X, Liu C, Xiang J, Chu KH (2015) High-density linkage mapping aided by transcriptomics documents ZW sex determination system in the Chinese mitten crab Eriocheir sinensis. Heredity 115:206–215
Danzmann RG (1997) PROBMAX: a computer program for assigning unknown parentage in a pedigree analysis from known genotypic pools of parents and progeny. J Hered 88(4):333. https://omictools.com/search?q=probmax
Danzmann RG (2018) https://uoguelphca-my.sharepoint.com/:f:/g/personal/rdanzman_uoguelph_ca/EjW14Zxt43RAqoHe6_t5QvMBqwkT3dfYzmfzmMdnU7hJXA or https://omictools.com/search?q=linkmfex
Dolgin ES, Charlesworth B (2008) The effect of recombination rate on the distribution and abundance of transposable elements. Genetics 178:2169–2177
Dukic M, Berner D, Roesti M, Haag CR, Ebert D (2016) A high-density genetic map reveals variation in recombination rate across the genome of Daphnia magna. BMC Genet 17:137
Everitt RG, Didelot X, Batty EM, Miller RR, Knox K, Young BC, Bowden R, Auton A, Votintseva A, Larner-Svensson H, Charlesworth J, Golubchik T, Ip CLC, Godwin H, Fung R, Peto TEA, Walker S, Crook DW, Wilson DJ (2014) Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat Commun 5:3956
Eyun S-I (2017) Phylogenomic analysis of Copepoda (Arthropoda, Crustacea) reveals unexpected similarities with earlier proposed morphological phylogenies. BMC Evol Biol 17:23
Felsenstein J (1981) Skeptacism towards Santa Rosalia, or why are there so few species of animals. Evolution 35(1):124–138
Foley BR, Rose CG, Rundle DE, Leong W, Moy GW, Burton RS, Edmands S (2011) A gene-based SNP resource and linkage map for the copepod Tigriopus californicus. BMC Genom 12:568
Fontnallas P, Hartl DL, Reuter M (2007) Genome organization and gene expression shape the transposable element distribution in the Drosophila melanogaster euchromatin. PLoS Genet 3(11):e210
Harrison JS, Edmands S (2006) Chromosomal basis of viability differences in Tigriopus californicus interpopulation hybrids. J Evol Biol 19:2040–2051
Heuch PA, Schram TA (1996) Male mate choice in a natural population of the parasitic copepod Lernaeocera branchialis (Copepoda: Pennellidae). Behaviour 133:221–239
Hey J (2004) What’s so hot about recombination hotspots. PLoS Biol 2(6):730–733
Hull MQ, Pike AW, Mordue-Luntz AJ, Rae GH (1998) Patterns of pair formation and mating in an ectoparasitic caligid copepod Lepeophtheirus salmonis (Kroyer 1837): implications for its sensory and mating biology. Phil Trans R Soc Lond B 353:753–764
Jorden CY, Charlesworth D (2012) The potential for sexually antagonistic polymorphism in different genomic regions. Evolution 66:505–516
Kaiser VB, Bachtrog D (2010) Evolution of sex chromosomes in insects. Annu Rev Genet 44:91–112
Kang S, Ahn D-H, Lee JH, Lee SG, Shin SC, Lee J, Min G-S, Lee H, Kim H-W, Kim S, Park H (2017) The genome of the Antarctic-endemic copepod, Tigriopus kingsejongensis. Gigascience. https://doi.org/10.1093/gigascience/giw010
Kang S, Ahn D, Lee JH, Lee SG, Shin SC, Lee J, Min G, Lee H, Kim H, Kim S, Park H (2016) Supporting data for: The genome of the Antarctic-endemic copepod, Tigriopus kingsejongensis. Giga-Science Database. https://doi.org/10.5524/1000249
Lenormand T, Dutheil J (2005) Recombination differences between the sexes: a role for haploid selection. PLoS Biol 3(3):e63
Li W (2018) Lab. https://i5k.nal.usda.gov/Tigriopus_californicus
Lichten M, Goldman AS (1995) Meiotic recombination hotspots. Annu Rev Genet 29:423–444
Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, Hvidsten TR, Leong JS, Minkley DR, Zimin A, Grammes F, Grove H, Gjuvsland A, Walenz B, Hermansen RA, von Schalburg K, Rondeau EB, Di Genova A, Samy JKA, Vik JO et al (2016) The Atlantic salmon genome provides insights into rediploidization. Nat Commun 533:17164
Lima TG (2014) Higher levels of sex chromosome heteromorphism are associated with markedly stronger reproductive isolation. Nat Commun 5:4743
Margarido GRA, Souza AP, Garcia AAF (2007) Onemap: software for genetic mapping in outcrossing species. Hereditas 144:78–79
McKinnon C, Drouin G (2013) Chromatin diminution in the copepod Mesocyclops edax: elimination of both highly repetitive and nonhighly repetitive DNA. Genome 56:1–8
Messmer AM, Leong JS, Rondeau EB, Mueller A, Despins CA, Minkley DR, Kent MP, Lien S, Boyce B, Morrison D, Fast MD, Norman JD, Danzmann RG, Koop BF (2018) A 200K SNP chip reveals a novel Pacific salmon louse genotype linked to differential efficacy of emamectin benzoate. Mar Genom 40:45–57. https://doi.org/10.1016/j.margen.2018.03.005
Mustafa A, Conboy GA, Burka JF (2001) Life-span and reproductive capacity of sea lice, Lepeophtheirus salmonis, under laboratory conditions. Aquacult Assoc Canada Spec Publ 4:113–114
Myers S, Bottolo L, Freeman C, McVean G, Donnelly P (2005) A fine-scale map of recombination rates and hotspots across the human genome. Science 310:321–324
Nachman MW (2002) Variation in recombination rate across the genome: evidence and implications. Curr Opinion Genet Develop 12:657–663
Nei M (1969) Linkage modification and sex differences in recombination. Genetics 63:681–699
Van Os H, Stam P, Visser RGF, Van Eck HJ (2005) RECORD: a novel method for ordering loci on a genetic linkage map. Theor Appl Genet 112:30–40
Perez F, Erazo C, Zhinaula M, Volckaert F, Calderon J (2004) A sex-specific linkage map of the white shrimp Penaeus (Litopenaeus) vannamei based on AFLP markers. Aquaculture 242:105–118
Pike AW, Wadsworth SL (2000) Sea lice on salmonids: their biology and control. Adv Parasitol 44:233–337
Poley JD, Braden LM, Messmer AM, Whyte SK, Koop BF, Fast MD (2016a) Cypermethrin exposure induces metabolic and stress-related gene expression in copepodid salmon lice (Lepeophtheirus salmonis). Comp Biochem Physiol D 20:74–84
Poley JD, Sutherland BJG, Jones SRM, Koop BF, Fast MD (2016b) Sex-biased gene expression and sequence conservation in Atlantic and Pacific salmon lice (Lepeophtheirus salmonis). BMC Genom 17:483
Pritchard VL, Dimond L, Harrison JS, Velazquez CCS, Zieba JT, Burton RS, Edmands S (2011) Interpopulation hybridization results in widespread viability selection across the genome in Tigriopus californicus. BMC Genet 12:54
Qiu G-F, Xiong L-W, Han Z-K, Liu Z-Q, Feng J-B, Wu X-G, Yan Y-L, Shen H, Huang L, Chen L (2017) A second generation SNP and SSR integrated linkage map and QTL mapping for the Chinese mitten crab Eriocheir sinensis. Sci Rep 7:39826
Qvanström A, Bailey RI (2009) Speciation through evolution of sex-linked genes. Heredity 102:4–15
Rastas P, Paulin L, Hanski I, Lehtonen R, Auvinven P (2013) Lep-MAP: fast and accurate linkage map construction for large SNP datasets. Bioinformatics 29(24):3128–3134
Ritchie MG (2007) Feathers, females, and fathers. Science 318:54–55
Ritchie G, Mordue AJ, Pike AW, Rae GH (1996a) Observations on mating and reproductive behaviour of Lepeophtheirus salmonis, Krøyer (Copepoda: Caligidae). J Exp Mar Biol Ecol 201:285–298
Ritchie G, Mordue AJ, Pike AW, Rae GH (1996b) Morphology and ultrastructure of the reproductive system of Lepeophtheirus salmonis (Krøyer, 1837)(Copepoda: Caligidae). J Crustacean Biol 16(2):330–346
Saether SA, Saetre G-P, Borge T, Wiley C, Svedin N, Andersson G, Veen T, Haavie J, Servedio MR, Bures S, Kral M, Hjernquist MB, Gustafsson L, Traff J, Qvarnström A (2007) Sex chromosome-linked species recognition and evolution of reproductive isolation in flycatchers. Science 318:95–97
Salmon louse genome in ensembl (2018) https://metazoa.ensembl.org/Lepeophtheirus.salmonis/info/Index); in NCBI (https://www.ncbi.nlm.nih.gov/genome/genomes/2713
Skilbrei OT, Espedal PG, Nilsen F, Perez Garcia E, Glover KA (2015) Evaluation of emamectin benzoate and substance EX against salmon lice in sea-ranched Atlantic salmon smolts. Dis Aquat Org 113:187–194
Standiford DM (1989) The effects of chromatin diminution on the pattern of C-banding in the chromosomes of Acanthocyclops vernalis Fischer (Copepoda: Crustacea). Genetica 79:207–214
Stapper AP, Beerli P, Levitan DR (2015) Assortative mating drives linkage disequilibrium between sperm and egg recognition protein loci in the sea urchin Strongylocentrotus purpuratus. Mol Biol Evol 32(4):859–870
The salmon louse project (2018) https://sealouse.imr.no/
Todd CD, Stevenson RJ, Reinardy H, Ritchie MG (2005) Polyandry in the ectoparasitic copepod Lepeophtheirus salmonis despite complex precopulatory and postcopulatory mate-guarding. Mar Ecol Prog Ser 303:225–234
Trivers RL (1988) Sex differences in rates of recombination and sexual selection. In: Meicho RD, Levin BR (eds) The evolution of sex. Sinnauer Assoc. Inc., Sunderland, pp 270–286
Voordouw MJ, Robinson HE, Anholt BR (2005) Paternal inheritance of the primary sex ratio in a copepod. J Evol Biol 18:1304–1314
Willett CS, Lima TG, Kovaleva I, Hatfield L (2016) Chromosome-wide impacts on the expression of incompatibilities in hybrids of Tigriopus californicus. Genes Genom Genet G3, 6:1739–1749
Wyngaard GA, Gregory TR (2001) Temporal control of DNA replication and the adaptive value of chromatin diminution in copepods. J Exp Zool (Mol Dev Evol) 291:310–316
Yang W-X, Dahms H-U, Hwang J-S (2008) A review of the karyological studies on the cyclopoida (Copepoda). Crustaceana 81(10):1229–1240
Yasuike M, Leong J, Jantzen SG, von Schalburg KR, Nilsen F, Jones SRM, Koop BF (2012) Genomic resources for sea lice: analysis of ESTs and mitochondrial genomes. Mar Biotechonol 14:155–166
Yeaman S (2013) Genomic rearrangements and the evolution of clusters of locally adaptive loci. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1219381110
You E-M, Lin K-F, Huang S-W, Chen M, Groumellec ML, Fann SJ, Yu H-T (2010) Construction of integrated genetic linkage maps of tiger shrimp (Penaeus monodon) using microsatellites and AFLP markers. Anim Genet 41:365–376
Acknowledgements
This work was funded by Elanco Fish Health Research Chair; NSERC Discovery (610108); ACOA-AIF TREAT2 (199308); Innovation PEI-Development and Commercialization grant DCFG (210205-70). The authors wish to thank Chris Bulger (Huntsman Marine Science Center) for help with lice mating and progeny production, as well as Drs. K. P. Ang and Leighanne Hawkins from Cooke Aquaculture for access to field sites for lice collections; and Tim Sheehan from NOAA for access to lice from wild salmon off of Greenland waters.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
: Markers possessing assignments to more than one linkage group across the mapping panels surveyed (XLSX 11 KB)
Online Resource 2
: List of genetic markers assigned to all male linkage groups and their assignment to recombination bins or steps within each linkage group, along with the integrated composite map distances. Map lengths are portrayed according to the reference male map. To find the adjusted composite map distance correction factor refer to the NOTES page (XLSX 3636 KB)
Online Resource 3
: Map recombination clusters overlap matrix for the male maps. All marker positions are listed in the same ‘head-tail’ orientation among all the mapping panel males and numbered sequentially from 1…N map positions. Markers indicated with an S or M symbol are singleton or markers with missing genotypes, respectively, that were excluded from the map build. Markers designated with a 0 were not assigned to the respective linkage group being examined. Male parent designations are given in row 1. The SNP identification (Affymetrix SNP-chip) corresponding to each SNP listed in sequential rows is given in the SNP_ID worksheet. Note: each male-specific worksheet contains 101,622 rows of data corresponding to the 101,622 SNPs listed in the SNP_ID worksheet (XLSX 44167 KB)
Online Resource 4
: List of genetic markers assigned to all female linkage groups and their assignment to recombination bins or steps within each linkage group, along with the integrated composite map distances. Map lengths are shown separately for each female map (XLSX 1787 KB)
Online Resource 5
: Map recombination clusters overlap matrix for the female maps. All marker positions are not listed in the same ‘head-tail’ orientations (see text for explanation). Markers indicated with an S or M symbol are singleton, or markers with missing genotypes, respectively, that were excluded from the map build. Markers designated with a 0 were not assigned to the respective linkage group being examined. Female parent designations are given in row 1. The SNP identification (Affymetrix SNP-chip) corresponding to each SNP listed in sequential rows is given in the SNP_ID worksheet. Note: each female-specific worksheet contains 101,622 rows of data corresponding to the 101,622 SNPs listed in the SNP_ID worksheet (XLSX 43866 KB)
Online Resource 6
: Recombination step overlaps of comparison male parent maps in relation to the map order established in the reference male map. Reference male map positions containing 2 or more recombination steps from one or more comparison parents are indicated in the middle of the output file. The distribution of all total markers assigned to the linkage group is summarized at the bottom of each linkage group file (XLSX 1796 KB)
Online Resource 7
: The left-hand panels depict the total number of crossovers detected within each recombination domain for a given linkage group. Domains roughly correspond to 10 cM intervals in the map and represent bins encompassing at least 3 recombination steps in the reference map (see text for explanation). The right-hand panel depicts the total number of genetic polymorphisms recorded among all mapping progeny across all the recombination domains surveyed. The abscissa for both plots should be interpreted as: “Recombination Domains”. The ordinate for the left-hand panels should be read as: “Total number of crossovers”, and for the right-hand panels as: “Total number of heterozygous loci” (PDF 615 KB)
Online Resource 8
: Revised male and female genetic linkage maps for SL15 (sex linkage group). Updated maps include hemizygous markers linked to SL15 (XLSX 5101 KB)
Online Resource 9
: Assignment of markers to LOD = 4.0 linkage group assemblies (SL01 – SL14) that match hemizygous patterns of inheritance (XLSX 19 KB)
Online Resource 10
: Assignment of markers to LOD = 4.0 linkage group assemblies that match homozygous null patterns of inheritance (XLSX 14 KB)
Online Resource 11
: Heterozygous markers detected within each of the recombination domains used to assess crossover event among the various sea lice linkage groups. Linkage groups are based upon male-specific maps. Polymorphisms were recorded for both sexes such that Parent_1 and Parent_2 = male- and female-specific polymorphisms, respectively (XLSX 12714 KB)
Online Resource 12
: Relationship between the total number of heterozygous loci recorded within a recombination domain versus the number of crossover events observed in the domain. The strength of the relationship within each linkage group is shown by the regression equation provided (PDF 309 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Danzmann, R.G., Norman, J.D., Rondeau, E.B. et al. A genetic linkage map for the salmon louse (Lepeophtheirus salmonis): evidence for high male:female and inter-familial recombination rate differences. Mol Genet Genomics 294, 343–363 (2019). https://doi.org/10.1007/s00438-018-1513-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-018-1513-7