
Abstract
Ticks and tick-borne diseases (TTBDs) are considered major causes of economic loss in the livestock sector which incur an annual control cost estimated at US$ 498.7 million in India. Among these diseases, babesiosis, theileriosis and anaplasmosis are listed among the top ten livestock diseases in India and cause significant mortality and morbidity among cattle. However, molecular characterization of bovine Babesia and Anaplasma species are scant; thus, the aim of this study is to perform molecular characterization of field isolates of Babesia spp. and Anaplasma spp. infecting bovines in Kerala, South India. Blood smears and whole blood samples were collected from a total of 199 apparently healthy adult female cattle in Kerala. Based on microscopy, Babesia spp., Theileria orientalis and Anaplasma spp. organisms were detected in 9 (4.5%), 40 (20%) and 6 (3%) samples, respectively. Genus-specific polymerase chain reactions for amplification of 18S rRNA of Babesia spp. and 16S rRNA of Anaplasma spp. revealed positive results with 18 (9%) and 14 (7%) samples. The phylogenetic analysis of 18S rRNA gene sequences of Babesia spp. confirmed the existence of two different populations of Babesia spp. circulating in the blood of infected cattle viz., Babesia bigemina and a Babesia sp. genetically related to Babesia ovata. Further phylogenetic analysis using rap-1a sequences of isolates of B. bigemina revealed higher levels of genetic heterogeneity. However, the field isolates of B. bigemina displayed only slight heterogeneity when the rap-1c gene was examined. Polymerase chain reaction followed by sequencing and phylogenetic analysis of 16S rRNA gene of Anaplasma spp. revealed the existence of Anaplasma marginale, Anaplasma bovis and Anaplasma platys in bovines in South India. Based on msp4 gene sequences, all the field isolates of A. marginale from Kerala were clustered in a single clade with others isolated from around the world. To our knowledge, this study forms the first report on occurrence of B. ovata-like parasites and A. platys in cattle from India.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among all animal diseases, ticks and tick-borne diseases (TTBDs) are considered major causes of economic loss in the livestock sector which directly or indirectly hamper the development of the dairy industry in India (Ghosh et al. 2006). It is estimated that approximately 80% of the world’s cattle population is at risk of infection with TTBDs (de Castro 1997). During 2014–15, babesiosis, theileriosis and anaplasmosis were listed among the top ten livestock diseases in India (Annual Report, PD-ADMAS 2014–15) with the annual control cost estimated at US$ 498.7 million, of which babesiosis alone attributed to approximately US$ 57.2 million (Minjauw and McLeod 2003).
Babesia spp. are the tick-transmitted intra-erythrocytic apicomplexan haemoparasites that are the second most prevalent blood-borne parasites of mammals in the world after trypanosomes (Telford et al. 1993). In cattle, Babesia spp. cause substantial mortality and morbidity and large economic loss due to infection with the causative agents Babesia bovis, Babesia bigemina, Babesia divergens, Babesia major, Babesia ovata, Babesia occultans and Babesia jakimovi. In India, babesiosis was first reported by Walker and Edward (1927) and it is thought that B. bigemina is the predominant pathogenic species in the country (Kolte et al. 2017; Ravindran and Ghosh 2017). However, there are also a few reports of B. bovis-associated babesiosis in Indian cattle and buffaloes (Indani 1938; Setty and Rao 1972; Gautam and Chhabra 1983; Muraleedharan et al. 1984, 1991; Shastri et al. 1991; Kolte et al. 2017).
The bovine anaplasmosis is a tick-borne haemo-rickettsial disease of ruminants caused by Anaplasma spp. (order Rickettsiales and family Anaplasmataceae), an obligate intracellular, Gram-negative bacteria parasitizing mammalian blood cells (Dumler et al. 2001). Infection due to Anaplasma marginale can result in the development of mild to severe anaemia and icterus without haemoglobinuria and haemoglobinaemia. Clinical symptoms may include fever, weight loss, abortion, lethargy, icterus and often death (Kocan et al. 2003). Anaplasmosis in cattle was first reported by Patnaik (1963) in Odisha, India, and subsequently, the pathogenic species A. marginale and Anaplasma bovis have been identified affecting cattle and buffaloes in India (Nair et al. 2013; Sharma et al. 2015; George et al. 2017).
Traditionally, the identification, description and classification of Babesia and Anaplasma species are based on the morphological characteristics using microscopic examination of Giemsa-stained thin or thick blood smears with or without serology. However, recent studies have demonstrated that Babesia and related piroplasmids are not monophyletic and can co-infect hosts; therefore, they require the use of molecular tools for the identification and characterization of these cryptic and polyphyletic species (Lack et al. 2012). To date, the small subunit ribosomal RNA (18S rRNA) gene is widely used for detection and phylogenetic analysis of protozoan organisms due to its high level of conservation (Van de Peer et al. 2000). Molecular phylogeny of Babesia spp. based on the sequence information of the 18S rRNA gene has been useful for the identification of major clade groups (Lack et al. 2012). Additionally, the conserved rhoptry-associated protein-1 (RAP-1) gene, which plays a key role in parasite invasion (Mosqueda et al. 2002), is considered as an informative marker for a phylogenetic group which includes Babesia motasi, B. bigemina, B. major, B. ovata and Babesia crassa (Niu et al. 2014). The RAP-1 locus is encoded by a family of 11 genes and is characterized by five tandemly arranged copies of rap-1a and rap-1b and a single polymorphic gene rap-1c (Suarez et al. 2003). Previous studies have determined that the multiple-copy rap-1a gene is highly conserved and is an informative marker for a broader phylogenetic analysis within the Babesia genus. Conversely, the highly conserved rap-1b and the single-copy polymorphic rap-1c gene are present only in B. bigemina and B. motasi (Niu et al. 2015). Hence, rap-1c is considered an eligible candidate for the molecular characterization of B. bigemina from the field isolates of different geographical areas (Hilpertshauser et al. 2007; Thompson et al. 2014).
Similarly, detection and characterization of Anaplasma spp. are performed using molecular tools. Most of these techniques target the 16S rRNA (Parola et al. 2003; Strik et al. 2007), 23S rRNA (Dahmani et al. 2015), major surface protein (msp) (de la Fuente et al. 2001, 2002; Shimada et al. 2004), heat-shock protein groEL (Park et al. 2005) and citrate synthase gltA (Inokuma et al. 2002b) genes for the detection of Anaplasma spp. Further differentiation of various Anaplasma spp. and strains typically relays on the genes msp1α, msp1β, msp4 and groEL with the msp4 gene and protein sequences proving the most useful for the genetic characterization due to high levels of inter-species variation (Quiroz-Castañeda et al. 2016). These specific major surface proteins (msp1α, msp1β, msp2 and msp4) are of further interest as they are expressed on infected erythrocytes, and therefore, variation in these msp genes could provide valuable information that could be used to identify potential targets for vaccine development (de la Fuente et al. 2005a, b; Contreras et al. 2017).
Even though babesiosis and anaplasmosis have a great impact on the livestock sector in India, previous studies on the molecular characterization of bovine Babesia and Anaplasma species are scant. Hence, the present communication focuses on the molecular characterization and phylogenetic analysis of the field isolates of Babesia spp. and Anaplasma spp. of Kerala, South India, based on the sequence polymorphism of 18S rRNA, rhoptry-associated proteins (rap-1a, rap-1c) of Babesia and 16S rRNA, msp4 genes of Anaplasma.
Materials and methods
Study area, animals and blood samples
Thin peripheral blood smears (from tip of the ear) and heparinized blood samples (~ 2 mL, from the jugular vein using 18-gauge needle) were randomly collected from a total of 199 apparently healthy adult female cattle from three different zones of Kerala, South India, viz., Northern Kerala (44 from Wayanad, 10 from Kozhikode), Central Kerala (30 from Thrissur) and Southern Kerala (28 from Thiruvananthapuram, 87 from Idukki). All the blood samples were collected as part of a normal farming/veterinary practice for regular disease screening. A map showing different locations within Kerala where blood samples were collected is depicted in Fig. 1.

Map of Kerala showing different places of collection of blood samples from three zones of Kerala viz., Northern Kerala (Kozhikode and Wayanad districts), Central Kerala (Thrissur district) and Southern Kerala (Thiruvananthapuram and Idukki districts) (National Agricultural Research Project Zones, Kerala Agro-Climatic Zones)
Staining technique
Thin peripheral blood smears were fixed in methanol and stained using 1:10 dilution (stain:water) of Giemsa’s stain (Merck Life Science, Mumbai) for 45 min. Stained blood smears were examined microscopically for the presence of parasites under the oil immersion (100× magnification) objective of the light microscope (Leica, Germany). The presence of a single piroplasm was considered as a positive case, and a minimum of 5000 red blood cells (RBCs) was screened before proclaiming the sample negative for any blood parasites.
Genomic DNA extraction and quantification
Genomic DNA was isolated from the heparinized blood samples (100 μL) using DNeasy® blood and tissue kit (Qiagen, Germany), according to the manufacturer’s protocol. Extracted DNA was eluted in 100 μL of DNA elution buffer and stored at − 20 °C for further analysis. DNA concentration was determined using a NanoDrop® 2000C spectrophotometer (Thermo Scientific, USA).
Positive and negative control
DNA isolated from the whole blood of a B. bigemina-infected bovine animal (from Teaching Veterinary Clinical Complex, College of Veterinary and Animal Sciences, Pookode, Kerala) was used as the positive control for B. bigemina-specific and Babesia genus-specific PCRs. Similarly, the DNA isolated from the whole blood of a A. marginale-infected bovine animal (from Teaching Veterinary Clinical Complex, College of Veterinary and Animal Sciences, Pookode, Kerala) was used as a positive control for A. marginale-specific and Anaplasma genus-specific PCR.
The leucocyte DNA from a 3-day old calf served as negative control for both Babesia and Anaplasma species.
Polymerase chain reaction for Babesia and Anaplasma species
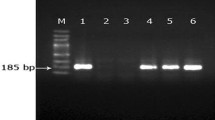
All polymerase chain reactions (PCRs) for Babesia and Anaplasma spp. were carried out in a final reaction volume of 25 μL containing 0.25 mM dNTPs (Genei, Bangalore), 1 U DyNAzyme II DNA polymerase (Thermo Scientific, USA), 1× PCR buffer (containing MgCl2 at a final concentration of 1.5 mM), 20–40 ng of template DNA and 20 pmol each of forward and reverse primers. The details of primers used for the amplification of different genes of Babesia and Anaplasma species are shown in Table 1. All the reactions were conducted as previously described (Table 1), without modification, in an automated thermal cycler with heated lid (Eppendorf, Germany). The PCR products were electrophoresed on a 2% agarose gel containing ethidium bromide (0.5 μg/mL) and visualized under UV light using a gel documentation system (Uvitech, Cambridge). All the blood samples of host genomic DNA positive for 18S rRNA (Babesia spp.) and 16S rRNA (Anaplasma spp.) were represented as isolates (Table 2).
Initially, amplification of the 18S rRNA gene (Hilpertshauser et al. 2007) specific for the genus Babesia was carried out. All the samples which revealed PCR amplification for Babesia genus-specific 18S rRNA gene were then used for PCR amplification of the SpeI-AvaI restriction fragment specific for B. bigemina (Figueroa et al. 1992), the apocytochrome b gene specific for B. bovis (Fahrimal et al. 1992), the apical membrane antigen-1 (ama-1) gene and the β-tubulin gene specific for B. ovata (Sivakumar et al. 2014), in order to confirm respective parasites. The confirmed B. bigemina isolates were further used for the molecular characterization by nested PCR amplification for rap-1a and rap-1c (Molad et al. 2015).
Additionally, for the identification of Anaplasma spp., PCR amplification of the 16S rRNA gene (Aktas et al. 2011) specific for the genus Anaplasma was carried out on all samples. All the positive samples were further characterized by PCR amplification of the A. marginale-specific msp4 gene (de la Fuente et al. 2001, 2002).
Sequencing and sequence analysis
Positive polymerase chain reaction products (18S rRNA, 16S rRNA, rap-1a, rap-1c, msp4) were purified using NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel, Germany) as per the manufacturer’s protocol. Each sample was sent to the AgriGenome Labs Private Ltd., Cochin, Kerala, for automated nucleotide sequencing by Sanger dideoxy method with both the forward and reverse primers. The resulting sequences were examined for overlapping peaks suggestive of co-infection using Bioedit software (Hall 1999) before consensus sequences of each isolate were compared to other published sequences available in GenBank using NCBI-BLAST (http://www.ncbi.nlm.nih.gov/BLAST). Unique sequences were deposited in GenBank database (Table 2).
Phylogenetic analysis
For the phylogenetic analysis of the Babesia and Anaplasma isolates, the nucleotide sequences were aligned using ClustalW (Thompson et al. 1994) with the previously published sequences in the GenBank. Aligned sequences were trimmed to the same length (with gaps) from which phylogenetic trees were constructed based on the maximum likelihood method, using the programme MEGA 7.0 (Kumar et al. 2016) with the suitable models [18S rRNA: Kimura 2-parameter (K2); rap-1a: Kimura 2-parameter (K2); rap-1c: Jukes-Cantor (JC); 16S rRNA: Kimura 2-parameter (K2) and msp4: Kimura 2-parameter (K2)]. The reliability of the topologies was tested by bootstrapping with 1000 replications. Genetic divergence analysis among the field isolates of Babesia and Anaplasma species was performed using MEGA7 (Kumar et al. 2016).
Results
Microscopical examination
The piroplasms of Babesia spp. were observed in nine (4.5%) of 199 blood smears with morphological variations like paired pear-shaped or larger single pear-shaped piroplasms or single oval/irregular amoeboid forms within the erythrocytes (Fig. 2). The piroplasms of Theileria orientalis and inclusions of A. marginale were observed in 40 (20%) and 6 (3%) out of 199 blood smears (images not shown).

Blood smears with various morphological appearances of the Babesia spp. piroplasms (1000×). a Paired pear-shaped piroplasms; b single pear-shaped piroplasms; c–f single oval/irregularly shaped amoeboid-form piroplasms (indicated by red arrows)
Molecular characterization of Babesia spp.
The Babesia genus-specific PCR amplifying the 18S rRNA gene revealed specific amplification in 18 (9%) of 199 samples tested. The specific PCRs targeting SpeI-AvaI restriction fragment, rap-1a and rap-1c genes of B. bigemina, were amplified in six (33%) of 18 samples. The PCRs targeting apocytochrome b gene of B. bovis, apical membrane antigen-1 (ama-1) and β-tubulin genes of B. ovata failed to amplify specific products in the present study.
The NCBI-BLAST and phylogenetic analysis of 18S rRNA gene sequences of the field isolates of Babesia spp. revealed that the South Indian field isolates occupied three different clades out of total six clades (Fig. 3). Twelve of our isolates showed 91–99% identity to B. bigemina (GenBank KU206297, KU206296) with five of these isolates clustered in clade 1, which also comprised B. bigemina isolates from other countries (India, Uganda, Argentina, Kenya, Mozambique, China, Brazil, Australia and Switzerland), and the further seven isolates clustered together forming a separate unique clade 2 (Fig. 5). The remaining isolates showed 98–99% identity to B. ovata (GenBank KU947081) and a Babesia sp. Hue-1 strain (GenBank LC125456) and clustered in clade 3 (Fig. 3). It should be noted that the amplicon sequence was too short for one isolate, Thrissur-1 (GenBank MG062769); therefore, it was not included in the phylogenetic tree. The B. major, B. occultans and B. bovis 18S rRNA sequences formed the clades 4, 5 and 6, respectively, in which there were no isolates from this study represented. The intraspecies divergence among B. bigemina isolates in clades 1 and 2 ranged between 1.3 and 3.4%. Similarly, the intraspecies divergence among B. ovata isolates within clade 3 was 0–7.6%.

Phylogenetic tree constructed using the 18S rRNA gene sequences of Babesia spp. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter (K2) model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3096). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 41 nucleotide sequences comprising of field isolates from the current study (indicated by red circle) and previously published sequences in the GenBank. Evolutionary analyses were conducted in MEGA7
The NCBI-BLAST analysis of rap-1a gene sequences of B. bigemina field isolates in the present study revealed 98 to 100% homology to the published B. bigemina isolates from Israel (GenBank KP822955, KP671751). The phylogenetic tree constructed based on rap-1a gene sequences of B. bigemina formed two distinct clades (Fig. 4). The isolates of B. bigemina from Mexico, Africa (Nigeria and Kenya), Egypt, Thailand, Philippines and Turkey were clustered in clade 1. All the six B. bigemina rap-1a sequences of field isolates from South India in the present study were clustered in clade 2 which also comprised isolates from USA and Israel (field and vaccine isolates). Of note, the isolates within clade 2 showed two subclades with Babesia bigemina isolates viz., Wayanad-1 and -8, forming a distinct subclade from the other isolates.

Phylogenetic tree constructed using the rap-1a gene sequences of Babesia bigemina. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter (K2) model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 25 nucleotide sequences comprising of field isolates from the current study (indicated by red circle) and previously published sequences in the GenBank. Evolutionary analyses were conducted in MEGA7
The NCBI-BLAST and phylogenetic tree based on rap-1c sequences indicated that the field isolates of B. bigemina from South India in the present study were included in two separate clades viz., clade 1 and clade 2 (Fig. 5). Among the five rap-1c isolates included in the phylogenetic analysis, two isolates (Wayanad-1 and TVM-1) had 98% homology with B. bigemina rap-1c sequences from China (GenBank KT312802) and clustered in clade 1, while three isolates (Wayanad-3, Wayanad-12 and Thrissu-2) showed 98–99% identity with rap-1c sequence of B. bigemina from Brazil (GenBank AY146985) and were distributed in clade 2 comprising isolates from Brazil (GenBank AY146985), Mexico (GenBank AY146986, AF026272) and Puerto Rico (GenBank AY146987) (Fig. 5). The Wayanad-8 isolate (GenBank MG191291) was not included for making tree as sequence length was too short despite showing BLAST homology with Wayanad-1 (GenBank MG191289) and Chinese isolates (GenBank KT312800) clustered in clade 1. The interclade divergence between B. bigemina isolates belonging to clades 1 and 2 was 0–3%.

Phylogenetic tree constructed using the rap-1c gene sequences of Babesia bigemina. The evolutionary history was inferred by using the Maximum Likelihood method based on the Jukes-Cantor (JC) model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The analysis involved 19 nucleotide sequences comprising of field isolates from the current study (indicated by red circle) and previously published sequences in the GenBank. Evolutionary analyses were conducted in MEGA7
Molecular characterization of Anaplasma spp.
Of the 199 samples tested, 14 (7%) were positive for the presence of the Anaplasma spp. based on successful amplification of the 16S rRNA genus-specific primers. The NCBI-BLAST and phylogenetic analysis of 16S rRNA gene sequences revealed that the sequences clustered into one of three distinct clades. Ten isolates were in clade 1 with 99–100% identity to A. marginale (GenBank KT264188, KU585985), one isolate was in clade 3 with 99% identity to A. bovis (GenBank KP314251) and three isolates were in clade 5 with 98–99% identity to A. platys (GenBank KU586124, KU586006) (Fig. 6). Intraspecific divergence of 0–0.7% and 1–1.2% was observed among the field isolates of A. marginale (n = 10) and A. platys (n = 3), respectively.

Phylogenetic tree constructed using the 16S rRNA gene sequences of Anaplasma spp. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter model (K2). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The analysis involved 36 nucleotide sequences comprising of field isolates from the current study (indicated by red circle) and previously published sequences in the GenBank. Evolutionary analyses were conducted in MEGA7
The phylogenetic analysis using NCBI-BLAST of A. marginale msp4 gene sequences confirmed the presence of A. marginale in the 10 isolates. However, only eight sequences could be analyzed further as two of the sequence products for TVM-3 and Wayanad-15 were too short. The remaining eight field isolates (Trissur-4, -5, -6, -7, -8, Wayanad-18, -20 and TVM-2) of Kerala clustered together along with isolates from around the world, including isolates from North America, South America and Asia, and revealed only minor levels of genetic diversity with intra-clade divergence of 0–0.9% within the msp4 gene (Fig. 7).

Phylogenetic tree constructed using the msp4 gene sequences of Anaplasma marginale. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter (K2) model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.1702)). The analysis involved 48 nucleotide sequences comprising of field isolates from the current study (indicated by red circle) and previously published sequences in the GenBank. There were a total of 714 positions in the final dataset. Evolutionary analyses were conducted in MEGA7
Discussion
Babesia spp.
Species differentiation is challenging among the large Babesia parasites of bovines because of the morphological similarity of B. bigemina and B. ovata. Babesia bigemina organisms appear as larger pear-shaped piroplasms in pairs joined at their narrow ends, placed centrally in the erythrocytes at acute angle, occupying at least 3/4 of the area within the cell or single larger pear-shaped, oval and ring forms (Muraleedharan 2015). Theileria orientalis are the most prevalent haemoprotozoan parasites in Kerala, which could be easily differentiated from B. bigemina; they are smaller with thin and thick rod shaped or annular with light staining trailing cytoplasm. In the present study, microscopy revealed that 40 samples contained T. orientalis, while Babesia piroplasms were detected in nine out of 199 samples.
Previous studies from Kerala, South India, have reported a very high seroprevalence of 67.6% based on indirect fluorescent antibody test (IFAT) (Ravindran et al. 2002) and 70.9% with slide enzyme-linked immunosorbent assay (SELISA) (Ravindran et al. 2007) against bovine babesiosis. However, more recently, Nair et al. (2011) reported a very low incidence (0.6%) of B. bigemina based on molecular tools in the same region. This difference in prevalence could be attributed to the cross-reactivity of antibodies against piroplasms of Babesia spp. and Theileria spp. present in the same geographical area in serological tests. In addition, genetic variation among the B. bigemina field isolates of Southern and Northern India has been reported (Ravindran et al. 2010), although it is unclear whether the observed genetic heterogeneity impacts on the virulence of the B. bigemina isolates.
Based on 18S rRNA sequences, all the B. bigemina field isolates used in the present study were grouped into two different clades, in which, clade 1 and clade 2 comprised of five and seven field isolates, respectively. Babesia bigemina-specific SpeI-AvaI restriction fragment, rap 1a and rap 1c genes were amplified from only three isolates of clade 1 (Wayanad-1, Wayanad-8 and Thrissur-2) and three isolates of clade 2 (Wayanad-3, Wayanad-12 and TVM-1). This may be due to the low template concentration or lack of sensitivity of the PCR methods. Despite this technical difficulty, B. bigemina types do appear to be clustered in two clades (18Sr RNA), although analysis of full-length gene sequences will give a more complete picture of the diversity of B. bigemina in India. However, this genetic heterogeneity observed among the B. bigemina isolates of the present study could be explained based on the heterogeneity among the rap-1a and 1c sequences of these isolates except Thrissur 2 (for rap-1a) and TVM-1 (for rap-1c). The reason for their altered behaviour could not be explained based on the results of the present study.
The phylogenetic classification of B. bigemina rap-1c gene sequences in the present study is in agreement with the previously described phylogenetic tree of B. bigemina rap-1c gene (Niu et al. 2015). Thus, a lower rate of polymorphism/heterogeneity of B. bigemina rap-1c gene sequences exists among the South Indian isolates and with the other isolates from different geographical locations of the world. Niu et al. (2015) reported that, due to the low genetic variation in B. bigemina rap-1c gene sequence, RAP-1C protein is often considered as a potential antigen for developing immuno-diagnostic techniques (like ELISA) for specific detection of B. bigemina. However, when RAP-1c protein is used as an antigen for diagnosis or as a vaccine candidate in South India, care should be taken to include both types of rap-1c variants (belonging to clades 1 and 2).
The existence of genetic diversity among B. bigemina isolates may be explained due to the varying geographical and climatic conditions affecting hosts carrying these parasites and the parasite’s interaction with different hosts (domestic and wild bovines). Among the places of sample collection, Wayanad and Idukki districts are hilly areas with lower environmental temperature compared to Kozhikode, Thrissur and Thiruvananthapuram, which are in plains with higher atmospheric temperature. All the districts surveyed have very good forest cover especially Wayanad and Idukki, where sizable deer population in the forest interacts with domestic livestock population frequently. Detailed studies are required for confirming these associations.
Results of the present study support those of Nair (2008) who speculated the occurrence of unidentified piroplasms in the cattle of Northern Kerala. In the present study, the evidence for the existence of Babesia species other than B. bigemina was provided by phylogenetic analysis of 18S rRNA gene sequences, which showed that six isolates were 98–99% identical to and cluster with B. ovata. Until now, B. ovata was not reported from the Indian subcontinent. Babesia ovata was reported only from the eastern Asian countries viz., China (Bai et al. 1990; Niu et al. 2015; Tian et al. 2015), Japan (Minami and Ishihara 1980; Yoshinari et al. 2013), Thailand and Mongolia (Yoshinari et al. 2013), including Vietnam (Weerasooriya et al. 2016) and Korea (Suh 1987). Indeed, the phylogenetic analysis of 18S rRNA gene sequences revealed that the five field isolates under this study shared the same clade 3 along with B. ovata isolates from Bangladesh and Babesia sp. Hue-1 strain from Vietnam. The PCR assay using B. ovata-specific primers targeting ama-1 and β-tubulin genes did not amplify the specific products. Hence, further study is warranted for the confirmation of the existence of B. ovata in South India.
In this study, Babesia bovis parasites could not be detected based on 18S rRNA gene sequence analysis or by B. bovis-specific PCR. However, B. bovis has been previously reported in India from Karnataka (Indani 1938; Setty and Rao 1972; Muraleedharan et al. 1984, 1991; Shastri et al. 1991) based on microscopical examination and from Maharashtra (Kolte et al. 2017) based on PCR technique.
Anaplasma spp.
Anaplasmosis is one of the most prevalent and economically important rickettsial diseases throughout the country. The higher prevalence of anaplasmosis in clinically normal animals indicates that there are subclinical or carrier animals in Kerala, South India (Nair et al. 2013). These carrier animals can act as the source of infection for naive hosts (Radostits et al. 2000). Anaplasmosis was previously reported from different regions of the country (Garg et al. 2004; Muraleedharan et al. 2005; Harish et al. 2006; Soundararajan and Rajavelu 2006), including Kerala (Devada et al. 1996; Gopinath 2004; Nair 2008; Nair et al. 2013). Though anaplasmosis is highly prevalent in India, reports on the molecular characterization of Anaplasma spp. in cattle are scarce. Hence, molecular characterization of Anaplasma spp. was carried out in the present study.
In the present study, microscopy revealed Anaplasma organisms only in six out of 199 samples, while PCR targeting 16S rRNA gene revealed 14 samples positive. The NCBI BLAST analysis and phylogenetic analysis of 16S rRNA gene sequences of field isolates of Anaplasma spp. confirmed the existence of A. marginale, A. bovis and A. platys infecting bovines in Kerala, South India. There have been no previous reports on A. platys infecting bovines in India. However, A. marginale and A. bovis have been previously reported in India based on both microscopy and molecular techniques (Devada et al. 1996; Sreekumar et al. 2000; Gopinath 2004; Nair 2008; Nair et al. 2013).
Genetic heterogeneity among the population of A. marginale was previously studied from two South Indian states viz., Seemandhra and Telangana (George et al. 2017). Based on phylogeny using msp4, it was reported that most of the strains (21/24) from these South Indian states showed close proximity with strains from Mexico and Brazil. In the present study, it was revealed that minimal heterogeneity exists within 16S rRNA and msp4 genes among the field isolates from Kerala. The results of phylogeny using msp4 in the present study are similar to those of George et al. (2017). Hence, it could be inferred that isolates of A. marginale of South India are genetically closer.
Anaplasma bovis (formerly referred as Ehrlichia bovis, Dumler et al. 2001) is a monocytotrophic Ehrlichia sp. which was first described in 1931 (Donatien and Lestoquard 1936) and later in cattle and buffalo mainly from Africa, Asia and Middle East countries. Rhipicephalus appendiculatus, Amblyomma variegatum, Hyalomma anatolicum and Hyalomma truncatum may act as vectors for A. bovis (Radostits et al. 2000). Anaplasma bovis was previously reported from various different states in the country mainly by microscopical examination (Devada et al. 1996; Sreekumar et al. 1996; Vijayalakshmi and Sreekrishnan 2005; Prasath et al. 2016) and by molecular techniques (Nair et al. 2013). However, the full extent of genotypic variability and phylogenetic relationships of the Indian A. bovis strains is still to be assessed.
Anaplasma platys is assumed to be transmitted by Rhipicephalus sanguineus (Inokuma et al. 2000; Dyachenko et al. 2012), which is the common dog tick in India (Abd-Rani et al. 2011). Previously, A. platys was described in dogs from USA (French and Harvey 1983), Brazil (Ferreira et al. 2007), China (Hua et al. 2000), Thailand (Suksawat et al. 2001), Taiwan (Chang et al. 1996), Japan (Inokuma et al. 2002a), Australia (Brown et al. 2006), Africa (Sanogo et al. 2003), Italy (de la Fuente et al. 2006), Spain (Aguirre et al. 2006), France (Beaufils et al. 2002), Turkey (Ulutas et al. 2007) and many other countries. Anaplasma platys-like parasites were also reported from wild ruminants viz., Brazilian marsh deer (Sacchi et al. 2012), white-tailed deer (Munderloh et al. 2003) and brown brocket deer and marsh deer (Silveira et al. 2012). However, this is the first study to identify A. platys in ruminants in India and there is no conclusive evidence for the presence of A. platys in dogs. Thus, a detailed systematic study is essential for confirming A. platys prevalence and pathogenicity in both carnivores and herbivores.
Conclusion
The present study confirmed the existence of two different populations of Babesia spp. circulating in the blood of bovines in Kerala viz., B. bigemina and Babesia sp., closely related to B. ovata/Babesia sp. Hue-1 strain (unidentified piroplasms). The B. bovis parasites could not be detected in cattle from Kerala, South India. The phylogenetic analysis based on rap-1a revealed the existence of higher levels of genetic heterogeneity among B. bigemina isolates while using rap-1c only, slight heterogeneity was observed. The present study also confirmed the existence of A. marginale, A. bovis and A. platys infecting bovines in Kerala. Anaplasma marginale isolates of Kerala were genetically conserved based on phylogenetic analysis of msp4. To our knowledge, this study forms the first report of B. ovata and A. platys in Kerala, South India. Further confirmation on the existence and pathogenicity of unconfirmed B. bigemina, B. ovata/B. ovata-like parasites, A. bovis and A. platys is urgently warranted.
References
Abd-Rani PAM, Irwin PJ, Coleman GT, Gatne M, Traub RJ (2011) A survey of canine tick-borne diseases in India. Parasit Vectors 4:141–148
Aguirre E, Tesouro MA, Ruiz L, Amusategui I, Sainz A (2006) Genetic characterization of Anaplasma (Ehrlichia) platys in dogs in Spain. J Vet Med B Infect Dis Vet Public Health 53:197–200
Aktas M, Altay K, Dumanli N (2011) Molecular detection and identification of Anaplasma and Ehrlichia species in cattle from Turkey. Ticks Tick-borne Dis 2:62–65
Annual Report (2014–15) Project directorate of animal disease monitoring and surveillance, Indian Council of Agricultural Research, New Delhi
Bai Q, Liu GY, Zhang L, Zhang JY (1990) Discovery and isolation of Babesia ovata in China. Chin J Vet Sci Technol 20:2–4
Beaufils JP, Inokuma H, Martin-Granel J, Jumelle P, Barbault-Jumelle M, Brouqui P (2002) Anaplasma platys (Ehrlichia platys) infection in a dog in France: description of the case, and characterization of the agent. Rev Med Vet 153:85–90
Brown GK, Canfield PJ, Dunstan RH, Roberts TK, Martin AR, Brown CS, Irving R (2006) Detection of Anaplasma platys and Babesia canis vogeli and their impact on platelet numbers in free-roaming dogs associated with remote aboriginal communities in Australia. Aust Vet J 84:321–325
Chang AC, Chang WL, Lin CT, Pan MJ, Lee SC (1996) Canine infectious cyclic thrombocytopenia found in Taiwan. J Vet Med Sci 58:473–476
Contreras M, Alberdi P, Mateos-Hernández L, Fernández de Mera IG, García-Pérez AL, Vancová M, Villar M, Ayllón N, Cabezas-Cruz A, Valdés JJ, Stuen S, Christian Gortazar C, de la Fuente J (2017) Anaplasma phagocytophilum MSP4 and HSP70 proteins are involved in interactions with host cells during pathogen infection. Front Cell Infect Microbiol 7:307
Dahmani M, Davoust B, Benterki MS, Fenollar F, Raoult D, Mediannikov O (2015) Development of a new PCR-based assay to detect Anaplasmataceae and the first report of Anaplasma phagocytophilum and Anaplasma platys in cattle from Algeria. Comp. Immunol Microbiol Infect Dis 39:39–45
de Castro JJ (1997) Sustainable tick and tick-borne disease control in livestock improvement in developing countries. Vet Parasitol 71:77–97
de la Fuente J, Van Den Bussche RA, Kocan KM (2001) Molecular phylogeny and biogeography of North American isolates of Anaplasma marginale (Rickettsiaceae: Ehrlichieae). Vet Parasitol 97:65–76
de la Fuente J, Bussche RAVD, Garcia-Garcia JC, Rodríguez SD, García MA, Guglielmone AA, Mangold AJ, Friche Passos LM, Blouin EF, Kocan K (2002) Phylogeography of New World isolates of Anaplasma marginale (Rickettsiaceae: Anaplasmataceae) based on major surface protein sequences. Vet Microbiol 88:275–285
de la Fuente J, Lew A, Lutz H, Meli ML, Hofmann-Lehmann R, Shkap V, Molad T, Mangold AJ, Almazán C, Naranjo V, Gortázar C, Torina A, Caracappa S, Garcıa-Perez AL, Barral M, Oporto B, Ceci L, Carelli G, Blouin EF, Kocan KM (2005a) Genetic diversity of Anaplasma species major surface proteins and implications for anaplasmosis serodiagnosis and vaccine development. Anim Health Res Rev 6:75–89
de la Fuente J, Torina A, Caracappa S, Tumeno G, Furla R, Almazan C, Kocan KM (2005b) Serologic and molecular characterization of Anaplasma species infection in farm animals and ticks from Sicily. Vet Parasitol 133:357–362
de la Fuente J, Torina A, Naranjo V, Nicosia S, Alongi A, La MF, Kocan KM (2006) Molecular characterization of Anaplasma platys strains from dogs in Sicily, Italy. BMC Vet Res 2:24
Devada K, Saseendranath MR, Abraham MJ, Tresamol PV (1996) Occurrence of Ehrlichia bovis in a cow. J Vet Anim Sci 27:66–67
Donatien A, Lestoquard F (1936) Rickettsia bovis, novelle espece pathogene pour le boeuf. Bull Soc Pathol Exot 29:1057–1061
Dumler JS, Barbet AF, CPJ B, Dasch A, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR (2001) Reorganization of the genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and “HE agent” as subjective synonyms of Ehrlichia phagocytophilia. Int J Syst Evol Microbiol 51:2145–2165
Dyachenko V, Pantchev N, Balzer J, Straubinger RK (2012) First case of Anaplasma platys infection in a dog from Croatia. Parasit Vectors 5:49
Fahrimal Y, Goff WL, Jasmer DP (1992) Detection of Babesia bovis carrier cattle by using polymerase chain reaction amplification of parasite DNA. J Clin Microbiol 30:1374–1379
Ferreira RF, Figueiredo Cerqueira AM, Pereira AM, Guimaraes CM, Sa AG, Abreu FS, Massard CL, Pereira Almosny NR (2007) Anaplasma platys diagnosis in dogs: comparison between morphological and molecular tests. Int J Appl Res Vet Med 5:113–119
Figueroa JV, Chieves LP, Johnson GS, Buening GM (1992) Detection of Babesia bigemina infected carriers by polymerase chain reaction amplification. J Clin Microbiol 30:2576–2582
French TW, Harvey JW (1983) Serologic diagnosis of infectious cyclic thrombocytopenia in dogs using an indirect fluorescent antibody test. Am J Vet Res 44:2407–2411
Garg R, Banerjee PS, Yadav CL, Garg A (2004) Sub-clinical babesiosis and anaplasmosis in cross bred cattle in an organized farm. J Vet Parasitol 18:151–153
Gautam OP, Chhabra MB (1983) Babesiosis: recent advances with special reference to India. Trop Vet Anim Sci 1:201–207
George N, Bhandari V, Sharma P (2017) Phylogenetic relationship and genotypic variability in Anaplasma marginale strains causing anaplasmosis in India. Inf Genet Evol 48:71–75
Ghosh S, Azhahianambi P, de la Fuente P (2006) Control of ticks of ruminants, with special emphasis on livestock farming system in India: present and future possibilities for integrated control—a review. Exp Appl Acarol 40:49–66
Gopinath VR (2004) Clinical investigations on parasitic anaemia in cattle. M.V.Sc. thesis. Kerala Agriculture University, Thrissur. 84p
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Harish BR, Chandranaik BM, Renukaprasad C, Jayakumar SR, Krishnappa G (2006) Incidence of haemoprotozoan diseases in Karnataka. Indian J Vet Med 26:30–31
Hilpertshauser H, Deplazes P, Meli ML, Hofmann-Lehmann R, Lutz H, Mathis A (2007) Genotyping of Babesia bigemina from cattle from a non-endemic area (Switzerland). Vet Parasitol 145:59–64
Hua P, Yuhai M, Shide T, Yang S, Bohai W, Xiangrui C (2000) Canine ehrlichiosis caused simultaneously by Ehrlichia canis and Ehrlichia platys. Microbiol Immunol 44:737–739
Indani JA (1938) Babesia bovis as cause of red water in an Indian buffalo. Indian J Vet Sci Anim Husb 9:99–101
Inokuma H, Raoult D, Brouqui P (2000) Detection of Ehrlichia platys DNA in brown dog ticks (Rhipicephalus sanguineus) in Okinawa Island, Japan. J Clin Microbiol 38:4219–4221
Inokuma H, Fujii K, Matsumoto K, Okuda M, Nakagome K, Kosugi R, Hirakawa M, Onishi T (2002a) Demonstration of Anaplasma (Ehrlichia) platys inclusions in peripheral blood platelets of a dog in Japan. Vet Parasitol 110:145–152
Inokuma H, Fujii K, Okuda M, Onishi T, Beaufils JP, Raoult D, Brouqui P (2002b) Determination of the nucleotide sequences of heat shock operon groESL and the citrate synthase gene (gltA) of Anaplasma (Ehrlichia) platys for phylogenetic and diagnostic studies. Clin Diagn Lab Immunol 9:1132–1136
Kocan KM, de la Fuente J, Gulielmone AA, Melendez RD (2003) Antigens and alternatives for control of Anaplasma marginale infection in cattle. Clin Microbiol Rev 16:698–712
Kolte SW, Larcombe SD, Jadhao SG, Magar SP, Warthi G, Kurkure NV, Glass EJ, Shiels BR (2017) PCR diagnosis of tick-borne pathogens in Maharashtra state, India indicates fitness cost associated with carrier infections is greater for crossbreed than native cattle breeds. PLoS ONE 12. https://doi.org/10.1371/journal.pone.0174595
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lack JB, Reichard MV, Van Den Bussche RA (2012) Phylogeny and evolution of the Piroplasmida as inferred from 18S rRNA sequences. Int J Parasitol 42:353–363
Minami T, Ishihara T (1980) Babesia ovata sp.n. isolated from cattle in Japan. Natl Inst Anim Health Q (Tokyo) 20:101–113
Minjauw B, McLeod A (2003) Tick-borne diseases and poverty: the impact of ticks and tick-borne diseases on the livelihoods of small-scale and marginal livestock owners in India and eastern and southern Africa. DFID Animal Health Programme, Centre for Tropical Veterinary Medicine, UK, p.124
Molad T, Erster L, Fleiderovitz L, Roth A, Leibovitz B, Wolkomirsky R, Mazuz ML (2015) Molecular characterization of the Israeli Babesia bigemina vaccine strain and field isolates. Vet Parasitol 7675:1–9
Mosqueda J, McElwain TF, Stiller D, Palmer GH (2002) Babesia bovis merozoite surface antigen 1 and rhoptry-associated protein 1 are expressed in sporozoites, and specific antibodies inhibit sporozoite attachment to erythrocytes. Infect Immun 70:1599–1603
Munderloh UG, Tate CM, Lynch MJ, Howerth EW, Kurtti TJ, Davidson WR (2003) Isolation of an Anaplasma sp. organism from white-tailed deer by tick cell culture. J Clin Microbiol 41:4328–4335
Muraleedharan K (2015) Babesia and babesiosis in livestock of Karnataka state, India—an overview. Vet Res Int 3:81–88
Muraleedharan K, Ziauddin KS, Swamy KG, Muraleedhar T, Seshadri SJ (1984) Some observations on clinical cases of Babesia bovis (Babes, 1888) Starcovici, 1893, in buffaloes (Bubalus bubalis). Indian Vet J 61:76–77
Muraleedharan K, Ziauddin KS, Hussain PM, Puttabyatappa B, Seshadri SJ (1991) Prevalence of haemoprotozoan parasites among buffaloes. Cheiron 20:79–82
Muraleedharan K, Ziauddin KS, Hussain PM, Puttabyatappa B, Mallikarjuna GB, Seshadri SJ (2005) Incidence of Anaplasma sp., Babesia sp. and Trypanosoma sp. in cattle of Karnataka. J Vet Parasitol 19:135–137
Nair AS (2008) Surveillance of haemoprotozoan and haemorickettsial diseases of cattle of Northern Kerala. M.V.Sc thesis, Kerala Agricultural University, Thrissur, Kerala. 126p
Nair AS, Ravindran R, Lakshmanan B, Kumar SS, Tresamol PV, Saseendranath MR, Senthilvel K, Rao JR, Tewari AK, Ghosh S (2011) Haemoprotozoa of cattle in Northern Kerala, India. Trop Biomed 28:68–75
Nair AS, Ravindran R, Lakshmanan B, Sreekumar C, Kumar SS, Remya R, Tresamol PV, Vimalkumar MB, Saseendranath MR (2013) Bovine carriers of Anaplasma marginale and Anaplasma bovis in South India. Trop Biomed 30:105–112
Niu Q, Valentin C, Bonsergent C, Malandrin L (2014) Strong conservation of rhoptry-associated-protein-1 (RAP-1) locus organization and sequence among Babesia isolates infecting sheep from China (Babesia motasi-like phylogenetic group). Infect Genet Evol 28:21–32
Niu Q, Liu Z, Yu P, Yang J, Abdallah MO, Guan G, Liu G, Luo J, Yin H (2015) Genetic characterization and molecular survey of Babesia bovis, Babesia bigemina and Babesia ovata in cattle, dairy cattle and yaks in China. Parasit Vectors 8:518
Park HS, Lee JH, Jeong EJ, Park TK, Kim TY, Chae JS, Park JH, Klein TA, Jang WJ, Park KH, Lee SH (2005) Differentiation of Anaplasmataceae through partial groEL gene analysis. Microbiol Immunol 49:655–662
Parola P, Cornet JP, Sanogo YO, Miller RS, Thien HV, Gonzalez JP, Raoult D, Telford IS, Wongsrichanalai C (2003) Detection of Ehrlichia spp., Anaplasma spp., Rickettsia spp., and other Eubacteria in ticks from the Thai-Myanmar border and Vietnam. J Clin Microbiol 41:1600–1608
Patnaik MM (1963) A note on bovine anaplasmosis. Indian Vet J 40:655–657
Prasath N, Selvaraj J, Jeyathilakan N, Saravanan M, Saravanan M, Ahamad DB, Sasikala M (2016) Occurrence of Anaplasma bovis (Ehrlichia bovis) with varying morphology in a crossbred cow in Tamilnadu, India. Indian J Vet Pathol 40:165–167
Quiroz-Castañeda RE, Amaro-Estrada I, Rodríguez-Camarillo SD (2016) Anaplasma marginale: diversity, virulence, and vaccine landscape through a genomics approach. BioMed Res. Int. 2016:Article ID 9032085, 18 pages. https://doi.org/10.1155/2016/9032085
Radostits OM, Gay CC, Blood DC, Hinchkliff HW (2000) Veterinary medicine—a text book of the diseases of cattle, sheep, pigs, goats and horses, Ninth edn. W.B. Saunders Company Ltd., New York, p 1812
Ravindran R, Ghosh S (2017) Botanicals for treatment of babesiosis and theileriosis. Ann Pharmacol Pharm 2:1105
Ravindran R, Mishra AK, Rao JR (2002) On the high seroprevalence of bovine babesiosis in Wayanad district of Kerala. J Appl Anim Res 30:142–145
Ravindran R, Mishra AK, Rao JR (2007) Slide enzyme-linked immunosorbent assay for the diagnosis of Babesia bigemina infection in bovines. Vet Res Commun 30:142–145
Ravindran R, Sreekumar C, Saravanan BC, Udaykumar M, Tewari AK, Kumar S, Rao JR, Mishra AK (2010) Genetic variation among Indian isolates of Babesia bigemina. J Vet Parasitol 24:159–163
Sacchi ABV, Duarte JMB, André MR, Machado RZ (2012) Prevalence and molecular characterization of Anaplasmataceae agents in free-ranging Brazilian marsh deer (Blastocerus dichotomus). Comp Immunol Microbiol Infect Dis 35(4):325–334
Sanogo YO, Davoust B, Inokuma H, Camicas JL, Parola P, Brouqui P (2003) First evidence of Anaplasma platys in Rhipicephalus sanguineus (Acari: Ixodida) collected from dogs in Africa. J Vet Res 70:205–212
Setty DRL, Rao NSK (1972) The pattern of asexual development of Babesia as seen in peripheral blood of a cow. Mysore J Agric Sci 6:474–480
Sharma A, Singla LD, Tuli A, Kaur P, Bal MS (2015) Detection and assessment of risk factors associated with natural concurrent infection of Trypanosoma evansi and Anaplasma marginale in dairy animals by duplex PCR in eastern Punjab. Trop Anim Health Prod 47:251–257
Shastri U, Degloorkar N, Kulkarni G (1991) Bovine babesiosis due to Babesia bovis at Parbhani (Maharashtra), India. J Vet Parasitol 5:29–34
Shimada MK, Yamamura MH, Kawasaki P, Tamekuni K, Igarashi M, Vidotto O, Vidotto MC (2004) Detection of Anaplasma marginale DNA in larvae of Boophilus microplus ticks by polymerase chain reaction. New York Acad Sci 1026:95–102
Silveira JA, Rabelo EM, Ribeiro MF (2012) Molecular detection of tick-borne pathogens of the family Anaplasmataceae in Brazilian brown brocket deer (Mazama gouazoubira, Fischer, 1814) and marsh deer (Blastocerus dichotomus, Illiger, 1815). Transbound Emerg Dis 59:353–360
Sivakumar T, Tattiyapong M, Okubo K, Suganuma K, Hayashida K, Igarashi I, Zakimi S, Matsumoto K, Inokuma H (2014) PCR detection of Babesia ovata from questing ticks in Japan. Ticks Tick Borne Dis 5:205–310
Soundararajan C, Rajavelu G (2006) Prevalence of haemoprotozoan among cattle and buffaloes. Indian Vet J 83:1258–1260
Sreekumar C, Anandan R, Balasundaram S, Rajavelu G (1996) Morphology and staining characteristics of Ehrlichia bovis. Comp Immunol Microbiol Infect Dis 19:79–83
Sreekumar C, Anandan R, Balasundaram S, John L (2000) Detection of E. bovis—like organism in cultured buffalo monocytes. Trop Anim Health Prod 32:67–72
Strik NI, Alleman AR, Barbet AF, Sorenson HL, Wamsley HL, Gaschen FP, Luckschander N, Wong S, Chu F, Foley JE, Bjoersdorff A, Stuen S, Knowles DP (2007) Characterization of Anaplasma phagocytophilum major surface protein 5 and the extent of its cross-reactivity with A. marginale. Clin Vaccine Immunol 14:262–268
Suarez CE, Palmer GH, Florin-Christensen M, Hines SA, Hötzel I, McElwain TF (2003) Organization transcription, and expression of rhoptry associated protein genes in the Babesia bigemina rap-1 locus. Mol Biochem Parasitol 127:101–112
Suh MD (1987) Pure isolation and identification of Babesia ovata by morphological characteristics and complement fixation test in Korea. Korean J Vet Res 27:307–316
Suksawat J, Pitulle C, Arraga-Alvarado C, Madrigal K, Hancock SI, Breitschwerdt EB (2001) Coinfection with three Ehrlichia species in dogs from Thailand and Venezuela with emphasis on consideration of 16S ribosomal DNA secondary structure. J Clin Microbiol 39:90–93
Telford SR, Gorenflot A, Brasseur P, Spielman A (1993) Babesial infection in human and wildlife. In: Kreier JP (ed) Parasitic protozoa, vol 5. Academic Press, San Diego, pp 1–47
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Thompson C, Baravalle ME, Valentini B, Mangold A, de Echaide TS, Ruybal P, Farber M, Echaide I (2014) Typification of virulent and low virulence Babesia bigemina clones by 18S rRNA and rap-1c. Exp Parasitol 141:98–105
Tian J, Du J, Yang Z, Liu A, Liu X, Liu G, Yin H (2015) A PCR-RFLP assay targeting RPS8 gene for the discrimination between bovine Babesia and Theileria species in China. Parasit Vectors 8:475
Ulutas B, Bayramli G, Karagenc T (2007) First case of Anaplasma (Ehrlichia) platys infection in a dog in Turkey. Turk J Vet Anim Sci 31:279–282
Van de Peer Y, Baldauf SL, Doolittle WF, Meyer A (2000) An updated and comprehensive rRNA phylogeny of (crown) eukaryotes based on rate-calibrated evolutionary distances. J Mol Evol 51:565–576
Vijayalakshmi P, Sreekrishnan R (2005) Occurrence, clinical manifestations and treatment of bovine ehrlichiosis. Indian J Vet Med 25:134–135
Walker GK, Edward JT (1927) Some diseases of cattle in India. Government of India, Calcutta, p 29
Weerasooriya G, Sivakumar T, Lan DTB, Long PT, Takemae H, Igarashi I, Inoue N, Yokoyama N (2016) Epidemiology of bovine hemoprotozoa parasites in cattle and water buffalo in Vietnam. J Vet Med Sci 78:1361–1367
Yoshinari T, Sivakumar T, Asada M, Battsetseg B, Huang X, Lan DT, Inpankaew T, Ybañez AP, Alhassan A, Thekisoe OM, De Macedo AC, Inokuma H, Igarashi I, Yokoyama N (2013) A PCR based survey of Babesia ovata in cattle from various Asian, African and South American countries. J Vet Med Sci 75:211–214
Acknowledgements
We thank all the veterinary doctors and local farming community for their cooperation.
Funding
This work was supported financially by the Indian Council of Agricultural Research (NAIP/C2066, NFBSFARA/BSA-4004/2013-14, NASF/ABA-6015/2016-17), the Kerala State Council for Science, Technology and Environment (022/YIPB/KBC/2013/CSTE, 010-14/ SARD/13/CSTE) and the Department of Animal Husbandry, Kerala [B2.3858/04/Plg(3) dt.2/2/07, B2.8401/08/Plg dt.19/18/2008].
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Larissa Howe
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pradeep, R.K., Nimisha, M., Sruthi, M.K. et al. Molecular characterization of South Indian field isolates of bovine Babesia spp. and Anaplasma spp.. Parasitol Res 118, 617–630 (2019). https://doi.org/10.1007/s00436-018-6172-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-018-6172-4