
Abstract
The development of new molecular methods has significantly improved the detection and identification of avian haemosporidian parasites (Plasmodium, Haemoproteus and Leucocytozoon) compared to microscopic examination. Very large numbers of previously hidden Haemosporida species of a wide range of avian hosts have thus been discovered in the last two decades. However, test parameters of the various detection methods remain largely unevaluated. In this study, the merits of microscopy, multiplex PCR, and nested PCR were compared to identify the infection status of three Malagasy bird species. A total of 414 blood samples of Hypsipetes madagascariensis, Foudia omissa and F. madagascariensis, as well as 147 blood smears, were examined for haemosporidian infection. Thirty-four lineages of haemosporidian parasites could be identified, of which six have been detected for the first time. Microscopy, multiplex and nested PCR showed differences in detection rate, most likely due to low parasitemia of chronically infected birds. The combination of both PCR methods yielded the best results. In particular, detection of multiple infections could be greatly improved and will enable more precise prevalence estimates of individual haemosporidian species in wild birds in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Haemosporidian parasites of the genera Plasmodium, Haemoproteus and Leucocytozoon are common vector-transmitted blood-parasites of birds. Parasite prevalence varies extremely among host species, e.g. from 0.9% prevalence in shorebirds (Soares et al. 2016) to almost 100% prevalence in feral pigeons (Nebel et al. 2020). The parasites are globally distributed with the exception of polar regions (Clark et al. 2014) and infect a great variety of bird species. More than 2,000 bird species have been described as hosts for haemosporidian parasites so far (MalAvi-database (Bensch et al. 2009) as of May 2021). However, estimates of high undetected parasite richness (Clark et al. 2014) suggest that parasite prevalence and host ranges remain underrated.
One reason for this is due to a sampling bias of the host birds. Some bird species are sampled very frequently while others are not sampled at all. This inevitably leads to erroneous estimates of parasite abundance, requiring locally adapted sampling strategies (Lotta et al. 2019).
An additional bias is the infection of the birds itself. In most field studies, mist nets are used to catch wild birds, which select for active individuals. During the short acute phase of haemosporidian infection, parasites usually appear in the blood at high densities whereas in the chronic phase of infection, parasites persist at lower abundance, usually less than one parasite per 1000 erythrocytes (Valkiunas 2005). Especially during primary parasitemia, the parasite infection impacts on birds’ health and thus activity (Mukhin et al. 2016). Therefore, the use of mist nets usually selects against birds in the acute phase of haemosporidian infection.
The third bias concerns the detection methods. Microscopic methods often appear to have less sensitivity at low levels of parasitemia during chronic infections, leading to underestimation of parasite abundance and prevalence (e.g. Jarvi et al. 2002; Durrant et al. 2006; Schumm et al. 2021). Since nested PCR allows detection of haemosporidians with a parasitemia of < 10–5, which is equivalent to less than one infected cell per 100,000 (Waldenström et al. 2004), this method appears to be the best choice for wild bird testing.
However, current PCR methods (eg. Hellgren et al. 2004; Beadell et al. 2004; Martinsen et al. 2008) also have limitations—especially in the detection of multiple infections. A number of wildlife studies suggest that multiple infections with haemosporidian parasites are common (Pérez-Tris and Bensch 2005; Palinauskas et al. 2015), with a high between-species variation in prevalence (Palinauskas et al. 2020). Thus, haemosporidian species with low abundance in mixed infections are frequently undetected. Nested PCR methods are particularly ineffective at detecting mixed infections that include the genera Plasmodium and Haemoproteus due to a low specificity of primers (Martínez et al. 2009). Differences in parasitemia possibly favor amplification of the parasite with the highest DNA concentration in the sample (Bernotiene et al. 2016). However, even when DNA from different parasites of a mixed infection is amplified and visualized by double bands in the chromatogram, the identity of the lineages can often not be determined further (Dimitrov et al. 2013). A newly established multiplex PCR assay (Ciloglu et al. 2019), based on the simultaneous amplification of gene fragments of the three haemosporidian genera, allows improved detection of mixed infections in birds. Although the multiplex PCR method has been used frequently in recent years (Dimitrov et al. 2019; Inumaru et al. 2020; Ellis et al. 2020; Schumm et al. 2021; Meister et al. 2021), only one paper has addressed the issue of mixed infections in more detail (Neto et al. 2020).
In this study, three bird species endemic to Madagascar (Foudia omissa, Foudia madagascariensis (Ploceidae) and Hypsipetes madagascariensis (Pycnonotidae)) were examined for haemosporidian infections using microscopic and molecular detection methods. The aims of the study were (1) to characterize haemosporidian parasite abundance and diversity present in the examined bird species, (2) to compare the methods used and (3) to identify mixed haemosporidian infections.
Material and methods
Study sites and blood samples
Blood samples were collected in the Maromizaha rainforest located in the eastern part of Madagascar (18°56′49″S, 48°27′33″E) 30 km from Moramanga city at an altitude between 943 and 1213 m. Fieldwork was done in the months September–January, with the majority of samples taken in November and December (2003–2007, 2010, 2012, 2014, 2016, and 2018). A total of 113 individuals of the bird species Madagascar Bulbul (Hypsipetes madagascariensis, Pycnonotidae), as well as 301 samples of Fodies (Forest Fody, Foudia omissa n = 207; Madagascar Fody, Foudia madagascariensis n = 42; Foudia sp. n = 52), were collected. Under the designation Foudia sp., birds were counted that could not be clearly assigned to one species and probably represent hybrids of both species. Hybrids between Foudia omissa and F. madagascariensis seem to be very common in the wild (Hawkins et al. 2015). The birds were caught in mist nets and a blood sample was taken by puncturing the brachial vein before the bird was released. Blood was immediately stored in lysis buffer (Wink 2006).
Preparation and examination of blood smears
Blood smears (n = 147) were prepared in the years 2007, 2012, 2014, 2016 and 2018 and fixed with 99% methanol for 10 min in the field. Using the Hemacolor® staining set (Merck KGaA, Germany), the blood smears were stained with Giemsa following the manufactures protocol. Every slide was screened for 20 min under high magnification (× 100 oil immersion objective, × 10 ocular) using an AxioImager M2 (Carl Zeiss AG, Germany). Pictures were taken and edited with the Zen software (Carl Zeiss AG, Germany). Using the morphology-based identification key of Valkiunas (2005), parasites were determined to genus or species level when possible.
Extraction of DNA, PCR, and sequencing
Total DNA was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) following the producer’s instructions and stored at –20 °C until further use. Different PCR protocols were used for haemosporidian parasite detection. To visualize possible contamination with target DNA, a negative control (nuclease-free water) was included in each test run as well as a positive control. DNA of Plasmodium berghei was used as positive control for multiplex PCR and nested PCR targeting Plasmodium and Haemoproteus spp., whereas an internal sample of Hypsipetes madagascariensis containing DNA of Leucocytozoon lineage FOUOMI02 was used as positive control for the nested PCR targeting Leucocytozoon spp.
For simultaneous detection of the three haemosporidian genera, we used the multiplex PCR assay of Ciloglu et al. (2019) with the three primer sets PMF/PMR, HMF/HMR and LMF/LMR (Ciloglu et al. 2019). The reactions were set up in a total volume of 10 µl containing 5 µl of 2 × Multiplex PCR Master-Mix (Quiagen, Hilden, Germany), 0.2 µl of each primer (10 µM) and 3.8 µl of DNA template. If the DNA concentration was higher than 10 ng/µl, the DNA template was diluted with nuclease-free water. The PCR amplification protocol started with an initial denaturation step of 95 °C for 15 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 59 °C for 90 s and extension at 72 °C for 30 s. The final extension occurs at 72 °C for 10 min.
Amplification products (10 µl) of the multiplex PCR were mixed with GelRed™ stain (BIOTREND, Köln, Germany) and electrophoretically resolved after 45 min at 90 V in 2% agarose gels. The different parasite genera present in the samples were determined by identifying the size of the resulting amplification products.
Parasite identification to lineage level was done using the nested PCR protocols developed by Bensch et al. (2000) and Hellgren et al. (2004). PCR reactions of the first run were carried out in a total volume of 25 µl containing 2.5 µl GeneAmp™ 10X PCR Buffer II (Applied Biosystems, Carlsbad USA), 2 µl MgCl2 (25 mM), 1 µl of each primer (HaemNF/HaemNR3; 10 mM), 0.5 µl of each dNTP (10 µmol), 0.125 µl AmpliTaq™ DNA Polymerase (5 U/µl; Applied Biosystems, Carlsbad USA), 5 µl template DNA (10–100 ng/µl) and 12.875 µl nuclease-free water. The PCR amplification protocol started with an initial denaturation step of 94 °C for 5 min, followed by 20 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s and extension at 72 °C for 45 s. The final extension occurs at 72 °C for 5 min. The reaction mixture of the nested PCRs consisted of 5 µl GeneAmp™ 10X PCR Buffer II (Applied Biosystems, Carlsbad USA), 4 µl MgCl2 (25 mM), 2 µl of each primer (HaemF/HaemR2 for Plasmodium/Haemoproteus detection and HaemFL/HaemR2L for Leucocytozoon detection; 10 mM), 1 µl of each dNTP (10 µmol), 0.25 µl AmpliTaq™ DNA Polymerase (5 U/µl; Applied Biosystems, Carlsbad USA), 2 µl amplification product of the initial PCR and 33.75 µl nuclease-free water in a total volume of 50 µl. The cycling conditions included an initial denaturation step of 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C (HaemFL/HaemR2L) or 55 °C (HaemF/HaemR2) for 30 s and extension at 72 °C for 45 s. The final extension occurred at 72 °C for 5 min. Amplification products (5 µl) of the nested PCR were also mixed with GelRed™ stain and then visualized on a 1.5% agarose gel after 20 min at 90 V. Amplification products were then purified using the PCR Product Purification Kit (Roche, Mannheim, Germany) and after sequencing (Microsynth AG, Switzerland), the resulting sequence data were edited using Geneious v. 2021.1.1. The final sequences were then distinguished by identifying their closest matches in GenBank (Benson et al. 2013) using the NCBI nucleotide BLAST search and a BLAST search of the MalAvi database (Bensch et al. 2009); as of August 2021. All newly detected lineages in our study were deposited in GenBank (accession numbers MZ852010—MZ852014 and OL804019).
Phylogenetic and statistical analyses
A phylogenetic tree was reconstructed using a maximum likelihood (ML) approach in MEGA X (Kumar et al. 2018). Phylogenies were generated by implementing the best fitting model (GTR + G) using 1000 pseudo-replicates. The dataset used consisted of haemosporidian lineages obtained in this study as well as homologous cytochrome b sequences of Hepatocystis sp. (FJ168565) and Theileria annulata (KF732030.1) as outgroups. The resulting phylogram was viewed and edited with MEGA X.
For each sample, the infection status was determined using (1) microscopy (if available), (2) the multiplex PCR approach, (3) the nested PCR approach and (4) a combination of all methods. The resulting prevalences were compared using chi-square tests using R-4.2.1 to draw conclusions about the sensitivity of the different methods.
Results
Haemosporidians were found in 58 of 147 blood smears (H. madagascariensis: 18/38; Foudia spp.: 40/109). In 16 cases, haemosporidian parasites could be identified to species level by morphology. Haemoproteus micronuclearis (lineage RBQ11, Fig. 1) was identified in ten blood smears of Foudia spp. and Haemoproteus sanguinis (BUL2, Fig. 2) in six blood smears of Hypsipetes madagascariensis.

Haemoproteus micronuclearis (RBQ11) from a Forest Fody (Foudia omissa, Ploceidae) sampled in the Maromizaha rainforest, Madagascar. Macrogametocyte in erythrocyte. Malaria pigment (hemozoin) is marked by a black arrow. Nucleus of host cell is marked by a white arrowhead. Giemsa stained blood smear. Scale bar = 10 µm

Haemoproteus sanguinis (BUL2) from a Madagascar Bulbul (Hypsipetes madagascariensis, Pycnonotidae) sampled in the Maromizaha rainforest, Madagascar. Macrogametocyte in erythrocyte. Malaria pigment (hemozoin) is marked by a black arrow. Nucleus of host cell is marked by a white arrowhead. Giemsa stained blood smear. Scale bar = 10 µm
All other Haemosporida could be morphologically assigned to genus level, but not to any described species. Gametocytes of the lineages Plasmodium BUL07 (n = 7), Haemoproteus FOUMAD02 (n = 14), and Leucocytozoon FOMAD01 (n = 2), as well as HYPMA02 (n = 6), were detected (Fig. 3). Species description for those lineages are not yet available.

Haemosporidian parasites detected in Giemsa stained blood smears of Malagasy birds. A Gametocytes of Plasmodium BUL07 in erythrocytes of Hypsipetes madagascariensis. B Microgametocyte (black arrowhead) and macrogametocyte (white arrowhead) of Haemoproteus FOUMAD02 in erythrocytes of Foudia omissa. C Gametocyte of Leucocytozoon FOMAD01 in roundish host cell of H. madagascariensis. D Gametocyte of Leucocytozoon HYPMA02 in roundish host cell of F. omissa. Scale bar = 10 µm
In H. madagascariensis, multiplex PCR detected at least one parasite species or lineage in 83/113 blood samples (73.45%) while nested PCR method detected 91/113 infected individuals (80.53%; χ2 = 1.599, df = 1, P = 0.21). The number of Plasmodium positives (χ2 = 0.073, df = 1, P = 0.79) and Haemoproteus positives (χ2 = 2.319, df = 1, P = 0.13) was identical between both methods, but differed significantly for Leucocytozoon (χ2 = 7.094, df = 1, P = 0.008): multiplex PCR found 49 individuals infected with Leucocytozoon (43.4%) whereas nested PCR detected 69 infections (61.1%). Nested PCR recovered more double infections than multiplex PCR, but the difference was not statistically significant (χ2 = 0.471, df = 1, P = 0.49). However, multiplex PCR detected significantly more triple infections than nested PCR (χ2 = 4.665, df = 1, P = 0.03).
In total, 16 different haemosporidian lineages (six Plasmodium, two Haemoproteus, and eight Leucocytozoon lineages) were thus found in the Hypsipetes madagascariensis blood samples (Table 1), all homologous to isolates deposited in GenBank. In the 93 positive birds, 146 infections with individual lineages were detected. The most abundant Plasmodium lineage was BUL07 (EU810628; n = 19), the most abundant Haemoproteus lineage was BUL2 (DQ847195; n = 23), and the most abundant Leucocytozoon lineages were HYPMA02 (MF442609; n = 38) and FOMAD01 (JN032605, n = 27). Two birds were recaptured in subsequent years: CC72543, captured in 2010 and 2014, contained Haemoproteus sp. BUL2 in both years. Bird CC72575, 2012 and 2014, was infected with Leucocytozoon sp. HYPMA02 in both years and had additionally acquired Haemoproteus BUL2 in 2014.
In Foudia spp., multiplex PCR detected 188/301 infected blood samples (62.5%), while nested PCR detected 207/301 (68.8%) (χ2 = 2.658, df = 1, P = 0.1). Prevalence of infection among the Foudia species did not differ (F. omissa/F. madagascariensis: χ2 = 0.065, df = 1; P = 0.8; F. madagascariensis/ Foudia sp.: χ2 = 0.141, df = 1, P = 0.7; Foudia sp./F. omissa: χ2 = 0.626, df = 1, P = 0.43). Both PCR methods found a similar number of samples containing Plasmodium and Haemoproteus DNA (Plasmodium: χ2 = 0.995, df = 1, P = 0.32; Haemoproteus: χ2 = 1.574, df = 1, P = 0.21). As with H. madagascariensis, Leucocytozoon spp. were detected significantly more often with nested PCR (χ2 = 50.271, df = 1, P = < 0.001). Likewise, nested PCR was able to detect significantly more double infections (χ2 = 14.898, df = 1, P = < 0.001). However, triple infections were detected more frequently by multiplex PCR (χ2 = 2.315, df = 1, P = 0.13). But the results of either method (nested PCR: n = 3; multiplex PCR: n = 8) underestimated the number of triple infections as determined by combining the results of both methods (n = 24; χ2 = 21.46, df = 2, P = < 0.001).
Foudia spp. samples contained 25 haemosporidian lineages (nine Plasmodium, three Haemoproteus, and 13 Leucocytozoon lineages; Table 1). One Plasmodium lineage (FOUOMI05) and five Leucocytozoon lineages (FOUOMI06-10) were detected for the first time and sequences were deposited in GenBank (Acc. Nos. MZ852010-MZ852014, OL804019). Multiple infections included, a total of 309 infections with individual lineages were detected in the 210 positive birds. The most abundant Plasmodium lineage was COLL7 (DQ368376; n = 65) and the most abundant Haemoproteus lineage was FOUMAD02 (JN661941; n = 48). Foudia madagascariensis was more often infected with Haemoproteus lineage RBQ11 (EF117229, n = 8) than with FOUMAD02 (n = 1). The most abundant Leucocytozoon lineages were HYPMA02 (MF442609; n = 72) and FOMAD01 (JN032605, n = 19). One recaptured individual of Foudia madagascariensis (FB52004) was infected with Plasmodium sp. COLL7 both in 2012 and 2014.
Combining the results of all detection methods, 82.3% (n = 93) of the H. madagascariensis samples and 69.8% (n = 210) of the Foudia spp. samples were positive for at least one haemosporidian parasite. In both bird genera (H. madagascariensis and Foudia spp.), double infections were detected most frequently, followed by single infections, and triple infections were detected least frequently (Table 2). However, the difference between single and double infections was not significant (H. madagascariensis: χ2 = 1.914, df = 1, P = 0.17; Foudia spp.: χ2 = 2.521, df = 1, P = 0.11). The most abundant combination was a double infection with Plasmodium and Leucocytozoon (23.9% for H. madagascariensis; 20.3% for Foudia spp.).
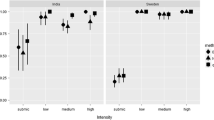
In comparison to both PCR methods, the detection rate of parasites using microscopy was very low (Fig. 4). For H. madagascariensis, 52.6% of the birds were negative on microscopy, whereas only 5.3–7.9% were PCR negative. For Foudia spp., the differences were less pronounced with 63.3% negative on microscopy and 28.4–35.8% PCR negative. Based on blood smears of H. madagascariensis, Plasmodium spp. was detected most often, followed by Haemoproteus and Leucocytozoon species. Leucocytozoon was significantly less detected by microscopy than by multiplex PCR (χ2 = 11.12, df = 1, P = < 0.001) and nested PCR (χ2 = 33.474, df = 1, P = < 0.001). Based on blood smears of Foudia spp., the genus Haemoproteus was detected most often, followed by Plasmodium and Leucocytozoon. Leucocytozoon was detected microscopically less frequently than with PCR methods, the difference with nested PCR was particularly significant (χ2 = 52.982, df = 1, P = 0). Within our dataset, nested PCR missed 23% of Plasmodium and Haemoproteus infections in comparison to combined results of all methods in H. madagascariensis samples, whereas multiplex PCR only missed < 10%. Within Foudia spp. samples, the nested PCR failed to detect 18.75% and the multiplex PCR 7.5% of all Plasmodium and Haemoproteus infections. The difference between nested PCR and combined result was significant for both bird families (H. madagascariensis: χ2 = 9.356, df = 1, P = 0.002; Foudia spp.: χ2 = 16.781, df = 1, P = < 0.001).

Comparison of the percentage of individuals infected by haemosporidian parasites detected (P: Plasmodium, H: Haemoproteus, L: Leucocytozoon, neg: negative) detected using microscopy, multiplex PCR (Ciloglu et al. 2019), and nested PCR method (Bensch et al. 2000; Hellgren et al. 2004). The total number of individuals screened in this analysis was 147 (H. madagascariensis: n = 38; Foudia spp.: n = 109)
The phylogeny of lineages found in this study is shown in Fig. 5. The phylogenetic analyses support the three haemosporidian genera as monophyletic groups. Seven of all 34 lineages (20.6%) were detected in both H. madagascariensis and Foudia spp.. Interestingly, no Haemoproteus lineage was shared between the two avian groups. The two most abundant Leucocytozoon lineages (FOMAD01 and HYPMA02) belong to separate clades. For Haemoproteus, a clear separation exists between lineages from Foudia spp. (FOUMAD02, FOUOMI01 and RBQ11) and the lineage BUL2 from H. madagascariensis. Plasmodium is divided into five distinct clades with GRW04, FOUMAD03 and COLL7 in one cluster, GRW09 and FOUOMI04 standing separate, a cluster of rarely detected lineages (FOUOMI05, WW3 and COLL4) and a cluster that almost exclusively infected H. madagascariensis (NEWAM05, BUL07, HYPMA01 and HYPMA04).

Evolutionary relationship among haemosporidian cytochrome b lineages estimated using maximum likelihood approach implemented in MEGA. Bootstrap support is shown above branches. Bootstrap values below 70% are not reported. The blue branch represents Leucocytozoon, the orange branch Haemoproteus, and the green one Plasmodium lineages. Newly detected lineages are marked with an asterisk. Squares to the right of lineage names indicate the host species in which the lineage was recovered (H: Hypsipetes madagascariensis, Fo: Foudia omissa, Fm: F. madagascariensis, Hy: Foudia sp.). Numbers within the squares indicate the number of host individuals infected
Discussion
We compared the blood parasite infection status of three common passerine bird species from Madagascar—the Madagascar bulbul (Hypsipetes madagascariensis, Pycnonotidae) and two species of fody (Ploceidae): The Forest Fody (Foudia omissa) and the Madagascar Fody (F. madagascariensis) with a special focus on mixed infections. Three different diagnostic methods were used to identify haemosporidian parasites and estimate prevalence of infection.
The results showed a difference of total prevalence between H. madagascariensis (82.3%) and Foudia spp. (69.8%). Prevalence was determined by combining two molecular methods (multiplex and nested PCR), which probably corresponds most closely to the real infection status. Microscopically, in the subset of samples for which blood smears were available, significantly fewer infections were found compared with PCR methods. Although known as a convenient tool for haemosporidian detection and essential for haemosporidian species description, microscopy has also been shown to be less sensitive than PCR methods in previous studies (Jarvi et al. 2002; Durrant et al. 2006; Schumm et al. 2021). However, it is also known that sensitivity of microscopy strongly depends on the quality of blood smears and the skill of the investigator, approaching the test parameters of PCR under favourable conditions (Valkiunas et al. 2008; Ciloglu et al. 2019; Nebel et al. 2020). Due to the preparation in the field in a humid rainforest and long storage before staining, the quality of our blood smears was often not sufficient for the description of species but appeared suitable for screening purposes in all cases. If the blood smears had been of excellent quality, additional infections might have been detected in single cases; however, the significance of the differences would probably not have been changed. Nevertheless, the good quality of blood smears is of outmost importance to exclude aforesaid doubts.
The most feasible explanation for the low sensitivity in our panel would be extremely low parasitemia during the haemosporidian infections. As all samples were collected from birds caught in the wild with mist nets, they were most probably healthy enough to be mobile and therefore in very early or chronic stage of disease when only few parasites are found in the blood (Valkiunas 2005). It has been shown that Haemoproteus reaches higher parasitemias than Plasmodium (Fallon and Ricklefs 2008; Asghar et al. 2011; Mukhin et al. 2016). This would explain that the detection rate of Haemoproteus in this study was very similar for all methods used. There are no reliable, detailed data on the parasitemia levels of Leucocytozoon, but the low detection rate in the blood smears suggests that it may be similar to Plasmodium. To test this hypothesis, blood smears should in future first be examined for the presence of Leucocytozoon at × 40 magnification, as recommended by Valkiunas (2005), in order to be able to determine parasitemia more specifically.
Similar to the data of Ciloglu et al. (2019), the multiplex PCR and the nested PCRs showed comparative sensitivity levels regarding Plasmodium and Haemoproteus. Leucocytozoon was detected significantly more frequently with nested PCR than with the other methods. This fact can be partly explained by low parasitemia. Samples containing DNA of the most abundant Leucocytozoon lineages HYPMA02 and FOMAD01 had tested negative several times with multiplex PCR method before employing nested PCR. For Foudia spp., 72% of HYPMA02 infections and 58% of FOMAD01 infections were missed, and for H. madagascariensis samples, 21% of HYPMA02 and 22% of FOMAD01 infections were not detected by multiplex PCR. The parasites may reach higher levels of parasitemia in H. madagascariensis as in Foudia spp. during chronic phase of infection, leading to a lower detection probability in Foudia spp. samples. Difference in sensitivity between the two PCR methods may also be due to different specificities. Within Foudia spp. samples, many Leucocytozoon lineages were not detected at all by multiplex PCR method. The sequence of cytochrome c oxidase subunit 1 (COX1) targeted by the primers used in the multiplex PCR might be not homologous enough for functional amplification. Since COX1 sequences of these lineages have not yet been described, this hypothesis cannot be tested so far.
Valkiunas et al. (2008) already highlighted the benefit of combining molecular and microscopic approaches in Avian malaria studies when possible. Furthermore, compared different screening protocols (microscopy, qPCR, nested PCR, restriction enzyme-based assay) and emphasized the benefits of a wider range of diagnostic tools combined with statistical ones when estimating prevalence of avian haemosporidians in natural populations. Therefore, this study attempted to approach the real prevalence of avian haemosporidian parasites by combining microscopy, multiplex PCR, and nested PCR methods. Microscopy remains the only direct evidence of infection, but low parasitemia and quality requirements for the blood smear preparation limits its value under certain conditions. The multiplex PCR assay (Ciloglu et al. 2019) is a very useful and important tool in detecting mixed haemosporidian infections. Cytochrome b, used in our nested PCR (Bensch et al. 2000; Hellgren et al. 2004), remains the sequence of choice for identifying avian haemosporidian parasites, as the MalAvi database (Bensch et al. 2009) provides a massive collection of comparative data. However, the nested PCR is quite ineffective at detecting mixed infections between parasites of the genera Plasmodium and Haemoproteus (Bernotiene et al. 2016). Within our dataset, nested PCR missed some 20% of Plasmodium and Haemoproteus infections in comparison to combined results of both PCR methods in the two bird families, whereas multiplex PCR only missed < 10% (Fig. 6). For house sparrows, Neto et al. (2020) already found that Plasmodium is barely detected in the presence of Haemoproteus, resulting in the non-detection of over 50% of Plasmodium infections. Further studies are needed to determine the level of underestimation of Plasmodium and Haemoproteus in different bird species.

The results clearly show that the real prevalence in bird populations is underestimated by any PCR method alone. This is particularly true in case of triple infections. In Hypsipetes madagascariensis, only one triple infection was detected with nested PCR, with multiplex PCR as many as seven, but the combination of the results yielded 11 triple infections. In the Foudia samples, the difference was even more significant, with three triple infections by nested PCR, eight by multiplex PCR, and 24 by combined results. Again, both PCR methods alone prevalence in both groups. From our data, the combination of the methods is clearly the best approach to determine the actual prevalence in bird populations.
Three individuals (two H. madagascariensis and one Foudia madagascariensis) were recaptured in different years. In the case of H. madagascariensis CC72543, it is likely that a chronic infection with Haemoproteus sanguinis BUL2 had persisted for at least 4 years. Since H. madagascariensis is known to live up to at least 10 years (Woog et al. 2018), this means that the infection with BUL2 probably persisted or existed for at least half of the life. The same picture emerges with F. madagascariensis FB52004. It was captured the first time 2012 and recaptured in 2014. Both samples contained DNA of Plasmodium sp. COLL7. However, no blood stages of Plasmodium sp. COLL7 were found in the blood smears of either sample, probably indicating low parasitemia during chronic infection. Since F. madagascariensis has been reported to live up to at least 4 years (Woog et al. 2018), the infection could have lasted at least half of the life. Another individual of H. madagascariensis (CC72575) contained DNA of Leucocytozoon HYPMA02 in 2012 and in 2014 DNA of the same lineage as well as DNA of Haemoproteus sanguinis (BUL2). In no case infections disappeared. These findings are in accordance with the assumption that infections with haemosporidian parasites can persist for years or even a lifetime (Valkiunas 2005).
Our data showed a higher proportion of double infections than single infections in general. It has been recently shown that the probability of being infected with haemosporidian parasites increases with host age (Slowinski et al. 2022). An accumulation of parasites in the hosts could also take place over time, which would increase the proportion of multiple infections. Further studies should therefore examine the probability of multiple infections in correlation with age more closely in order to test this hypothesis.
This study was able to collect additional information about the most common lineages (Plasmodium COLL7, GRW09 and BUL07; Haemoproteus FOUMAD02, RBQ11 and BUL2; Leucocytozoon FOMAD01 and HYPMA02). Plasmodium COLL7 has been detected in previous studies (e.g. Beadell et al. 2006; Ishtiaq et al. 2012; Lutz et al. 2015; Lauron et al. 2015). It has been reported that the lineage might act as a specialist on Madagascar, whereas it seems to be generalized on mainland Africa (Musa et al. 2019). Since we did not find Plasmodium COLL7 in the Hypsipetes madagascariensis samples even with an additional PCR method used in the current study, our data support this hypothesis. Plasmodium lineage GRW09 has been isolated earlier from 87 different host species and six different vector mosquito species (MalAvi as of May 2022). In each sample set, Foudia spp. and Hypsipetes madagascariensis, this lineage was detected in about 9% of the samples in this study. Similar to Plasmodium COLL7, the lineage BUL07 seems to be specialized on Hypsipetes madagascariensis, whereas on mainland Africa it is a generalist (Musa et al. 2019) infecting numerous bird species of various families including Ploceidae (Lutz et al. 2015). In the phylogenetic analysis BUL07 clusters with HYPMA01 and HYPMA04 which was also exclusively isolated from H. madagascariensis in this study. However, host specialization has not been reported for HYPMA01 and HYPMA04 in a previous study (Musa et al. 2019). Haemoproteus FOUMAD02 has been exclusively isolated from Malagasy Foudia spp. so far (Ishtiaq et al. 2012; Musa et al. 2019). The lineage seems to be endemic on Madagascar and highly specialized on Foudia spp. Only Haemoproteus RBQ11 (H. micronuclearis) and BUL2 (H. sanguinis) have so far been described morphologically. Lineages allocated to Haemoproteus sanguinis were found to be monophyletic (Martinsen et al. 2006) and the species is reported to infect mainly birds belonging to the Pycnonotidae (Valkiunas 2005). Gametocytes of H. sanguinis were found in Hypsipetes madagascariensis throughout his study (Fig. 2). Haemoproteus micronuclearis was described in 2011, the species seems to be widespread in sub-Saharan Africa (Iezhova et al. 2011). Transmission occurs among birds belonging to Quelea and Ploceus (Ploceidae) (Iezhova et al. 2011). The detection of gametocytes of H. micronucelaris in blood smears of Foudia omissa, as well as the amplification of the barcoding sequence from F. omissa and F. madagascariensis (Ploceidae), indicates that these two species are also suitable hosts for this parasite. Furthermore, a specialization of the lineage on African Ploceidae is suggested (Musa et al. 2019). In the phylogenetic tree, RBQ11 clusters with FOUMAD02 indicate that specialization can be mapped genetically. However, more extensive phylogenetic studies will be necessary to support this hypothesis. The most abundant Plasmodium and Haemoproteus lineages except GRW09 are specialised whereas both of the most abundant Leucocytozoon lineages (HYPMA02 and FOMAD01) seem to be generalists. These two lineages account for 78.4% (Foudia spp.) and 84.4% (H. madagascariensis) of all infections with Leucocytozoon and are genetically widely separated (Fig. 5). HYPMA02 has been detected exclusively in Malagasy birds so far (Musa et al. 2019) whereas FOMAD01 has been detected in Malagasy birds (Ivanova et al. 2018; Musa et al. 2019) and additionally in two Foudia madagascariensis on La Réunion (Cornuault et al. 2012), belonging to the Mascarenes. Both lineages appear to be endemic to Madagascar and, in the case of FOMAD01, additionally to islands in the region. Although these lineages seem to be very common on Madagascar, there has been little evidence of them in previous studies, as most studies on avian malaria on Madagascar have so far either been based purely on morphology (Savage et al. 2009) or have excluded Leucocytozoon (Ishtiaq et al. 2012). None of the two lineages has been assigned to a morphologically described species so far (MalAvi, as of May 2022). Savage et al. (2009) detected Leucocytozoon bouffardi, L. brimonti, and L. pycnonoti in blood smears of F. omissa and H. madagascariensis. However, L. bouffardi and L. brimonti had earlier been included in the common species Leucocytozoon fringillinarum and L. pycnonoti seems to be a synonym of L. majoris (Valkiunas 2005). Based on the morphological characteristics, the lineages could rather be assigned to L. fringillinarum, as the nucleus of the host cell does not extend more than half of the circumference of the gametocyte. But neither HYPMA02 nor FOMAD01 are homologous to sequences of Leucocytozoon fringillinarum (ZOLEU02, MG726144.1) or L. majoris (CB1, FJ168564), respectively. Taxonomic descriptions of Leucocytozoon species have been based on two different assumption so far. Several investigations have provided empirical evidence that each bird family has its own Leucocytozoon species (Solis 1973; Fallis et al. 1974; Bennett et al. 1991). Therefore, new parasites are named when previously unstudied host families are examined. Others argue that this is not a valid approach and insist on morphological features for describing Leucocytozoon species (Valkiunas 2005). Both extremes are difficult to support. Neither are all Leucocytozoon species specialized on one bird family (see data of this study), nor do all morphologically identical individuals belong to one species (Galen et al. 2018). Further studies are needed to clarify the taxonomic status of HYPMA02 and FOMAD01 as well as Leucocytozoon in general.
In conclusion, the combination of all methods used in this study (microscopy, multiplex PCR, and nested PCR) appears to be the best approach to determine the actual infection status of avian haemosporidian parasites in bird populations. Direct detection of parasites by microscopy is still an indispensable tool for species identification and description. Mixed haemosporidian infections are neglected in most studies because the detection is difficult. Only by combining the results of both PCR methods, we were successful to detect the majority of multiple infections. Given their frequency, this approach will be indispensable in future studies to estimate the frequency of haemosporidian lineages in birds with any precision.
References
Asghar M, Hasselquist D, Bensch S (2011) Are chronic avian haemosporidian infections costly in wild birds? J Avian Biol 42:530–537. https://doi.org/10.1111/j.1600-048X.2011.05281.x
Beadell JS, Gering E, Austin J et al (2004) Prevalence and differential host-specificity of two avian blood parasite genera in the Australo-Papuan region. Mol Ecol 13:3829–3844. https://doi.org/10.1111/j.1365-294X.2004.02363.x
Beadell JS, Ishtiaq F, Covas R et al (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proceedings of the Royal Society b: Biological Sciences 273:2935–2944. https://doi.org/10.1098/rspb.2006.3671
Bennett GF, Earlé RA, Peirce MA et al (1991) Avian Leucocytozoidae: the leucocytozoids of the Phasianidae sensu lato. J Nat Hist 25:1407–1428. https://doi.org/10.1080/00222939100770891
Bensch S, Stjernman M, Hasselquist D et al (2000) Host specificity in avian blood parasites: a study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc Royal Society b: Biol Sci 267:1583–1589. https://doi.org/10.1098/rspb.2000.1181
Bensch S, Hellgren O, PÉrez-Tris J (2009) MalAvi: A public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour 9:1353–1358. https://doi.org/10.1111/j.1755-0998.2009.02692.x
Benson DA, Cavanaugh M, Clark K et al (2013) GenBank. Nucleic Acids Research 41. https://doi.org/10.1093/nar/gks1195
Bernotiene R, Palinauskas V, Iezhova T et al (2016) Avian haemosporidian parasites (Haemosporida): A comparative analysis of different polymerase chain reaction assays in detection of mixed infections. Exp Parasitol 163:31–37. https://doi.org/10.1016/j.exppara.2016.01.009
Ciloglu A, Ellis VA, Bernotienė R et al (2019) A new one-step multiplex PCR assay for simultaneous detection and identification of avian haemosporidian parasites. Parasitol Res 118:191–201. https://doi.org/10.1007/s00436-018-6153-7
Clark NJ, Clegg SM, Lima MR (2014) A review of global diversity in avian haemosporidians (Plasmodium and Haemoproteus: Haemosporida): new insights from molecular data. Int J Parasitol 44:329–338. https://doi.org/10.1016/j.ijpara.2014.01.004
Cornuault J, Bataillard A, Warren BH et al (2012) The role of immigration and in-situ radiation in explaining blood parasite assemblages in an island bird clade. Mol Ecol 21:1438–1452. https://doi.org/10.1111/j.1365-294X.2012.05483.x
Dimitrov D, Valkiunas G, Zehtindjiev P et al (2013) Molecular characterization of haemosporidian parasites (Haemosporida) in yellow wagtail (Motacilla flava) with description of in vitro ookinetes of Haemoproteus motacillae. Zootaxa 3666:369–381. https://doi.org/10.11646/zootaxa.3666.3.7
Dimitrov D, Marinov MP, Bobeva A et al (2019) Haemosporidian parasites and leukocyte profiles of pre-migratory rosy starlings (Pastor roseus) brought into captivity. Animal Migration 6:41–48. https://doi.org/10.1515/ami-2019-0005
Durrant KL, Beadell JS, Ishtiaq F et al (2006) Avian hematozoa in South America: a comparison of temperate and tropical zones. Ornithol Monogr 60:98–111. https://doi.org/10.2307/40166831
Ellis VA, Huang X, Westerdahl H et al (2020) Explaining prevalence, diversity and host specificity in a community of avian haemosporidian parasites. Oikos 129:1314–1329. https://doi.org/10.1111/oik.07280
Fallis AM, Desser SS, Khan RA (1974) On species of Leucocytozoon. Adv Parasitol 12:1–67. https://doi.org/10.1016/S0065-308X(08)60386-3
Fallon SM, Ricklefs RE (2008) Parasitemia in PCR-detected Plasmodium and Haemoproteus infections in birds. J Avian Biol 39:514–522. https://doi.org/10.1111/j.2008.0908-8857.04308.x
Galen SC, Nunes R, Sweet PR, Perkins SL (2018) Integrating coalescent species delimitation with analysis of host specificity reveals extensive cryptic diversity despite minimal mitochondrial divergence in the malaria parasite genus Leucocytozoon. BMC Evolutionary Biology 18. https://doi.org/10.1186/s12862-018-1242-x
Hawkins F, Safford R, Skerrett A (2015) Birds of Madagacar and the Indian Ocean Islands. Christopher Helm, London
Hellgren O, Waldenstrom J, Bensch S (2004) A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol 90:797–802. https://doi.org/10.1645/GE-184R1
Iezhova TA, Dodge M, Sehgal RNM et al (2011) New avian Haemoproteus species (Haemosporida: Haemoproteidae) from African birds, with a critique of the use of host taxonomic information in hemoproteid classification. J Parasitol 97:682–694. https://doi.org/10.1645/GE-2709.1
Inumaru M, Aratani S, Shimizu M et al (2020) Penguins are competent hosts of Haemoproteus parasites: the first detection of gametocytes, with molecular characterization of Haemoproteus larae. Parasites and Vectors 13. https://doi.org/10.1186/s13071-020-04176-1
Ishtiaq F, Beadell JS, H.warren B, Fleischer RC (2012) Diversity and distribution of avian haematozoan parasites in the western Indian Ocean region: a molecular survey. Parasitol 139:221–231. https://doi.org/10.1017/S0031182011001831
Ivanova K, Zehtindjiev P, Mariaux J et al (2018) Avian haemosporidians from rain forests in Madagascar: molecular and morphological data of the genera Plasmodium, Haemoproteus and Leucocytozoon. Infect Genet Evol 58:115–124. https://doi.org/10.1016/j.meegid.2017.12.017
Jarvi SI, Schultz JJ, Atkinson CT (2002) PCR diagnostics underestimate the prevalence of avian malaria (Plasmodium relictum) in experimentally-infected passerines. J Parasitol 88:153–158. https://doi.org/10.1645/0022
Kumar S, Stecher G, Li M et al (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lauron EJ, Loiseau C, Bowie RCK et al (2015) Coevolutionary patterns and diversification of avian malaria parasites in African sunbirds (Family Nectariniidae). Parasitol 142:635–647. https://doi.org/10.1017/S0031182014001681
Lotta IA, Valkiūnas G, Pacheco MA et al (2019) Disentangling Leucocytozoon parasite diversity in the neotropics: descriptions of two new species and shortcomings of molecular diagnostics for leucocytozoids. Int J Parasitol Parasites Wildl 9:159–173. https://doi.org/10.1016/j.ijppaw.2019.05.002
Lutz HL, Hochachka WM, Engel JI et al (2015) Parasite prevalence corresponds to host life history in a diverse assemblage of afrotropical birds and haemosporidian parasites. PLoS One 10(4):e0121254. https://doi.org/10.1371/journal.pone.0121254
Martínez J, Martínez-De La Puente J, Herrero J et al (2009) A restriction site to differentiate Plasmodium and Haemoproteus infections in birds: on the inefficiency of general primers for detection of mixed infections. Parasitology 136:713–722. https://doi.org/10.1017/S0031182009006118
Martinsen ES, Paperna I, Schall JJ (2006) Morphological versus molecular identification of avian Haemosporidia: an exploration of three species concepts. Parasitology 133:279–288. https://doi.org/10.1017/S0031182006000424
Martinsen ES, Perkins SL, Schall JJ (2008) A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): evolution of life-history traits and host switches. Mol Phylogenet Evol 47:261–273. https://doi.org/10.1016/j.ympev.2007.11.012
Meister SL, Richard OK, Hoby S et al (2021) Fatal avian malaria in captive Atlantic puffins (Fratercula arctica) in Switzerland. Int J Parasitol Parasites Wildl 14:97–106. https://doi.org/10.1016/j.ijppaw.2020.12.007
Mukhin A, Palinauskas V, Platonova E et al (2016) The strategy to survive primary malaria infection: an experimental study on behavioural changes in parasitized birds. PLoS ONE 11. https://doi.org/10.1371/journal.pone.0159216
Musa S, Mackenstedt U, Woog F, Dinkel A (2019) Avian malaria on Madagascar: prevalence, biodiversity and specialization of haemosporidian parasites. Int J Parasitol 49:199–210. https://doi.org/10.1016/j.ijpara.2018.11.001
Nebel C, Harl J, Pajot A et al (2020) High prevalence and genetic diversity of Haemoproteus columbae (Haemosporida: Haemoproteidae) in feral pigeons Columba livia in Cape Town, South Africa. Parasitol Res 119:447–463. https://doi.org/10.1007/s00436-019-06558-6
Neto JM, Mellinger S, Halupka L et al (2020) Seasonal dynamics of haemosporidian (Apicomplexa, Haemosporida) parasites in house sparrows Passer domesticus at four European sites: comparison between lineages and the importance of screening methods. Int J Parasitol 50:523–532. https://doi.org/10.1016/j.ijpara.2020.03.008
Palinauskas V, Žiegyte R, Ilgunas M et al (2015) Description of the first cryptic avian malaria parasite, Plasmodium homocircumflexum n. sp., with experimental data on its virulence and development in avian hosts and mosquitoes. Int J Parasitol 45:51–62. https://doi.org/10.1016/j.ijpara.2014.08.012
Palinauskas V, la Puente JM, Hernández-Soto SR, Marzal A (2020) Experimental parasitology and ecoimmunology: concepts and opportunities in avian haemosporidian studies. In: Santiago-Alarcon D, Marzal A (eds) Avian Malaria and related parasites in the tropics. Springer International Publishing, Cham, Switzerland
Pérez-Tris J, Bensch S (2005) Dispersal increases local transmission of avian malarial parasites. Ecol Lett 8:838–845. https://doi.org/10.1111/j.1461-0248.2005.00788.x
Savage AF, Robert V, Goodman SM et al (2009) Blood parasites in birds from Madagascar. J Wildl Dis 45:907–920. https://doi.org/10.7589/0090-3558-45.4.907
Schumm YR, Bakaloudis D, Barboutis C et al (2021) Prevalence and genetic diversity of avian haemosporidian parasites in wild bird species of the order Columbiformes. Parasitology Research 1405–1420. https://doi.org/10.1007/s00436-021-07053-7/Published
Slowinski SP, Geissler AJ, Gerlach N, Heidinger BJ, Ketterson ED (2022) The probability of being infected with haemosporidian parasites increases with host age but is not affected by experimental testosterone elevation in a wild songbird. J Avian Biol 2022(1). https://doi.org/10.1111/jav.02819
Soares L, Escudero G, Penha VAS, Ricklefs RE (2016) Low prevalence of haemosporidian parasites in shorebirds. Ardea 104:129–141. https://doi.org/10.5253/arde.v104i2.a8
Solis J (1973) Nonsusceptibility of some avian species to turkey Leucocytozoon infection. Poult Sci 52:498–500. https://doi.org/10.3382/ps.0520498
Valkiunas G (2005) Avian malaria parasites and other Haemosporidia. CRC Press, Boca Raton
Valkiunas G, Iezhova TA, Križanauskiene A et al (2008) A comparative analysis of microscopy and PCR-based methods for blood parasites. J Parasitol 94:1395–1401. https://doi.org/10.1645/GE-1570.1
Waldenström J, Bensch S, Hasselquist D, Östman Ö (2004) A new nested polymerase chain reaction method very efficient in detecting Plasmodium and Haemoproteus infections from avian blood. J Parasitol 90:191–194. https://doi.org/10.1645/GE-3221RN
Wink M (2006) Use of DNA markers to study bird migration. J Ornithol 147:234–244. https://doi.org/10.1007/s10336-006-0065-5
Woog F, Ramanitra N, Rasamison AS, Tahiry RL (2018) Longevity in some Malagasy rainforest passerines. Ostrich 89:281–286. https://doi.org/10.2989/00306525.2018.1502693
Acknowledgements
We are indebted to the Malagasy authorities for granting all the relevant research and export permits and to the Groupe d’Étude et de Recherche sur les Primates de Madagascar (GERP), namely Jonah Ratsimbazafy and Rose Marie Randrianarison, for letting us work at Maromizaha. Hajanirina Rakotomanana and Daniel Rakotondravony at the Department of Animal Biology, University of Antananarivo supported us throughout. We thank Jean-Robert Lekamisi, Lova Tahiry Rasolondraibe, Onja Randriamalala, Nicola Lillich, Pia Reufsteck, Jean-Louis Berthoud, and all the other Malagasy and German assistants for their help in the field and students who contributed to the study, namely Lotta Schultz, Saskia Döhnert, Szilvia Csaba, and Regina Magaña Vazquez. We wish to thank Thomas Romig and anonymous reviewers for kindly providing comments on the paper.
Funding
Open Access funding enabled and organized by Projekt DEAL. This project was funded by the Deutsche Forschungsgemeinschaft (DFG, GermanResearch Foundation) – 457213393.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Section Editor: Nawal Hijjawi
Permission for taking blood samples from birds in Madagascar was granted by the Ministère de l`Environnement , de l`Ecologie et des Forêts (MEEF). No birds were harmed during this study.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Musa, S., Mackenstedt, U., Woog, F. et al. Untangling the actual infection status: detection of avian haemosporidian parasites of three Malagasy bird species using microscopy, multiplex PCR, and nested PCR methods. Parasitol Res 121, 2817–2829 (2022). https://doi.org/10.1007/s00436-022-07606-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-022-07606-4