
Abstract
Cyst-forming coccidia, Toxoplasma gondii and Neospora caninum, are recognised as important causes of animal disease. Molecular diagnostics based on the presence of DNA in animal tissue are required to specifically detect T. gondii and N. caninum while achieving high levels of analytical sensitivity. We optimised available single-plex probe base qPCR assays into a multiplexed qPCR panel to detect cyst-forming coccidia, i.e. T. gondii and N. caninum. The T. gondii assay is based on a 529-bp repetitive (REP) element and the N. caninum assay on the NC5 repetitive region. Using target sequence synthetic DNA, the limit of detection (LOD) was determined to be 100 copies, that is less than a single tachyzoite of either T. gondii or N. caninum. The T. gondii and N. caninum multiplexed qPCR assay optimised in this study can be used to effectively detect parasite DNA for diagnostic purposes in animal tissue.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyst-forming coccidia, such as Toxoplasma gondii and Neospora caninum, are recognised as important causes of animal and human disease (Dubey 2021; Dubey et al. 2017). Molecular diagnostics are based on the presence of parasite DNA in animal tissue. The differentiation of T. gondii and N. caninum is of utmost importance due to the clinical symptoms associated with toxoplasmosis and neosporosis. For this reason, PCR techniques based on repetitive regions found in the genomes of T. gondii and N. caninum have historically been selected as targets for amplification (Muller et al. 1996). Two T. gondii DNA markers commonly targeted by PCR-based diagnostics are the B1 gene and 529-bp repetitive (REP) element (Homan et al. 2000). For N. caninum, the NC5 region has become the standard target because of its multicopy nature (Muller et al. 1996). As technology progressed, conventional PCRs were replaced by quantitative PCR (qPCR) methods. qPCR allowed for the rapid and specific detection of DNA in organisms such as T. gondii and N. caninum (Fekkar et al. 2008; Ghalmi et al. 2008). Alongside T. gondii and N. caninum, animals can also be infected with Hammondia spp., an organism that is often considered non-pathogenic, yet some case reports suggest otherwise (Allan et al. 2022; Reichel et al. 2007; Schares et al. 2021; Šlapeta et al. 2002; Steffl and Nautscher 2019). A highly specific qPCR assay for Hammondia hammondi, based on the repetitive element Hhamm222, has recently been developed (Schares et al. 2021).
The aim of this study was to optimise and evaluate the performance of available single-plex probe-based qPCR assays into a multiplexed qPCR panel to detect cyst-forming coccidia, i.e. T. gondii and N. caninum in host tissue or fluid samples during suspected clinical disease.
Materials and methods
This study used the synthetic T. gondii DNA fragments B1 (AF179871) and REP (AF146527) (243 nt), a H. hammondi DNA fragment Hham222 (KC223619), and an N. caninum DNA fragment NC5 (X84238) targets (232nt). The synthetic DNA was obtained as gBlocks (Integrated DNA Technologies, Australia). The synthetic DNA was resuspended in distilled water and a stock solution of 109 copies/μL was made and stored at – 20 °C in clear plastic microtubes. Stock dilutions were discarded and made fresh after five thaw-freeze cycles.
Four probe-based qPCR assays were used throughout this study targeting T. gondii, H. hammondi, and N. caninum (Table 1). The qPCR assays were adopted from published studies (Ghalmi et al. 2008; Schares et al. 2021). In addition, probe-based qPCRs targeting the partial canine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) region were performed to verify the presence of mammalian DNA (Table 1). All probes and primers were obtained from Integrated DNA Technologies (IDT, Australia). The Myra Liquid Handling System (Bio Molecular Systems, Australia) was used to prepare and multiplex all PCR reactions. All qPCRs were run in 10 μl volume with 1 μl of template (106 to 1 synthetic DNA template copy per reaction), 5 μl of Luna® Universal Probe qPCR Master Mix (New England Biolabs, Australia), 400 nM of forward and reverse primers, and 100 nM of the appropriate probes. The cycling conditions for all runs were 95 °C for 3 min as initial denaturation followed by 40 cycles of 95 °C for 5 s and 60 °C for 15 s. All qPCRs were run using a CFX96 (BioRad, Australia) with runs analysed using CFX Maestro 3.2 (BioRad, Australia) to construct standard curves from serial dilutions and obtained efficacy (E) and R2 values. Cycle threshold (Ct) was auto-calculated. Assays were considered optimised if E = 90 to 110% and R2 > 0.94. This study defines analytical sensitivity as the limit of detection (LOD) being the lowest concentration of DNA (in copy numbers) detected in all three replicates. The assays were either run with respective synthetic controls only or further spiked with mammalian DNA (100 ng).
Results and discussion
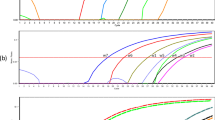
Four probe-based qPCR assays targeting cyst-forming coccidia (T. gondii, H. hammondi and N. caninum) were combined with or without the host DNA To determine the limit of detection (LOD) in the form of copy numbers (Fig. 1A). For example, both T. gondii qPCRs assays targeting either REP or/and B1 were able to detect a single copy of a synthetic DNA without the presence of host DNA either independently or multiplexed (Fig. 1A). When host DNA was added to each assay the limit of detection was 10 copies for both REP and B1 targets. Multiplexing T. gondii REP and B1 assays with the mammalian assay further decreased LOD to 100 copies (Fig. 1A). Combination of the H. hammondi Hham222 proved to be difficult as the multiplexed assay failed to reach optimal E and R2. Mammalian DNA was detected and successfully amplified in all reactions and for all the above assays the efficacy and R2 values were acceptable (E = 90–110%, R2 > 0.99). When comparing the amplification curves for T. gondii assays, the REP qPCR amplified approximately one cycle earlier in the same dilutions of the target DNA compared the B1 qPCR assay (Fig. 1B). Similar to Galvani et al. (2019) and Barry et al. (2019), this study uses synthetic DNA rather than biological DNA in multiplexed real-time qPCRs for the detection of T. gondii and N. caninum. Our study not only quantified each assay’s LOD and efficiency, but also simulated realistic samples through the addition of mammalian DNA. This is a key point as the effects of background mammalian DNA on the diagnostic assay were previously unknown.

A Determination of limit of detection using synthetic DNA for cyst-forming coccidia. Probe-based assays to detect Toxoplasma gondii DNA, Hammondia hammondi DNA and Neospora caninum DNA were run independently or in combination with/or without host DNA (spike). The multiplexed assay was further combined with internal control assay detecting the target host—mammal, using generic GAPDH assay. Assays used are indicated by black squares and presence of host DNA by blue square. Limit of detection (LOD) in copy numbers (#) is depicted below the checkerboard. Encircled numbers represent assays expanded in B and C panels. B Standard curve for T. gondii assays targeting REP and B1 in the presence of mammalian DNA. C Standard curve for T. gondii assay targeting REP and N. caninum assay targeting NC5 region in the presence of mammalian DNA
Taking the above results into an account, the T. gondii REP assay was selected with the N. caninum NC5 assay and combined with the host mammalian qPCR assay. Although a loss of LOD was observed compared to the single-plex assay, the multiplexed qPCR could detect and amplify 100 copies for both targets (T. gondii REP, N. caninum NC5) in all three replicates in the presence of host DNA (E = 90–110%, R2 > 0.94) (Fig. 1AC). The LOD for both is sufficient to detect a single organism (tachyzoite), because REP is present in 150–300 copies per T. gondii genome and NC5 is present in ~ 350 copies per genome of N. caninum (Okeoma et al. 2004). Attempts to add H. hammondi to the multiplexed qPCR were unsuccessful and such an assay will be required to be run separately as originally intended (Schares et al. 2021).
This study recorded a reduction in LODs when assays were combined. This issue has been noted in past literature where multiplexing was found to reduce assay analytical sensitivity due to primer interference and formation of nonspecific products (Choi et al. 2018; Peleg et al. 2010). Assuring consistent and reliable PCR performance is part of quality assurance in any diagnostic laboratory to deliver consistent, relevant, and accurate results (Bustin 2010). The present study took advantage of synthetic DNA targets to define analytical sensitivity as the smallest amount of the target measured in copy numbers in a sample that can accurately be measured. The analytical sensitivity expressed as LOD was used here to demonstrate the PCRs’ performance under a variety of conditions that the assays are intended for such as multiplexing in the presence of host DNA. This approach aligns with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) (Bustin 2010). Determination of analytical sensitivity enables informed decision when combining assays. While loss of analytical sensitivity may not be the critical issue for T. gondii and N. caninum assay, multiplexing with H. hammondi and applying MIQE guidelines exposed underperformance of the assay both in terms of analytical sensitivity as well as assay efficacy. Our study confirmed that T. gondii REP qPCR assay is preferred as it amplifies the target earlier yet is equal as far as analytical sensitivity when compared to the T. gondii B1 assay (Belaz et al. 2015; Pomares et al. 2020).
To conclude, the T. gondii and N. caninum multiplexed qPCR assay optimised in this study can be used to effectively detect parasite DNA for diagnostic purposes in animal tissue.
Data availability
All data are included in the manuscript.
References
Allan F, Blake D, Miller Z, Church D (2022) Biliary protozoa in a dog with acute cholangiohepatitis fed a raw food diet. J Vet Intern Med 36:2177–2180. https://doi.org/10.1111/jvim.16565
Barry R, Nissly RH, Feria W, Thirumalapura N, Tewari D, Jayarao BM, Kuchipudi SV (2019) A probe-based real-time PCR assay for the detection of Neospora caninum in clinical samples from cattle. Vet Parasitol 269:2–6. https://doi.org/10.1016/j.vetpar.2019.04.002
Belaz S, Gangneux J-P, Dupretz P, Guiguen C, Robert-Gangneux F (2015) A 10-year retrospective comparison of two target sequences, REP-529 and B1, for Toxoplasma gondii detection by quantitative PCR. J Clin Microbiol 53:1294–1300. https://doi.org/10.1128/JCM.02900-14
Bustin SA (2010) Why the need for qPCR publication guidelines?—The case for MIQE. Methods 50:217–226. https://doi.org/10.1016/j.ymeth.2009.12.006
Choi W, Yeom SY, Kim J, Jung S, Jung S, Shim TS, Kim SK, Kang JY, Lee SH, Cho I-J, Choi J, Choi N (2018) Hydrogel micropost-based qPCR for multiplex detection of miRNAs associated with Alzheimer’s disease. Biosensors Bioelectron 101:235–244. https://doi.org/10.1016/j.bios.2017.10.039
Dubey JP (2021) Toxoplasmosis of animals and humans (3rd ed.). CRC Press, Boca Raton, FL https://doi.org/10.1201/9781003199373
Dubey JP, Hemphill A, Calero-Bernal R, Schares G (2017) Neosporosis in animals (1st ed.). CRC Press, Boca Raton, FL https://doi.org/10.1201/9781315152561
Fekkar A, Bodaghi B, Touafek F, Le Hoang P, Mazier D, Paris L (2008) Comparison of immunoblotting, calculation of the Goldmann-Witmer coefficient, and real-time PCR using aqueous humor samples for diagnosis of ocular toxoplasmosis. J Clin Microbiol 46:1965–1967. https://doi.org/10.1128/JCM.01900-07
Galvani AT, Christ APG, Padula JA, Barbosa MRF, de Araújo RS, Sato MIZ, Razzolini MTP (2019) Real-time PCR detection of Toxoplasma gondii in surface water samples in São Paulo, Brazil. Parasitol Res 118:631–640. https://doi.org/10.1007/s00436-018-6185-z
Ghalmi F, China B, Kaidi R, Daube G, Losson B (2008) Detection of Neospora caninum in dog organs using real time PCR systems. Vet Parasitol 155:161–167. https://doi.org/10.1016/j.vetpar.2008.04.007
Homan WL, Vercammen M, De Braekeleer J, Verschueren H (2000) Identification of a 200-to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Int J Parasitol 30:69–75. https://doi.org/10.1016/s0020-7519(99)00170-8
Muller N, Zimmermann V, Hentrich B, Gottstein B (1996) Diagnosis of Neospora caninum and Toxoplasma gondii infection by PCR and DNA hybridization immunoassay. J Clin Microbiol 34:2850–2852. https://doi.org/10.1128/jcm.34.11.2850-2852.1996
Okeoma CM, Williamson NB, Pomroy WE, Stowell KM, Gillespie L (2004) The use of PCR to detect Neospora caninum DNA in the blood of naturally infected cows. Vet Parasitol 122:307–315. https://doi.org/10.1016/j.vetpar.2004.06.001
Orr B, Ma G, Koh WL, Malik R, Norris JM, Westman ME, Wigney D, Brown G, Ward MP, Šlapeta J (2020) Pig-hunting dogs are an at-risk population for canine heartworm (Dirofilaria immitis) infection in eastern Australia. Parasit Vectors 13:69. https://doi.org/10.1186/s13071-020-3943-4
Peleg O, Baneth G, Eyal O, Inbar J, Harrus S (2010) Multiplex real-time qPCR for the detection of Ehrlichia canis and Babesia canis vogeli. Vet Parasitol 173:292–299. https://doi.org/10.1016/j.vetpar.2010.06.039
Pomares C, Estran R, Press CJ, Bera A, Ramirez R, Montoya JG, Gangneux FR (2020) Is real-time PCR targeting Rep 529 suitable for diagnosis of toxoplasmosis inpatients infected with non-type II strains in North America? J Clin Microbiol 58:e01223-e1319. https://doi.org/10.1128/jcm.01223-19
Reichel MP, Ellis JT, Dubey JP (2007) Neosporosis and hammondiosis in dogs. J Small Anim Pract 48:308–312. https://doi.org/10.1111/j.1748-5827.2006.00236.x
Schares G, Globokar Vrhovec M, Tuschy M, Joeres M, Barwald A, Koudela B, Dubey JP, Maksimov P, Conraths FJ (2021) A real-time quantitative polymerase chain reaction for the specific detection of Hammondia hammondi and its differentiation from Toxoplasma gondii. Parasit Vectors 14:78. https://doi.org/10.1186/s13071-020-04571-8
Šlapeta JR, Koudela B, Votýpka J, Modrý D, Hořejš R, Lukeš J (2002) Coprodiagnosis of Hammondia heydorni in dogs by PCR based amplification of ITS 1 rRNA: differentiation from morphologically indistinguishable oocysts of Neospora caninum. Vet J 163:147–154. https://doi.org/10.1053/tvjl.2001.0599
Steffl M, Nautscher N (2019) Detection of Hammondia heydorni-like oocysts in feces of a dog with recurrent diarrhea. Tierarztl Prax Ausg 47:189–192. https://doi.org/10.1055/a-0890-2350
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The study was supported by the Betty & Keith Cook Canine Research Fund (University of Sydney).
Author information
Authors and Affiliations
Contributions
M.T. and J.Š. wrote the manuscript. M.T. performed the reseach. Both authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Xing-Quan Zhu
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Truong, M., Šlapeta, J. Analytical sensitivity of a multiplex quantitative PCR for Toxoplasma gondii and Neospora caninum. Parasitol Res 122, 1043–1047 (2023). https://doi.org/10.1007/s00436-023-07796-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-023-07796-5