
Abstract
The purpose of this study was to estimate the prevalence of equine piroplasmosis in Sudan. The presence of antibodies against Babesia caballi and Theileria equi was determined in serum samples obtained from 158 horses raised in different locations in Sudan by enzyme-linked immunosorbent assay (ELISA). The B. caballi 48-kDa and the T. equi EMA-2 purified recombinant proteins were used as antigens in the ELISA test. Results showed that seven (4.4%) were positive for B. caballi and 80 (63.5%) were positive for T. equi. Polymerase chain reaction (PCR) assays have been applied using primers targeting the B. caballi 48-kDa merozoite antigen, the T. equi SSUrRNA and the T. equi EMA-1 genes. PCR performed on 131 blood spots in filter paper revealed that 33 (25.2%) samples were positive for T. equi but no positives were found for B. caballi. It is concluded that equine piroplasmosis is endemic in the country. This is the first study on serological and molecular epidemiological diagnosis on equine piroplasmosis in Sudan.
Similar content being viewed by others


Introduction
Equine piroplasmosis, caused by Theileria equi and Babesia caballi, is globally distributed and poses a serious threat to the horse raising industry and international movement of horses (Friedhoff et al. 1990; Avarzed et al. 1997). The disease is widely distributed in the tropical and subtropical areas (Preston 2001; Uilenberg 2001). T. equi is considered more pathogenic and more consistent cause of hemoglobinuria and death in equines, while B. caballi causes a more persistent syndrome characterized by fever and anemia (Zaugg and Lane 1992). T. equi infection may be suppressed by chemotherapy but it cannot be completely eliminated (de Waal and van Heerden 1994). Equine piroplasmosis was firstly reported in Sudan by Oliver (1907) cited in Abdoon (1984), who studied the epidemiology of the disease in Khartoum State using blood smears and the complement fixation test (CFT). Of the investigated 80 horses, 16% showed typical clinical signs of piroplasmosis, 20% showed Babesia parasites in blood smears, and 70% were positive by CFT. Oliver’s findings also illustrated that T. equi was more common than B. caballi with a prevalence of 53.4% in the examined population.
It is not possible to differentiate between T. equi and B. caballi infections on the basis of clinical signs (de Waal 1992). Definitive diagnosis depends on the identification of B. caballi and T. equi in blood smears stained by Giemsa or by acridine orange (Ali et al. 1996). Although the method is simple, it is insufficient for accurate detection and identification of B. caballi and T. equi during mixed infections and in particular in carrier statuses with low parasitemia (Seifi et al. 2000; Krause 2003). For serological detection of equine piroplasmosis, CFT and the indirect immunofluorescent antibody test (IFAT) have been used as standard tests for the detection of equine piroplasmosis. Many problems have been reported for the CFT including low sensitivity and the yield of false-positive and false-negative results for B. caballi (Friedhoff and Soulé 1996; Ikadai et al. 2001). Thus, in several studies, the CFT has been replaced by the more sensitive IFAT, which has been used in serosurveillance for Theileria and Babesia infections in many countries. Researchers, however, have also reported on the inherent problems of IFAT that include cross reactivity, subjectivity, and impracticability especially for testing large numbers of samples (Brüning 1996; Papadopoulos et al.1996). Various forms of enzyme-linked immunosorbent assay (ELISA) have been standardized and reported to be more sensitive than the CFT in the diagnosis of equine piroplasmosis (Brüning et al. 1997; Bakheit et al. 2007). Several T. equi proteins have demonstrated immunoreactive antigens and used in ELISA, of which the merozoite surface proteins EMA-1 and EMA-2 have been identified as the most common immunodominant molecules of T. equi (Knowles et al. 1992; Kappmeyer et al.1993) and the 48-kDa rhoptry protein of B. caballi (Ikadai et al. 1999; Xuan et al. 2001). Moreover, it has been demonstrated that EMA-1 is geographically conserved among all T. equi isolates (Knowles et al. 1991), thus could provide a good diagnostic tool when used in ELISA. Besides the application of microscopy and serology in the diagnosis of equine piroplasmosis, in vitro culture of blood was used for the detection of carrier host animals with T. equi (Holman et al. 1997). Molecular techniques have been proven useful for the detection of equine piroplasmosis. These methods are based on species-specific polymerase chain reaction (PCR) assays, which mainly target the 18S rRNA gene (Caccio et al. 2000; Birkenheuer et al. 2003; Criado-Fornelio et al. 2003; Rampersad et al. 2003). PCR has been found sensitive enough to detect parasite DNA from 2.5 μl blood sample with parasitemia of 0.000001% (Xuan et al. 1998; Alhassan et al. 2007).
The objective of this study was to apply ELISA and PCR methods to determine the prevalence and distribution of T. equi and B. caballi infections in the equine population in Sudan.
Materials and methods
Description of study area
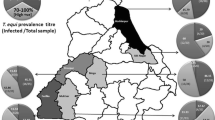
Samples were collected from different eco-climatic zones which included Khartoum, Khartoum North, and Omdurman (15°40′ N 32′28′ E), Atbara (17°42′ N 34′ E), Kosti (13°09′ N 32°40′ E), El Obied (13°11′ N 30°13′ E), and Gadarif (14° N 35°30′ E; Fig. 1). Whole blood was taken by jugular venipuncture using vacutainer tubes, and thereafter, serum samples were separated and stored at −20°C till use. Blood spots on filter papers were collected for PCR amplification. The blood spots were made in circles of 1/2 in. diameter, air dried, labeled, and fixed with 70% ethanol before being stored at 4°C.

Map of Sudan. Localities from where samples were collected are marked with filled circles
ELISA
The presence of antibodies against T. equi and B. caballi was determined in serum samples from 158 horses by ELISA. The B. caballi 48-kDa and the T. equi EMA-2 purified recombinant proteins were obtained from the National Research Centre for Protozoan Diseases, Obihiro, Japan and used as antigens in ELISA. Recombinant antigens were diluted to 7 μg/ml in carbonate bicarbonate buffer, pH 9.6 and 50 μl used to coat the wells of 96 wells micro-titer ELISA plate. The plates were kept at 4°C overnight. The plates were then washed six times with phosphate-buffered saline (PBS) pH 7.4, supplemented with 0.05% Tween-20 (PBS-T). The unoccupied sites in each well were blocked by adding 200 μl in each well of a blocking buffer composed of PBS, pH 7.4, 3% skimmed milk (PBS-SM) and incubated for 1 h at 37°C. The plates were again washed six times with PBS-T. Sera were diluted at 1:100 in PBS-SM and pipetted in a volume of 100 μl/well. Each plate was used to test 40 serum samples, with the first two columns used for controls including blank control (two wells), positive controls (four wells), and negative controls (ten wells). Plates were incubated for 1 h at 37°C then washed with PBS-T. Goat anti-horse IgG antibody, conjugated to horse radish peroxidase was added at a dilution of 1:10,000 in PBS-SM and incubated for 1 h at 37°C. Finally, a substrate solution [composed of 0.1 M citric acid, 0.2 M sodium phosphate, 0.003% H2O2, 0.3 mg of 2, 2′-azino-di-(3-ethylbenzthiazoline sulfonate) per milliliter] was freshly prepared and added in a volume of 100 μl/well and allowed to stand in dark at room temperature for 1 h. The development of a green color was obtained from positive control and positive samples. The plates were read at 450 nm in an ELISA plate reader (Titertek multiskan reader, Labsystems multiskan RC) and the mean (OD) of the negative control was calculated. Any sample showing an OD above the mean + (3 × standard deviation) of ten negative wells was considered positive.
DNA extraction and PCR amplification
Under clean conditions, the filter papers were put on a dry clean rapper pad cleaned with 70% ethanol. A 3-mm diameter from each spot was punched-out into 1.5 ml microcentrifuge tube using puncture as previously described (Alhassan et al. 2007). DNA of B. caballi and T. equi were separately extracted from the blood spots on filter paper using QIAamp DNA mini kit (Qiagen, Germany) as described by the manufacturer. The obtained DNA was then stored at −20°C till used.
Two sets of oligonucleotides were used to amplify T. equi DNA. The first PCR assay was performed according to Alhassan et al. (2005). This assay utilized a universal forward primer sequence (Bec-UF2) with the sequence 5′-TCGAAGACGATCAGATACCGTCG and a T. equi-specific reverse primer (Bec-R) with the sequence 5′-ATCGCAAGGAAGTTTAAGGCA. The second set of specific primers was designed based on the sequence of the T. equi merozoite antigen 1(EMA-1) gene with a forward primer EMA-1F: 5′-GCATCCATTGCCATTTCGAG and a reverse primer EMA-1R: 5′-GCTTCTCCGTCTATGGCGCA. To amplify DNA of B. caballi, a forward primer BC48-F: 5′-GGCTCCCAGCGACTCTG and reverse primer BC48-R: 5′-GCATCAAGAGGGCACTTAAG was used to amplify 610 bp from the B. caballi BC48 gene. EMA-1 is encoded by a single copy gene in T. equi (Knowles et al. 1997; Kappmeyer et al. 1993), while BC48 is a multi-copy gene encoding the 48-kDa rhoptry protein of B. caballi (Ikadai et al. 1999). PCR was performed in a final volume of 25 μl, which contained 3 μl template DNA, 2.5 μl of 10× PCR reaction buffer, 1.5 mM MgCl2, 0.2 mM each dNTP, 800 nM of each primer, and 1.25 U of Taq DNA polymerase (AmpliTaq Gold, Applied Biosystems, Japan). PCR cycling included an initial denaturation step at 94°C for 2 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s for both primer pairs used to amplify T. equi DNA, and 55°C of the primer pair to amplify B. caballi, extension at 72°C for 1 min. This was followed by a final extension step at 72°C for 7 min. PCR products were electrophoresed in 1.5% agarose in TBE buffer before being visualized under UV light. B. caballi-positive samples showed an expected band of 610 bp, while T. equi-positive samples showed a band of 435 bp for PCR amplifying the SSU rRNA and 744 bp for PCR amplifying the EMA-1 gene fragments.
Results
Serum antibodies detection
A total number of 80 out of 126 serum horse samples (63.5%) were found positive for T. equi antibodies. The highest prevalence was found in Khartoum North (100%) and Atbara (100%) followed by Omdurman (84.4%). Prevalence was lower in Kosti (45.5%), Khartoum (44.4%), and El Obied (39%; Table 1). Out of 158 samples, only seven (4.4%) were positive for B. caballi in Atbara (10%), Omdurman (8.2%), and El Obied (2.5%), and no positives were detected in Khartoum, Khartoum North, and Kosti areas (Table 1).
PCR detection of T. equi and B. caballi
One hundred thirty-one blood spots from horses were subjected to DNA isolation and PCR. All PCR assays used to amplify T. equi or B. caballi performed well in our hands, and expected bands of 435 bp (for the T. equi 18S gene), 744 bp (for the T. equi EMA-1 gene), and 610 bp (for B. caballi 48-kDa gene) were detected (shown exemplarily for T. equi in Fig. 2). Out of the 131 samples tested, none was positive for B. caballi, but 33 (25.2%) were positive for T. equi (Table 2). The highest prevalence was found in Kosti (62.5%), followed by Atbara (52.6%), Khartoum North (15.8%), Khartoum (15%), El Obied (14%), and Omdurman (0%).

Amplification of Theileria equi DNA using two primer sets: A primer set to amplify a fragment of 435 bp of the 18S gene (a) and another set to amplify a fragment of 744 bp of the T. equi EMA-1 gene (b). Lane M is a 100-bp ladder. Lanes 1–9 in (a) and lane 9 in (b) represent field samples obtained from apparently healthy horses in El Khwai District, Sudan. Lane C+ is a control positive DNA. Lane C− is a negative DNA control isolated from horse blood
Discussion
The objective of this study was to estimate the prevalence of equine piroplasmosis in Sudan using serological and molecular tools. Several reasons account for the application of these techniques for equine piroplasmosis compared to the microscopic detection. Microscopic examination has been shown insensitive to detect low parasitemia, especially in endemic areas of the disease (Calder et al. 1996). Moreover, microscopic detection is subjective; distinguishing Babesia species on the basis of host specificity appears to be less useful than once thought when B. microti has been shown to have broader host specificity (Edelhofer et al. 1998). Thus, in many instances, it has been pointed out that serological and molecular techniques represent a more objective tool for the diagnosis of equine piroplasmosis (Persing and Conrad 1995).
In this study, we applied recombinant protein-based ELISA to detect circulating antibodies against T. equi and B. caballi infections. These ELISAs utilized the EMA of T. equi and the 48-kDa rhoptry protein of B. caballi, their suitability for serodiagnosis has been shown before (Ikadai et al. 1999; Xuan et al. 2001; Knowles et al. 1992). Our findings indicated a high prevalence of antibodies against these parasites in all investigated areas, an indication that equine piroplasmosis is widespread in Sudan. On the other hand, the antibody prevalence of T. equi was found to be higher than that of B. caballi. This could possibly be attributed to the vector distribution. The most abundant ticks associated to horses are Hyalomma anatolicum anatolicum and they might be more potentially important in the transmission of T. equi than B. caballi in Sudan. Another possible reason for the low prevalence of B. caballi could be the earlier elimination of the parasite after a short period of infection (Frerichs et al. 1969). This is also supported by the findings that B. caballi is difficult to detect in blood smears at any stage of the disease except the early acute phase of infection (Todorovic and Carson 1981). In a previous study based on CF test alone, Abdoon (1984) found 54.3% T. equi and 45.7% B. caballi. The positive samples of B. caballi reported by Abdoon (1984) using blood smears may not reflect the natural situation as the samples were taken from clinical cases of animals brought to the veterinary clinics.
During this study, we noticed differences in the prevalence of equine piroplasms between the individual study areas. In Omdurman, for instance, there was a high titer of antibodies against both parasites, and 27 out of 32 serum samples (84.4%) were positive for T. equi and four out of 49 serum samples (8.2%) were positive for B. caballi. This is probably due to differences in the management of the sample animals pertaining to their nutrition and healthcare with tick control.
Using molecular tools on the other hand, T. equi was shown again to be more prevalent than B. caballi though the overall prevalence of both parasites by PCR was lesser than that detected by ELISA. Molecular detection of the parasites requires DNA isolation from parasites that are physically present in the blood sample to a detectable level above the sensitivity threshold of the particular detection method used. Usually, T. equi parasites are not completely eliminated from the blood of horses after treatment or natural recovery (de Waal and van Heerden 1994) as compared to B. caballi. In endemic countries, horses are sometimes known to adapt to infections, but stress and other factors that cause severe immuno-suppression may result in sub-clinical infections becoming overt and detectable. This perhaps explains the lesser prevalence of T. equi using PCR. In addition, there is evidence that animals infected with T. equi become lifelong carriers (Brüning 1996), while that of B. caballi may also persist in sub-clinical form for at least 1–4 years before being eliminated. Therefore, failure to detect B. caballi by PCR is most probably due to the parasites clearance from the circulating blood by the host or reduction to a level beyond the detection sensitivity of the PCR method used. In addition, all samples collected during this study were obtained from adult horses (4 years old and above) and this might be age dependent as observed by Rüegg et al. (2007).
To our knowledge, this is the first report on epidemiology of equine piroplasms using molecular techniques in Sudan. Accurate diagnosis of equine piroplasmosis is essential for providing baseline information about its epidemiology, distribution, and prevalence in the affected equine population and for effective control measures. However, considering the land mass of Sudan and the sample size used, further investigations on equine piroplasms are required.
References
Abdoon AMO (1984) Studies on some aspects of equine piroplasmosis in Khartoum district, Sudan. M.Sc. Dissertation University of Khartoum, pp 85
Alhassan A, Pumidonming W, Okamura M, Hirata H, Battsetseg B, Fujisaki K, Yokoyama N, Igarashi I (2005) Development of a single-round and multiplex PCR method for the simultaneous detection of Babesia caballi and Babesia equi in horse blood. Vet Parasitol 129:43–49
Alhassan A, Govind Y, Tam NT, Thekisoe OM, Yokoyama N, Inoue N, Igarashi I (2007) Comparative evaluation of the sensitivity of LAMP, PCR and in vitro culture methods for the diagnosis of equine piroplasmosis. Parasitol Res 100:1165–1168
Ali S, Sugiomoto C, Onuma M (1996) Equine piroplasmosis. J Equine Sci 7:69–70
Avarzed A, de Waal DT, Igarashi I, Saito A, Oyamada T, Toyoda Y, Suzuki N (1997) Prevalence of equine piroplasmosis in central Mongolia. Onderstepoort J Vet Res 64:141–145
Bakheit MA, Seitzer U, Mbati PA, Ahmed JS (2007) Serological diagnostic tools for the major tick-borne protozoan diseases of livestock. Parassitologia 49(Suppl 1):53–62
Birkenheuer AJ, Levy MG, Breitschwerdt EB (2003) Development and evaluation of a semi nested PCR for detection and differentiation of Babesia gibsoni (Asian genotype) and B. canis DNA in canine blood samples. J Clin Microbiol 41:4174–4177
Brüning A (1996) Equine piroplasmosis an up date on diagnosis, treatment and prevention. Br Vet J 152:139–151
Brüning A, Phipps P, Posnett E, Canning EU (1997) Monoclonal antibodies against Babesia caballi and Babesia equi and their application in serodiagnosis. Vet Parasitol 68:11–26
Caccio S, Camma C, Onuma M, Severini C (2000) The beta-tubulin gene of Babesia and Theileria parasites is an informative marker for species discrimination. Int J Parasitol 30:1181–1185
Calder JA, Reddy GR, Chieves LP, Courtney CH, Littell R, Livengood JR, Norval RA, Smith C Dame JB (1996) Monitoring Babesia bovis infections in cattle by using PCR based tests. J Clin Microbiol 11:2748–2755
Criado-Fornelio A, Martinez-Marcos A, Buling-Sarana A, Barba-Carretero JC (2003) Molecular studies on Babasia, Theileria and Hepatozoon in southern Europe. Part 11. Phylogenetic analysis and evolutionary history. Vet Parasitol 114:173–194
de Waal DT (1992) Equine piroplasmosis: a review. Br Vet J 148:6–14
de Waal DT, van Heerden J (1994) Equine babesiosis. In: Coetzer JAW, Thomson GR, Tustin RC (eds) Infectious diseases of livestock with special reference to South Africa. vol. 1. Oxford University Press, Cape Town, South Africa, pp 293–304
Edelhofer R, Kanout A, Schuh M, Kutzer E (1998) Improved disease resistance after Babesia divergens vaccination. Parasitol Res 84(3):181–187
Frerichs WM, Holbrook AA, Johnson AJ (1969) Equine piroplasmosis: production of antigens for the complement-fixation test. Am J Vet Res 30(8):1337–1341
Friedhoff KT, Soulé C (1996) An account on equine babesiosis. Rev Sci Tech Off Int Epizoot 15:1191–1201
Friedhoff KT, Tenter AM, Muller I (1990) Haemoparasites of equines: impact on international trade of horses. Rev Sci Tech 9(4):1187–1194
Holman PJ, Hietala SK, Kayashima LR, Olson D, Waghela SD, Wagner GG (1997) Case report: field acquired sub-clinical Babesia equi infection confirmed by in vitro culture. J Clin Microbiol 35:474–476
Ikadai H, Xuan X, Igarashi I, Tanaka S, Kanemaru T, Nagasawa H, Fujisaki K, Suzuki N, Mikami T (1999) Cloning and expression of a 48-kilodalton Babesia caballi merozoite rhoptry protein and potential use of the recombinant antigen in an enzyme-linked immunosorbent assay. J Clin Microbiol 37(11):3475–3480
Ikadai H, Nag A, Xuan X, Igarashi I, Kamio TK, Tsuji N, Oyamada T, Suzuki N, Fujisaki K (2001) Sero-epidemiologic Studies on Babesia caballi and Babesia equi infections in Japan. J Vet Med Sci 64:325–328
Kappmeyer LS, Perryman LE, Knowles DP Jr (1993) A Babesia equi gene encodes a surface protein with homology to Theileria species. Mol Biochem Parasitol 62:121–124
Knowles DP Jr, Kappmeyer LS, Stiller D, Hennager SG, Perryman LE (1992) Antibody to a recombinant merozoite protein epitope identifies horses infected with Babesia equi. J Clin Microbiol 30:3122–31226
Knowles DP Jr, Perryman LE, Goff WL, Miller CD, Harrington RD, Gorham JR (1991) A monoclonal antibody defines a geographically conserved surface protein epitope of Babesia equi merozoites. Infect Immun 59:2412–2417
Knowles DP, Kappmeyer LS, Perryman LE (1997) Genetic and biochemical analysis of erythrocyte-stage surface antigens belonging to a family of highly conserved proteins of Babesia equi and Theileria species. Mol Biochem Parasitol 90:69–79
Krause PJ (2003) Babesiosis diagnosis and treatment. Vector-borne Zoonotic Dis 3:45–51
Papadopoulos B, Brossard M, Perie NM (1996) Piroplasms of domestic animals in Macedonia region of Greece. 3. Piroplasms of small ruminants. Vet Parasitol 63:67–74
Persing DH, Conrad PA (1995) Babesiosis: new insights from phylogenetic analysis. Infect Agents Dis 4:182–195
Preston PM (2001) Theileriosis. In: Service MW (ed) Encyclopedia of arthopod-transmitted infections of man and domestic animals. CABI, Wallingford, pp 487–502
Rampersad J, Cesar E, Campbell MD, Samlal M, Ammons D (2003) A field evaluation of PCR for the routine detection of Babesia equi in horses. Vet Parasitol 114:81–87
Rüegg SR, Torgerson P, Deplazes P, Mathis A (2007) Age-dependent dynamics of Theileria equi and Babesia caballi infections in southwest Mongolia based on IFAT and/or PCR prevalence data from domestic horses and ticks. Parasitology 134:939–947
Seifi HA, Mohria M, Sardaria K (2000) A mixed infection of Babesia equi and Babesia caballi in a racing colt: a report from Iran. J Equine Vet Sci 20:858–860
Todorovic RA, Carson CA (1981) Methods for measuring the immunological response to Babesia. In: Ristic M, Kreier JP (eds) Babesiosis. Academic, New York, pp 381–410
Uilenberg G (2001) Babesiosis. In: Service MW (ed) Encyclopedia of arthropod-transmitted infections of man and domestic animals. CABI, Wallinford, pp 53–60
Xuan X, Igarashi I, Avarzed A, Ikadai N, Inoue N, Nagasawa H, Fujisakai K, Toyoda Y, Suzuki N, Milkami T (1998) Diagnosis of Babesia caballi infection in horses by polymerase chain reaction. J Protozool Res 8:85–89
Xuan X, Nagai A, Battsetseg B, Fukumoto S, Makala LH, Inoue N, Igarashi I, Mikami T, Fujisaki K (2001) Diagnosis of equine piroplasmosis in Brazil by serodiagnostic methods with recombinant antigens. J Vet Med Sci 63:1159–1160
Zaugg JL, Lane VM (1992) Efficacy of buparvaquone as a therapeutic and clearing agent of Babesia equi of European origin in horses. Am J Vet Res 53:1396–1399
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salim, B.O.M., Hassan, S.M., Bakheit, M.A. et al. Diagnosis of Babesia caballi and Theileria equi infections in horses in Sudan using ELISA and PCR. Parasitol Res 103, 1145–1150 (2008). https://doi.org/10.1007/s00436-008-1108-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-1108-z