
Abstract
Background
Nuclear pore complexes (NPCs) are sophisticated and dynamic protein structures that straddle the nuclear envelope and act as gatekeepers for transporting molecules between the nucleus and the cytoplasm. NPCs comprise up to 30 different proteins known as nucleoporins (NUPs). However, a growing body of research has suggested that NPCs play important roles in gene regulation, viral infections, cancer, mitosis, genetic diseases, kidney diseases, immune system diseases, and degenerative neurological and muscular pathologies.
Purpose
In this review, we introduce the structure and function of NPCs. Then We described the physiological and pathological effects of each component of NPCs which provide a direction for future clinical applications.
Methods
The literatures from PubMed have been reviewed for this article.
Conclusion
This review summarizes current studies on the implications of NPCs in human physiology and pathology, highlighting the mechanistic underpinnings of NPC-associated diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The nucleus is a membrane-bound organelle in eukaryotic cells that safeguards and preserves genetic material and coordinates vital cellular processes (Dey and Baum 2021). There are several components of the nucleus (I). Nuclear envelope: the nuclear envelope (NE) is composed of two lipid bilayers, namely the inner and outer membranes (INM and ONM), which are separated by a perinuclear space. The NE also contains nuclear pore complexes (NPCs) that penetrate the double membrane (Dey and Baum 2021; Schwarz and Blower 2016; Lin and Hoelz 2019), and the whole structure of the NE encloses the nucleus and separates the contents from the cytoplasm. NPCs in the NE allow the selective transport of molecules between the nucleus and cytoplasm (Lin and Hoelz 2019). Notably, the NE is not only a smooth outer boundary but is also interrupted by invagination, which reaches deep within the nucleoplasm and can traverse the nucleus completely (Malhas et al. 2011). (II). Nucleoplasm: the nucleoplasm is a gel-like matrix but is not homogenous within the nucleus, where various nuclear components are suspended (Galganski et al. 2017; Mancini et al. 1996). The nucleoplasm includes numerous types of nuclear bodies, also called nuclear domains or sub-compartments (Galganski et al. 2017). (III). Chromatin: chromatin is a complex of DNA and proteins that make up chromosomes. It consists of DNA wrapped around histone proteins to form nucleosomes, which condense to form higher-order structures. Chromatin is crucial for the regulation of gene expression, DNA replication, and repair.
Previous studies have shown that nuclear membrane proteins are involved in the pathogenesis of human diseases via transcriptional regulation, nuclear positioning, mechanotransduction, and signaling pathways. Morphological changes in the size and shape of the nucleus are widespread in cancer cells; however, little is known about the underlying molecular mechanisms and functional correlations. Hence, the relationship between NE and cancer is complex and multifaceted. For example, human NPCs serve as a gateway for the exchange of molecules between the nucleus and cytoplasm, including RNA molecules, proteins, and other macromolecules (Beck and Hurt 2017). Furthermore, many tumor suppressor proteins such as p53, breast cancer type 1 susceptibility protein (BRCA1), and phosphatase and tensin homolog (PTEN) rely on human NPCs for transport between the nucleus and cytoplasm (Ikliptikawati et al. 2023; Thompson 2010; Xie et al. 2021a). However, disruption of nucleocytoplasmic shuttling of these tumor suppressors can impair their normal functions, allowing uncontrolled cell growth and promoting tumor development. For instance, high expression levels of human nucleoporin NUP153, which facilitates nucleocytoplasmic transport, occur in hepatocellular carcinoma (HCC), probably leading to alterations in NPC composition and function (Gan et al. 2022; Nofrini et al. 2016). In addition, mutations in the human nucleoporin NUP98, which provides vital interaction sites for nucleocytoplasmic transport, have been found in hematologic malignancies, including leukemia, and contribute to the fusion of oncoproteins (Nofrini et al. 2016; McNeer et al. 2019; Chandra et al. 2022). Moreover, the human nucleoporins POM121 and importinβ mediate the nuclear import of a series of oncogenic transcription factors (e.g., such as the transcription factor E2F1 (E2F1), transcription factor p64 (c-Myc), and androgen receptor (AR), promoting the progression of prostate cancer (PCa) (Rodriguez-Bravo et al. 2018). Therefore, this review focuses on the link between nucleoporins in NPCs and diseases.
The composition of NPCs
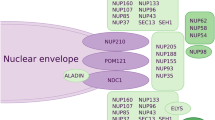
At the junction of the INM and ONM of the human nucleus, approximately 600 copies of approximately 30 proteins termed nucleoporins (NUPs) assemble into NPCs, with structural elements of stacked a-helical repeats and/or b-propellers; approximately one-third of these proteins also contain phenylalanine-glycin (FG) repeat sequences for the selective transport of cargoes (Nofrini et al. 2016). These NUPs assemble into distinct subcomplexes within the NPCs, contributing to their overall organization and function. Generally, NPCs are classified based on their composition and substructure: (I). Outer ring complex: the outer ring complex, including the cytoplasmic ring (CR) and nuclear ring (NR), which are anchored by the inner pore ring, forms the structural framework of the NPCs (Beck and Hurt 2017). It consists of several NUPs, including NUP93, NUP205, NUP188, and NUP155, among others (Nofrini et al. 2016), forming a circular structure at the periphery of the NPCs and serving as a docking site for other nucleoporin subcomplexes. (II). Inner ring complex: the inner ring complex containing NUPs such as NUP58, NUP54, and NUP62, among others, is located on the nuclear side of the NPCs and connects the outer ring complex to the central channel, helping anchor the nuclear basket and contributing to the selective permeability of the NPCs (Nofrini et al. 2016; Kosinski et al. 2016; Gaik et al. 2015). (III). Central channel: the central channel of NPCs forms the main transport gate for molecules, such as proteins, RNA, and ribosomal subunits, passing shuttling between the nucleus and cytoplasm, and consists of NUPs, including NUP62, NUP58, and NUP54 (Beck and Hurt 2017). (IV). Nuclear basket: the nuclear basket is a specialized structure within NPCs that extends into the nucleus, where it plays a role in the anchoring and organization of NPCs, as well as in the regulation of nucleocytoplasmic transport (Kosinski et al. 2016; Gaik et al. 2015; Cibulka et al. 2022). The nuclear basket contains NUPs such as NUP153, NUP50, zinc finger C3HC-type protein 1 (ZC3HC1), and translocated promoter region protein (TPR) (Beck and Hurt 2017; Appen et al. 2015; Gunkel et al. 2021). (V). Cytoplasmic filaments: cytoplasmic filaments containing several NUPs, such as NUP358, NUP214, NUP98, NUP88, ALADIN, mastermind-like domain-containing protein 1 (CG1), and the mRNA export factor RAE1 (RAE1), contribute to the anchoring of NPCs to the NE and are involved in interactions with cytoplasmic factors associated with transport regulation (Lin and Hoelz 2019; Hamada et al. 2011; Wälde et al. 2012). Specifically, cytoplasmic filaments are peripheral elements emanating from the nucleus and CR, and are extensions of NPCs that project into the cytoplasm (Beck and Hurt 2017; Nofrini et al. 2016). Cytoplasmic filament NUPs recruit cargo transport factor complexes for nucleocytoplasmic transport and orchestrate the export and remodeling of messenger ribonucleoprotein particles in preparation for translation (Bley et al. 2022). (VI). Transmembrane NUPs POM121, nuclear pore membrane glycoprotein 210 (GP210), and nuclear division cycle protein 1 (NDC1) enclose the central channel and play a gatekeeping role, and their stability affects the levels of several other NUPs (Coyne et al. 2020; Liu et al. 2009; Gomez-Cavazos and Hetzer 2015) (Fig. 1).

Human NPCs comprise seven parts, including cytoplasmic filaments, the outer ring complex (cytoplasmic rings and nuclear ring), transmembrane nucleoporins, the central channel, the inner ring complex, and the nuclear basket. On the left is a description of the general structure of the NPCs, and on the right is a description of the specific NUPs of each component
Outer ring complex
The CR contains 16 copies of the Y-shaped complex (Y-complex), encircling head to tail to form the inner and outer layers of eight Y-complexes (Fontana et al. 2022; Knockenhauer and Schwartz 2016; Kelley et al. 2015). The Y-complex is composed of a short arm (Nup160 and Nup37), long arm (Nup85, Nup43, and Seh1), and stem (Nup96, Sec13, Nup107, and Nup133) in Xenopus laevis (Fontana et al. 2022; Bilokapic and Schwartz 2012). Previous studies have shown that Seh1 and Nup43, although indispensable in mouse embryonic stem cells, are required for their average cell growth rates, viability upon differentiation, and maintenance of proper NPC density (Gonzalez-Estevez et al. 2021). In addition, Nup133, including TPR and Nup153, is required for proper nuclear basket assembly and dynamics in mouse embryonic stem cells (Souquet et al. 2018; Orniacki et al. 2023). Moreover, in Xenopus laevis oocytes, the CR contains two copies of Nup205, two copies of the Nup214-Nup88-Nup62 complex, one Nup155, and five copies of Nup358 (Fontana et al. 2022). Significantly, Nup358 contains FG repeats previously shown to form a gel-like condensate phase for selective cargo passage (Knockenhauer and Schwartz 2016; Frey et al. 2006; Lemke 2016). Additionally, four Nup358 copies clamp around the Y-complexes to stabilize CR (Fontana et al. 2022). Under the anchoring effect of Nup358, there are eight cytoplasmic filaments, namely, Nup88, Nup214, Nup98, the mRNA export factor GLE1 (GLE1), CG1, ribonucleic acid export 1 (RAE1), and Nup62 (Beck and Hurt 2017; Sakiyama et al. 2016, 2017).
NUP160 and NUP37
In Xenopus laevis, full-length Nup160 consists of a seven-bladed β-propeller followed by an extended α-helical domain, and its C-terminal fragment is located at the vertex of the Y complex and sandwiched between Sec13 homolog 1 (Seh1) and SEC13-like protein 1 (Sec13) (Zhu et al. 2022). A previous study showed that Nup160 interacts with Nup98 and Nup153 and plays a role in mRNA export but not in protein import or export in Xenopus laevis (Vasu et al. 2001). Functionally, Nup160 knockdown inhibits cell proliferation; induces apoptosis, autophagy, and cell migration; and alters the expression and localization of podocyte-associated molecules in mouse podocytes (Xie et al. 2021b). Phenotypically, silencing of Drosophila Nup160 in nephrocytes (fly renal cells) led to functional abnormalities, reduced nuclear volume and cell size, and a disorganized nuclear membrane structure (NPC and nuclear lamin localization defects) (Zhao et al. 2019). NUP160 was found to be highly expressed in the peripheral blood or cell lines of patients with chronic myeloid leukemia (CML), inhibiting the sensitivity of CML to imatinib (Zhang et al. 2019). In human angiosarcoma, the frequent gene fusion NUP160-SLC43A3 causes truncation of NUP160 and stimulates cell proliferation (Shimozono et al. 2015). In addition to NUP160 mutations caused by gene fusion, compound heterozygous mutations in NUP160 (R1173× and E803K) have been reported in steroid-resistant nephrotic syndrome (SRNS) (Zhao et al. 2019). Notably, although Nup160 is initially associated with nucleoplasmic transport, it is also involved in autophagy, hybrid sterility, female sterility, abnormal morphology, and slow development in flies (Xie et al. 2021b; Maehara et al. 2012) (Fig. 2).

Summary of the physiological and pathological effects of each component of the Y complex in humans
Human NUP37, also known as P37 or MCPH24, is vital for kinetochore-microtubule interactions and mitosis (Loïodice et al. 2004). Recent bioanalysis studies have suggested that NUP37 is a potential prognostic biomarker and oncogene for non-small cell lung cancer (NSCLC), breast cancer (BC), glioma, and pan-cancer (He et al. 2022; Li and Liu 2021; Liu et al. 2021a). Additionally, tumor tissues with increased NUP37 expression exist in a relatively immunosuppressive microenvironment and are resistant to multiple anticancer drugs (He et al. 2022). Previous studies have shown that increased NUP37 expression in HCC acts as a positive regulator of Yes-associated protein 1/TEA domain family member (YAP1/TEAD) signaling, thereby promoting cancer progression (Luo et al. 2017). Similarly, maternal NUP37 contributes to the nuclear import of YAP1 and subsequently activates YAP1/TEAD signaling, which is involved in oocyte maturation and preimplantation embryo development in mammals (Peng et al. 2022; Guo et al. 2022). Moreover, the interaction between NUP37 and low-density lipoprotein-related receptor 5 (LRP5) enhances the stabilization of NUP37, thereby facilitating liver cancer (LC) cell proliferation (Chen et al. 2019). However, LRP5 is mainly expressed in cell membranes, suggesting that LRP5 is likely to enter the nucleus and interact with NUP37 in the nucleus (Chen et al. 2019). Interestingly, the epigenetic mechanism of elevated NUP37 expression in HCC is closely associated with the aberrant hypomethylation pattern of the NUP37 promoter (cg24826236, cg08316365, and cg08085165) (Tang et al. 2022a, b). In colorectal cancer (CRC) cell lines, upregulated expression of NUP37 promotes the activation of phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT) signaling, thereby facilitating the progression of CRC (Xiong et al. 2023). Similarly, overexpression of NUP37 activates PI3K/AKT signaling, promoting the proliferation, migration, and invasion of gastric cancer (GC) cells (Zhang et al. 2021a), and forms an immunosuppressive microenvironment in gliomas (He et al. 2022) (Fig. 2 and Table 1). Notably, several vital proteins involved in PI3K/AKT signaling, including AKT, mechanistic target of rapamycin complex 1 (mTORC1), PTEN, and 3-phosphoinositide-dependent kinase 1 (PDK1), can shuttle between the nucleus and cytoplasm (Chen et al. 2022; Gupta et al. 2022). However, the relationship between NUP37 and these proteins in terms of nucleoplasmic transport remains unclear.
NUP85, NUP43, and SEH1
In terms of biological function, human NUP85 (also known as FROUNT) plays a significant role in the development of tissues, particularly those of the brain and nervous system (Ravindran et al. 2021, 2023). In addition, NUP85 is a cytosolic regulator that mediates leukocyte and monocyte migration by binding to the chemokine receptor CC chemokine receptor 2/5 (CCR2/5) (Toda et al. 2014; Esaki et al. 2014), and it is worth noting that the interaction between NUP85 and these two chemokine receptors can be blocked by disulfiram (Terashima et al. 2020). Crucially, CCR2/5 are major chemotactic regulators of tumor-promoting macrophages at the tumor site, and blocking these receptors could impede tumor progression in experimental animal models (Zhang et al. 2013; Sanford et al. 2013; Popivanova et al. 2009) (Fig. 2).
Initially, Nup85 was associated with childhood-onset SRNS in four affected individuals with intellectual disabilities in Xenopus (Braun et al. 2018a). Recently, biallelic and heterozygous human NUP85 variants were found in primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders (MCPH–SCKS) (Ravindran et al. 2021, 2023). However, to date, mutations in NUP85 have not been found in cancer studies. NUP85 deficiency remarkably inhibits tumor progression in lung carcinoma and melanoma and decreases macrophage tumor-promoting activity by directly mediating chemokine receptor activity (Terashima et al. 2020). Similarly, blockade of Nup85 attenuates steatosis and monocyte recruitment caused by macrophage chaperone-mediated autophagy deficiency in nonalcoholic steatohepatitis (NASH) mice (Zhang et al. 2023). approximately 10–20% of NASH patients may progress to liver fibrosis, cirrhosis, and even LC (Kessoku et al. 2021), indicating that NUP85 is likely associated with the early tumorigenesis of LC.
Human NUP43 harbors a canonical β-propeller fold consisting of seven 40 repeats of Trp-Asp (WD) (13–73, 77–116,134–168, 175–212, 220–257, 264–344, and 353–38), and each WD40 repeat of NUP43 is composed of four antiparallel β-strands (Xu et al. 2015a). In addition, an ordered loop at the bottom surface of NUP43 WD40 may act as a bridge that directly binds to NUP85–SEH1 (Xu et al. 2015a). Spatially, NUP43 binds to NUP85 through its bottom surface and approaches NUP37 (Bui et al. 2013). The carcinogenic role of NUP43 has been confirmed in GC (Li et al. 2020; Yang et al. 2022) and BC (Ren et al. 2022; Tian et al. 2018; Zhang et al. 2021b; Jiang et al. 2021). In GC cells, the long noncoding RNA (lncRNA) NCK1-AS1 functions as a sponge for microRNA-137 (miR-137) to increase NUP43 expression, thereby contributing to GC malignant behaviors (Li et al. 2020). In addition, NUP43 was found to be abnormally highly expressed in luminal A and human epidermal growth factor receptor 2 (HER2)+ BC patients, and might independently predict poor prognosis in cancer patients (Tian et al. 2018). However, the upregulated expression of NUP43, which is targeted by the miR-409 gene, reduces the proliferative potential and inhibits cell cycle progression in CML cells (Liu et al. 2019) (Fig. 2, Table 1).
Human SEH1 is required for chromosome association (e.g., targeting GATOR2 and NUP153 to mitotic chromosomes), chromosome segregation (e.g., controlling Aurora B localization at centromeres), proper kinetochore function (e.g., stabilizing kinetochore‒microtubule interactions by recruiting chromosome region maintenance 1 (CRM1) and RanGTPase AP1 (RanGAP1)–NUP358), and efficient localization of the chromosomal passenger complex (Platani et al. 2009, 2018; Zuccolo et al. 2007). Additionally, SEH1 is a critical component of the GATOR2 complex (consisting of MIOS, WDR24, WDR59, SEH1, and SEC13), which acts as a switch for mTORC1 signaling depending on nutrient levels (Valenstein et al. 2022). In addition, the process of tumor metabolic reprogramming is frequently accompanied by upregulation of mTOR signaling, thereby facilitating enhanced nutrient acquisition for the production of metabolites essential for sustaining growth and proliferation (Szwed et al. 2021) (Fig. 2).
NUP96, SEC13, NUP107, and NUP133
Nup96 and Nup98 are encoded by the same gene, Nup98–Nup96, which is a protein precursor and is produced during autoproteolysis, and both proteins are associated with mRNA export in yeast and mammals (Fontoura et al. 1999; Enninga et al. 2002; Faria et al. 2006). Interestingly, the upstream region of human NUP96 is the NUP98 coding region, and NUP98 chromosomal translocation (fusion with the Homeobox A9 (HOXA9) gene) has been observed in patients with acute myeloid leukemia (AML) (Nakamura et al. 1996; Borrow et al. 1996). Conceivably, gene fusion disrupts the function of NUP96, which requires NUP98-dependent autocatalytic processing from the NUP98-96 precursor protein to be properly localized and available (Fontoura et al. 1999). In addition, human NUP96 differentially regulates the nucleocytoplasmic distribution and expression of key cell cycle regulators of mRNAs/proteins (e.g., Cyclin B1, Cyclin D3, and CDK6) in a cell cycle-dependent manner (Chakraborty et al. 2008). Phenotypically, deficits in Nup96 in mice lead to slightly enhanced proliferation of T cells and bone marrow-derived macrophages, suggesting a potential role for Nup96 as a haploinsufficient tumor suppressor (Chakraborty et al. 2008). However, another study showed that deficits in Nup96 in mice impaired antigen presentation and T-cell proliferation upon immunization (Faria et al. 2006), suggesting that Nup96 functions exhibit organizational differences. Moreover, human NUP96 stably interacts with the WD repeat region of SEC13 (another nucleoporin associated with the formation of coat protein complex II (COPII)-coated vesicles) during interphase, mediating the shuttling of SEC13 and likely coupling functions between the nucleus and cytoplasm (Enninga et al. 2003) (Fig. 2).
Several studies have shown that human SEC13 mainly plays roles in two distinct complexes, including a component of the NPCs and the GATOR2 complex (Beck and Hurt 2017; Valenstein et al. 2022). Functionally, SEC13 also primarily regulates the metaphase/anaphase transition and contributes to maintaining genomic stability during mitosis (Sihn et al. 2005); thus, SEC13 can maintain gene stability or impact cell growth by regulating the corresponding signaling pathways, reflecting its potential opposite effect on cancers. Furthermore, in mice, proper myelination and remyelination require Sec13-mediated autocrine pleiotrophin signaling and protein transport via Sec13 (Liu et al. 2022). In addition, SEC13 is involved in the VISA-mediated antiviral signaling pathway by increasing VISA aggregation and ubiquitination, thus enhancing the phosphorylation and dimerization of interferon regulatory factor 3 (IRF3) to facilitate IFN-β production and strengthen antiviral immune activity (Chen et al. 2018a). In GC patients, the expression of SEC13 is significantly decreased and negatively correlated with tumor stage, suggesting that SEC13 may act as a tumor suppressor in GC (Hussein et al. 2022). However, the gene and protein expression of SEC13 is dramatically increased in colon cancer (CC) patients, which may be related to its glycolytic function, suggesting that SEC13 has opposite effects in different cancer backgrounds (Liu et al. 2022). Notably, glycolysis is highly active in tumor cells and its reprogramming is closely related to tumor occurrence and progression (Pavlova and Thompson 2016) (Fig. 2, Table 1).
Human NUP107, located in the core scaffold of NPCs, is characterized by a leucine zipper motif and a large number of kinase consistency sites in its carboxy-terminal region but does not contain FG repeats (Banerjee et al. 2010). During cell mitosis, NUP107 plays a crucial role in driving the assembly of NPCs (Zuccolo et al. 2007). In glioblastoma, NUP107 functions in transport surveillance by tethering proteasome 26 s-mediated p53 degeneration, thereby promoting tumorigenesis (Ikliptikawati et al. 2023). In addition, depletion of a single nucleoporin, NUP107, induces apoptosis in human astrocytoma cells (Banerjee et al. 2010). Furthermore, overexpression of NUP107 confers resistance to oxidative stress but does not affect the migration or proliferation of cervical cancer cells (Shi et al. 2020). In terms of transport, apoptotic protease activating factor-1 (Apaf-1), an adaptor protein critically involved in mitochondrial cell death, binds to the central domain of Nup107 (457–618) through its CED-4 domain, leading to the nuclear accumulation of Apaf-1 under DNA damage in mouse (Jagot-Lacoussiere et al. 2015). Interestingly, the nuclear presence of Apaf-1 is a positive prognostic factor in NSCLC patients (Zermati et al. 2007; Besse et al. 2004). In rat, Nup107 also facilitates the nuclear export of sodium channel protein type 5 subunit alpha (Scn5a) mRNA and consequently induces the protein expression of Scn5a, regulating cardiac electrophysiology (Scn5a‐encoded INa channel) (Guan et al. 2019). Increased Scn5a expression is associated with more aggressive tumor characteristics (e.g., BC, CC, and ovarian cancer), including lymph node invasion, recurrence of metastasis, and reduced survival (Brackenbury 2012; Luiz and Wood 2016). In HCC cells, NUP107 is upregulated, portends poor prognosis, and can predict the survival of patients with HCC with reasonable accuracy (Nong et al. 2023). Moreover, a single nucleotide variant in NUP107 may be predictive of sensitivity to platinum chemotherapy in patients with ovarian cancer (Alanee et al. 2017). These findings imply that abnormal NUP107 expression is closely associated with tumor occurrence and progression. In addition, there is a strong association between childhood SRNS (dysfunction of podocytes is a hallmark of SRNS) and a high incidence of NUP107 mutations (Braun et al. 2018b; Rosti et al. 2017; Miyake et al. 2015; Park et al. 2017). These NUP107 mutations (c.303G>A, p.M101I; c.1021dupG, p.E341Gfs*3; c.2129_2131delAAG, p.E710del; c.2666A>G, p.Y889C) cause changes in the amino acid sequence of this gene, indicating that the function and structure of the NUP107 protein may change in SRNS (Braun et al. 2018b). However, the mechanism by which NUP107 mutations cause the glomerular phenotype in humans remains unknown. Previous studies have shown that NPCs are closely related to mechanical transduction signals (Swift and Discher 2014; Fedorchak et al. 2014), and mechanical stretching reduces podocyte proliferation and cell volume by reorganizing the actin cytoskeleton in vitro (Endlich et al. 2001; Petermann et al. 2002). Hence, increased postnatal capillary pressure may be more prone to damaging podocytes with NUP107 mutations, leading to mechanical cell stretching. NUP107 missense mutations (c.1063C>T, p.R355C) have also been found in hypogonadotropic hypogonadism, and are an important cause of morbidity (Ren et al. 2018). Similarly, a recessive missense mutation of NUP107 (c.1339G> a, p.D447N) occurs in XX female gonadal dysgenesis, characterized by underdeveloped, dysfunctional ovaries (Weinberg-Shukron et al. 2015). The impact of Drosophila Nup107 p.D364N (corresponding to human NUP107 p.D447N) in Drosophila females resulted in almost complete sterility, with a marked reduction in progeny, morphologically aberrant eggshells, and disintegrating egg chambers (Weinberg-Shukron et al. 2015). These results indicate a pivotal role of NUP107 in female reproductive development and more common conditions, such as premature ovarian failure (Fig. 2, Table 1).
Previous physiological studies have revealed that Nup133 is essential for embryonic development but not for mouse embryonic stem cell proliferation (Souquet et al. 2018). Similarly, deficiency or dysfunction of Nup133 impairs NPC assembly, maternal transcription factor (TF) nuclear transport, and zygotic genome activation, whereas Nup133 overexpression promotes these processes during zebrafish early embryogenesis (Shen et al. 2022). Furthermore, Nup133 deficiency impairs murine neural lineage differentiation (Lupu et al. 2008). Consistently, a Nup133-knockdown zebrafish model exhibited microcephaly, fewer neuronal cells, underdeveloped glomeruli, and fusion of the foot processes of podocytes (Fujita et al. 2018; Rogg et al. 2022). Pathologically, the human homozygous NUP133 mutation c.3335-11T>A results in the insertion of 9 bp of the intronic sequence between exons 25 and 26 in the mutant transcript, impairing the interaction between NUP133 and NUP107 and thus causing Galloway–Mowat syndrome (Fujita et al. 2018). In SRNS, these NUP133 mutations (c.691C>G, p.R231G; c.3164T>C, p.I1055S; c.2922T>G, p.S974R; c.182+387T>G; c.2898G>C, p.K966N) have been reported (Braun et al. 2018a; Wang et al. 2023a). Notably, there were no significant gene expression changes in Nup133-mutated podocytes, essentially excluding a direct loss- or gain-of-function mechanism, but the localization of NUP133 and NUP107 to the NE was mildly defective, leading to spreading defects (Rogg et al. 2022). This evidence suggests that this mild damage may only affect sensitive cell types, and changes in the levels of NUP133 protein may be the main consequence of the mutant variant. Indeed, a susceptible and critical signaling pathway in podocytes is small RhoGTPase signaling, and activation of small RhoGTPase cell division control protein 42 (Cdc42) is a common consequence of NUP133 deficiency (Braun et al. 2018a; Rogg et al. 2022) (Fig. 2, Table 1).
NUP214, NUP88 and NUP62
In Xenopus laevis, the Nup214–Nup88–Nup62 complex, located in the CR subunit, is associated with two copies of the Y complex (Huang et al. 2020). In humans, the interaction between NUP88 and NUP214 facilitates the localization of NUP88 to NPCs (Fornerod et al. 1997; Bonnin et al. 2018), whereas the interaction between NUP88 and NUP62 contributes to stabilizing NUP88 by inhibiting the proteasomal degradation of overexpressed NUP88 (Bonnin et al. 2018; Singh et al. 2023). A previous study showed that genetic disruption of human NUP88 results in pleiotropic developmental defects, including locomotor defects and neuromuscular junction dysregulation (Bonnin et al. 2018). Notably, biallelic mutations in nucleoporin NUP88 are a cause of a lethal fetal akinesia deformation sequence (FADS) characterized by impaired fetal movement, multiple congenital malformations, and a poor prognosis, which affects rapsyn, a vital regulator of the muscle nicotinic acetylcholine receptor at the neuromuscular junction (Bonnin et al. 2018) (Fig. 3).

Summary of the physiological and pathological effects of each component of the cytoplasmic filaments in humans
The expression of NUP88 is abnormally upregulated in cancers, including ovarian tumors (Martínez et al. 1999), cervical cancer (Makise et al. 2021), head and neck cancer (Singh et al. 2023), and CRC (Zhao et al. 2010). Previous studies have demonstrated that elevated levels of NUP88 confer proliferation and migration advantages to cancer cells by sequestering NF-κB partly into the nucleus of unstimulated cells and consequently activating the NF-κB pathway at the level of nucleocytoplasmic transport (Singh et al. 2023; Takahashi et al. 2008). Reasonably, the Nup88-Nup214 complex, in coordination with CRM1, is believed to promote the nuclear translocation of NF-κB and pre-ribosomal assembly in Drosophila and human (Takahashi et al. 2008; Xylourgidis et al. 2006; Bernad et al. 2006). In addition, overexpression of NUP88 promotes the early stage of tumorigenesis by inducing multinucleated cell formation in both HeLa cells and a mouse model (Hashizume et al. 2010; Barr et al. 2004), which may be caused by the NUP88-dependent multinucleated phenotype resulting from changes in the organization of vimentin and polo-like kinase 1 (PLK1) (Makise et al. 2018; Naylor et al. 2016) (Fig. 3, Table 2).
The human NUP214 protein, which possesses FG repeat domains and plays a critical role in nucleocytoplasmic transport, may serve as a docking site in receptor-mediated import of substrates across NPCs (Fichtman et al. 2019; Kraemer et al. 1994). Physiologically, NUP214 has been implicated in altering nucleocytoplasmic transport (Saito et al. 2016; Kindermann et al. 2019), cell differentiation (Mendes and Fahrenkrog 2019; Boer et al. 1998), and microbial infection (Cassany et al. 2015). For instance, a recent study revealed that nucleoporin fusions formed by NUP214-SET promote the accumulation of mixed-lineage leukemia 1 (MLL1), a histone methyltransferase essential for maintaining HOXA10 gene expression (Oka et al. 2023; Cigdem et al. 2021). Additionally, NUP214 interacts with factors such as the mRNA export receptor TAP/NXF1 and exportin-1 (XPO1)/CRM1, facilitating mRNA export and leucine-rich nuclear export signal (NES)-dependent protein export, respectively (Saito et al. 2016) (Fig. 3).
In normal cells, NUP214 is a component of NPCs and is involved in nucleocytoplasmic transport and regulation of molecules entering and exiting the nucleus. However, in leukemic cells, NUP214 can be disrupted by chromosomal translocations, leading to the formation of fusion genes that encode aberrant proteins. One of the well-studied fusion genes involving NUP214 is the NUP214-DEK fusion gene, resulting from a t(6;9) chromosomal translocation, which is associated with AML and myelodysplastic syndrome (Sandén and Gullberg 2015). The NUP214-DEK fusion protein can dysregulate transcription and contribute to leukemogenesis by promoting gene expression, which leads to uncontrolled cell proliferation and inhibition of differentiation and apoptosis (Saito et al. 2016; Ageberg et al. 2008). Another fusion protein involving NUP214 is NUP214-SET, which is created from a different chromosomal translocation and was identified in patients with T-acute lymphoblastic leukemia (T-ALL), implicating disparate leukemogenic driver mechanisms (Mendes and Fahrenkrog 2019). This fusion protein is known to be associated with leukemogenesis via aberrant activation of the HOXA gene cluster, which is crucial for the proliferation and survival of leukemic cells (Vlierberghe et al. 2008; Quentmeier et al. 2009). Specifically, chromosomal translocations in the 5' untranslated region (5'UTR) of NUP214, including NUP214-SET and NUP214-DEK, disrupt NUP214-dependent protein nuclear export by inhibiting the nuclear export receptor CRM1, which results in aberrant accumulation of CRM1 protein cargoes in the nucleus (Oka et al. 2019; Port et al. 2015). In addition, in T-ALL, the NUP214-ABL1 fusion gene is known to be an oncogene that leads to constitutive tyrosine kinase activity and activation of several downstream signaling pathways (e.g., the PI3K/AKT/mTOR and JAK/STAT pathways) that are crucial for cell cycle progression and survival, ultimately leading to the pathological proliferation of T cells (Simioni et al. 2016; Kleppe et al. 2011; Quintás-Cardama et al. 2008). Furthermore, in ALL, the less frequent fusion of NUP214-SQSTM1 also impairs the interaction between NUP214 and CRM1, therefore driving leukemogenesis (Lavau et al. 2020; Gorello et al. 2010) (Fig. 3, Table 2).
Human NUP62, which is located not only in the outer ring complex but also in the central channel, is one of several NUPs that forms the main plug and plays a role in facilitating the selective transport of molecules (Borlido and D'Angelo 2018). The known physiological functions associated with Nup62 mainly include the facilitation of selective transport, such as Cyclin B in Drosophila (Okazaki et al. 2020), mRNA in mammals (Ke et al. 2019), nuclear import of p63 (Hazawa et al. 2018), stress response (Kinoshita et al. 2014), and cell division in humans (Fukuhara et al. 2006; Hashizume et al. 2013). Specifically, it plays a role in mitotic cell cycle progression by regulating centrosome segregation, centriole maturation, and spindle orientation (Hashizume et al. 2013). Interestingly, it may be involved in protein recruitment to the centrosome after nuclear breakdown (Hashizume et al. 2013). NUP62 has been implicated in the development of various diseases, including cancers (Singh et al. 2023; Wang et al. 2021a; Borlido and D'Angelo 2018), neurological disorders (Gleixner et al. 2022; Basel-Vanagaite et al. 2006; Ding and Sepehrimanesh 2021; Harrer et al. 2023), and rheumatoid arthritis (Batliwalla et al. 2005; Senécal et al. 2014). In GC, high NUP62 expression influences cell migration and epithelial–mesenchymal transition (EMT) through Wnt/β-catenin and transforming growth factor beta (TGF-β) signaling pathways (Wang et al. 2021a). In squamous cell carcinomas (SCC), depletion of NUP62 inhibits proliferation and augments the differentiation of cells by mediating p63 nuclear import (Hazawa et al. 2018; Borlido and D'Angelo 2018). Moreover, previous studies have demonstrated that NUP62 can maintain the spindle assembly checkpoint downstream of kinetochores, thereby ensuring maintenance of chromosomal stability (Hashizume et al. 2013; Chien et al. 2020), indicating that NUP62 may participate in another potential mechanism of human cancer development. Emerging research suggests that NUP62 is involved in amyotrophic lateral sclerosis (ALS) (Gasset-Rosa et al. 2019; Khalil et al. 2022) and multiple sclerosis (Gasset-Rosa et al. 2019). One common characteristic of ALS is the abnormal aggregation of proteins, including the mislocalization and aggregation of transactive response DNA-binding protein 43 (TDP-43) (Gasset-Rosa et al. 2019; Khalil et al. 2022; Gleixner et al. 2022). NUP62 has been found to interacts with TDP-43 to regulate nucleocytoplasmic transport (Gasset-Rosa et al. 2019; Gleixner et al. 2022) (Fig. 3, Table 2).
NUP358
In mammals, Nup358 is structurally present in cytoplasmic annulate lamellae, with parallel membrane stacks closely resembling NE (Cordes et al. 1996). Human NUP358 is distinguished by its unique structure: several zinc finger motifs, a cyclophilin A homologous domain, four Ran-binding domains, and a SUMO E3 ligase domain in mammal (Prunuske et al. 2006; Wu et al. 1995; Pichler et al. 2002). NUP358 also binds to transport receptors and modulates the activity of various protein complexes via SUMOylation in mammal (Hutten et al. 2008; Engelsma et al. 2004). Research has indicated a link between NUP358 and mRNA processing bodies (P-bodies) and stress granules (SGs), which are cytoplasmic aggregates involved in mRNA turnover and stress response (Sahoo et al. 2017). NUP358 depletion disrupts P bodies and adversely affects the miRNA pathway, suggesting its potential regulatory role in mRNA surveillance and gene expression (Sahoo et al. 2017). Nup358 also appears to be a target of the host response to viral infections. For example, the compromised function of Nup358 affects SG formation, which is a defense mechanism against viral infection in Drosophila (Sadasivan et al. 2022). In particular, cricket paralysis virus (CrPV) exploits Nup358 for replication via selective degradation of Nup358 depending on the R146 residue of CrPV, linking NPC function to a viral infection strategy (Sadasivan et al. 2022). In addition, human NUP358 interacts with the cytoplasmic dynein bicaudal D cargo adaptor 2 (BicD2), which recruits the dynein machinery to the nucleus (Gibson et al. 2023). NUP358/BicD2 interaction is crucial for mitotic spindle assembly and has profound implications for brain and muscle development (Gibson et al. 2023, 2022). Moreover, the dynein adaptor BicD2 is a master regulator of selective cargo recognition for minus-end-directed transport on microtubules, binding to cargoes and linking them to dynein motors (Gibson et al. 2023).
NUP358, as an adapter, possesses a largely disordered C-terminal region with FG repeats for gel-like phase formation and selective transport and binding sites for Ran GTPase-activating protein, Ran, and other effectors (Knockenhauer and Schwartz 2016; Frey et al. 2006; Lemke 2016; Wu et al. 1995). In mammal, Nup358 has diverse functions involved in the regulation of nucleocytoplasmic transport, viral infection, and cancer progression (Jiang et al. 2022; Gloerich et al. 2011; Wang et al. 2018), notably resulting from the combination of the intrinsic properties of its protein domains (e.g., SUMO-interacting motifs) and the various subcellular localizations of Nup358 (e.g., NPCs and kinetochores) (Liu et al. 2020; Joseph et al. 2004; Forler et al. 2004; Cooper et al. 2005). Notably, NUP358 forms a complex with SUMOylated RanGAP1, a GTPase-activating protein that catalyzes the conversion of RanGTP to RanGDP, consequently establishing a RanGTP gradient across the NE and regulating the directionality and efficiency of the nucleocytoplasmic transport processes (Wu et al. 1995; Carlon-Andres et al. 2020). NUP358 acts as a cargo- and receptor-specific assembly platform, facilitating the efficient nuclear import of proteins, such as importinα/β-cargo deleted in breast cancer 1 (DBC-1) and DNA methyltransferase 1 associated protein 1 (DMAP-1), in a receptor-independent manner (Wälde et al. 2012). In addition, NUP358 interacts with cytoplasmic human telomerase reverse transcriptase (hTERT), which is bound to NPCs by the import receptor importin 7, thereby mediating its nuclear import (Frohnert et al. 2014). Intriguingly, NUP358 is also involved in GTP hydrolysis on Ran, allowing RanGDP and importin7 to be transported to the cytoplasm where they are kept near NPCs to initiate a new round of import (Frohnert et al. 2014). In addition to facilitating protein transport, recent studies have shown that NUP358 facilitates adenoviral genome import in a manner dependent on the transport receptor transportin1 (Carlon-Andres et al. 2020). In addition, it plays a role in mRNA metabolism and post-transcriptional events, including translation (Gong et al. 2023), export (Dawlaty et al. 2008; Culjkovic-Kraljacic et al. 2012), and mRNA stability and localization regulation in humans (Gong et al. 2023; Moreno-Oñate et al. 2020). Previous studies have demonstrated that the regions of NUP358 that participate in eukaryotic translation initiation factor 4E (eIF4E)-dependent and bulk mRNA export are associated with zinc fingers and leucine-rich domains, respectively (Dawlaty et al. 2008; Culjkovic-Kraljacic et al. 2012) (Fig. 3).
Mutations and dysfunction in NUP358 can be implicated in various diseases, including retinal diseases (Castagnet et al. 2003), neuropathies (Khalaf et al. 2019; Vyas et al. 2013), cancer (Wang et al. 2021b; Yang et al. 2017), viral infections (Jiang et al. 2022), and autoimmune disorders (Senécal et al. 2014). A previous study demonstrated that the cyclophilin-like domain of NUP358 associated with the S1 subunit selectively promotes the accumulation of properly folded proteins targeted for degradation through the ubiquitin-proteasome system (UPS) (Yi et al. 2007). Notably, regulation of the UPS is thought to underlie the molecular pathogenesis of retinal aging (Kapphahn et al. 2007). Similarly, the Ran-binding domains of NUP358 that interact with Ran-GTP are linked to an age-related increase in proteasome-related proteins in the retina and the degeneration of cone photoreceptor structures (Patil et al. 2019). Retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1) is a critical component of cone and rod photoreceptor cells and localizes to particular foci around NPCs, suggesting its role in nuclear transport (Castagnet et al. 2003; Roepman et al. 2005). Moreover, NUP358 mediates the nucleocytoplasmic shuttling of RPGRIP1s and their interaction with other partners in amacrine and 661 W neurons, indicating its role in the pathogenesis of the retina (Castagnet et al. 2003). Neurologically, NUP358 also associates selectively with the axon initial segment (AIS) of mature neurons through interactions with ankyrin-G, an essential AIS scaffold protein (Khalaf et al. 2019). Additionally, NUP358 interacts with dishevelled and atypical protein kinase C through its N-terminal region, which is crucial for neuronal polarization during cell migration (Vyas et al. 2013) (Fig. 3, Table 2).
The conflicting roles of NUP358, a tumor suppressor and carcinogenic protein, in cancer have been previously reported. It has been reported that NUP358 is upregulated in cervical cancer cells, which potentiates the growth, migration, and invasion of cervical cancer cells (Wang et al. 2021b). In HCC, NUP358 directly promotes CCAAT/enhancer-binding protein alpha (CEBPα) SUMOylation and degradation, affecting the balance of the O-GlcNAcylation enzymes O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), subsequently triggering more O-GlcNAcylation events for oncogenic proteins (Liu et al. 2021c). Moreover, NUP358 contributes to obesity-associated protein (FTO) SUMOylation at K216, which promotes FTO degradation and subsequently promotes HCC tumorigenesis (Liu et al. 2020). Similarly, NUP358 regulates cyclin-dependent kinase inhibitor 1 B (CDKN1B) nuclear-cytoplasmic translocation by promoting CDKN1B SUMOylation at K73, thereby facilitating cholangiocarcinoma cell proliferation (Yang et al. 2017). In contrast, NUP358 suppresses CIN and tumorigenesis by maintaining its proper localization to the inner centromeres of Topo IIα during mitosis via SUMOylation (Dawlaty et al. 2008). Furthermore, NUP358 facilitates SUMOylation of promyelocytic leukemia protein (PML) transcript variant IV, favoring PML nuclear body formation and PML-IV function, and therefore plays a tumor suppressive role in CRC cells (Satow et al. 2012). Similarly, NUP358 interacts with a small heterodimer partner (SHP) at NE and mediates its SUMOylation at K68, consequently maintaining bile acid homeostasis and preventing bile acid-related liver injury, suggesting its inhibition of HCC tumorigenesis in the early stage (Kim et al. 2016). Interestingly, Nup358 interacts with P-element-induced wimpy testis (Piwi) and promotes Piwi entry into the nucleus, thereby silencing transposons and regulating genomic instability in Drosophila (Munafò et al. 2021), which could reveal its contradictory effects on cancers. In addition, NUP358 has been found to have gene fusions (NUP358-ALK) in some cancers, such as inflammatory myofibroblastic tumors (Ma et al. 2003; Mariño-Enríquez et al. 2011) and myeloid malignancies (Röttgers et al. 2010; Murakami et al. 2018; Hergott et al. 2023). The NUP358-ALK fusion occurs because of a rearrangement within chromosome (chr) 2q13, resulting in constitutive, ligand-independent autophosphorylation and activation of the receptor tyrosine kinase ALK, which promotes uncontrolled cell growth and leads to tumor formation (Ma et al. 2003; Mariño-Enríquez et al. 2011; Lee et al. 2017) (Fig. 3 and Table 2).
To date, these mutations in NUP358 (c.3363G>T, p.K1121N; c.128A>T, p.D43V; c.5249C>G, p.P1750R; c.1754C>T, p.T585M; c.1350A>T, p.L450F) have been identified and are believed to contribute to the development of acute necrotizing encephalopathy type 1 (ANE1) (Neilson et al. 2009; Jiang et al. 2022; Bashiri et al. 2020; Chew and Ngu 2020). Mechanistically, these mutations could cause overproduction of proinflammatory cytokines, including interleukin 6 (IL6) and tumor necrosis factor-α (TNF-α), which are associated with ANE1 (Shen et al. 2021). Specifically, individuals with missense mutations in the N-terminal region of NUP358 (mainly T585M, T653I, I656V, and T681C) secrete excessive amounts of cytokines, leading to a cytokine storm in response to viral infections, which causes neuropathology, seizures, coma, and even mortality (Shen et al. 2021). Notably, these ANE1-associated mutations impair the interaction of ANE1 with TNRC6/GW182 proteins and impair miRNA function, which may contribute to the development of ANE1 (Shen et al. 2021; Deshmukh et al. 2021) (Fig. 3, Table 2).
NUP98
The human NUP98 gene encodes a 186 kDa precursor that undergoes autoproteolytic cleavage to generate NUP98 and NUP96 (Fontoura et al. 1999). One of the distinctive features of NUP98 is that it can undergo a process known as "autoproteolysis," indicating that it can self-cleave into two functionally distinct parts (Sun and Guo 2008), underscoring the complex function of NUP98 within the cell. The N-terminal portion is involved in the formation of NPCs (Chatel et al. 2012), whereas the C-terminal portion is involved in gene regulation and chromatin structure (Cross and Powers 2011). Physiologically, NUP98 participates in bidirectional transport across NPCs (Miorin et al. 2020), regulates gene expression in cooperation with DExH-Box helicase 9 (DHX9) (Capitanio et al. 2017), and restricts viral infection (Nunzio et al. 2013). Alterations in NUP98, including translocations and mutations, have been associated with various forms of leukemia and other cancers (Michmerhuizen et al. 2020). NUP98 fusion with various partners due to chromosomal translocations can lead to aberrant gene regulatory mechanisms, thereby contributing to oncogenesis.
NUP98 gene fusions are notable abnormalities associated with various hematologic malignancies, particularly AML, CML, MLL, and myelodysplastic syndromes (Struski et al. 2017; Shima et al. 2017; Hatano et al. 1999). The N-terminal portion of NUP98 and the C-terminal portion of its fusion partner are universally involved in NUP98 fusion oncoproteins (Michmerhuizen et al. 2020). Previous studies have summarized the role of NUP98 fusion in hematologic malignancies (Michmerhuizen et al. 2020; Sump and Brickner 2017). Similarly, NUP98 exerts a carcinogenic effect by promoting galectin-3 cytoplasmic translocation (Funasaka et al. 2013) and regulating protein synthesis (Pulianmackal et al. 2022). Although the oncogenic properties of NUP98 fusion proteins in hematologic malignancies are well characterized, NUP98 plays an opposite role in other cancers. In HCC, NUP98 regulates the post-transcriptional expression of select p53 target genes and exerts its cancer-inhibitory effects (Singer et al. 2012) (Fig. 3, Table 2).
GLE1
In human, GLE1 is a protein that shuttles between the nucleus and the cytoplasm and is known to regulate ATP-dependent DEAD-box RNA helicases (e.g., DDX1 and DDX19) in the cytoplasm; these proteins are involved in mRNA export, translation initiation, transcription termination, and stress granule dynamics (Alcázar-Román et al. 2010; Suntharalingam et al. 2004; Sharma and Wente 2020; Gray et al. 2022; Kendirgi et al. 2003) (258). Both human GLE1 isoforms exhibit diffuse cytoplasmic localization, but GLE1b is also localized to NE and NPCs (Rayala et al. 2004). In terms of transportation, GLE1b forms a heterotrimeric complex with Nup155 and CG1 in vitro and is required for the export of 70-kDa heat shock protein (HSP70) mRNA from the nucleus to the cytoplasm (Kendirgi et al. 2005, 2003). A previous study revealed that GLE1 aggregates through its N-terminal structural domain (a coiled-coil domain and a 10-amino acid aggregation-prone region) in a phosphorylation-dependent manner, and that the ability of GLE1 to self-polymerize is essential for it to perform multiple functions (Mason and Wente 2020). Interestingly, the coiled-coil structural domain and aggregation-prone region are additive for efficient mRNA export and stress granule formation, whereas the two self-polymerizing domains are independently required for the regulation of translation under cellular stress in the case of polymerization between GLE1a and GLE1b isoforms (Mason and Wente 2020). In contrast, GLE1 self-polymerization is not required for GLE1 phosphorylation or translation initiation under nonstress conditions (Mason and Wente 2020). Therefore, the polymerization state and localization of GLE1 endow it with diverse functions.
Mutations in GLE1 cause two recessive subtypes of arthrogryposis multiplex congenita (AMC), a condition characterized by joint contractures at birth (Nousiainen et al. 2008). The two AMC subtypes related to GLE1 are lethal congenital contracture syndrome 1 (LCCS1) and lethal arthrogryposis with anterior horn cell disease (LAAHD) (Nousiainen et al. 2008). LCCS1 is a severe autosomal recessive fetal motor neuron disease that leads to severe congenital contractures, which are joint deformities present at birth and often result in prenatal or early postnatal death (Nousiainen et al. 2008; Seytanoglu et al. 2016; Whittle et al. 2021). Individuals with LAAHD present with a similar, albeit milder, phenotype with diminished fetal mobility and contractures prenatally and postnatal respiratory failure, which can result in perinatal death (Vuopala and Herva 1994). The key difference in survival between LCCS1 and LAAHD may be due to the nature of GLE1 mutations and the resulting residual function of the GLE1 protein. The GLE1 mutations in the two AMC subtypes (c.432-10A >G, p.T144_E145ins; c.1706G4A, p.R569H; c.1849G4A, p.V617M; c.2051T4C, p.I684T) involve the coiled-coil domain and CG1-binding domain (Nousiainen et al. 2008; Paakkola et al. 2018). The T144_E145ins mutation in GLE1 is believed to abolish GLE1 oligomerization and shuttling, which disrupts the efficient nuclear export of mRNA at NPCs (Folkmann et al. 2013). Furthermore, other mutations (c.1706G4A, p.R569H; c.1849G4A, p.V617M; c.2051T4C, p.I684T) localized in the CG1-binding domain that interacts with CG1 to promote mRNA export (Kendirgi et al. 2005). Notably, the homozygous I684T mutation impaired the nuclear localization of GLE1, confirming its pathogenic role (Paakkola et al. 2018). Given that a previous study found that Gle1 plays a role in Schwann cells development (Seytanoglu et al. 2016), mutations in human GLE1 may also lead to defects in neuronal development. GLE1 is involved in the cellular response to stress through SGs, which temporarily store mRNAs when cells are under stress (Aditi et al. 2019, 2015). Presumably, mutations in GLE1 could impair this process, increasing the vulnerability of cells, particularly neurons, to stress and potentially contributing to the pathogenesis of AMC. Recently, the phenotypic spectrum of GLE1-related disorders has been extended to ALS and a mild congenital form resembling congenital myopathy (Kaneb et al. 2015; Tan et al. 2017). In addition, GLE1 may play a crucial role in the molecular etiology of giant cell tumors of the bone through direct interaction with miR-376a-3p (Fellenberg et al. 2016) (Fig. 3, Table 2).
RAE1
In the nucleus, human RAE1 interacts with other NUPs and mRNA export factors to facilitate the efficient export of mRNA molecules (Zheng et al. 2019; Addetia et al. 2021; Ren et al. 2010). Specifically, RAE1 directly contributes to the nuclear export of mRNAs by anchoring to a specific NUP98 motif (GLEBS-like motif) in NPCs (Ren et al. 2010; Pritchard et al. 1999). However, RAE1 can directly interact with other proteins (e.g., nuclear mitotic apparatus (NuMA), structural maintenance of chromosome protein 1 (SMC1), and importin β) not only in the nucleus but also in the cytoplasm, playing a critical role in mitotic bipolar spindle formation and assembly (Wong et al. 2006; Wong and Blobel 2008; Blower et al. 2005). In addition, RAE1 acts as a downstream regulatory target of the Hippo/SWH signaling pathway to promote mitotic cell cycle progression (Jahanshahi et al. 2016). Intriguingly, when the Hippo/SWH signaling pathway is inactive, RAE1 acts independently of the transcriptional coactivator Yorkie (Yki) gene to increase organ size by promoting mitotic S-phase entry and increasing cellular proliferation (Jahanshahi et al. 2016). When the Hippo/SWH signaling pathway is activated, it inhibits the activity of RAE1 in a WTS-dependent manner to restrict organ growth, suggesting its ability to regulate pathway homeostasis (Jahanshahi et al. 2016). Moreover, Drosophila Rae1 regulates synaptic growth in postmitotic motor neurons by binding to the evolutionarily conserved highwire (Hiw) and protecting Hiw from autophagy-mediated degradation (Tian et al. 2011). Similarly, another study showed that Rae-1 loss-of-function mutations cause axon and synapse defects by disrupting cooperation between GLO-4 and FSN-1 in Caenorhabditis elegans (Grill et al. 2012). Furthermore, one study showed that RAE1 and NUP98 maintain progenitor function through HDAC-dependent chromatin targeting to escape nucleolar localization (Neely et al. 2023).
Given the role of RAE1 in mRNA transport and spindle assembly, alterations in its expression or function may contribute to the genomic instability observed in cancer cells. There is evidence to suggest that RAE1, through its interaction with NuMA and the mitotic checkpoint protein Bub3, plays a role in maintaining proper chromosome segregation during mitosis, and disruptions in this process can lead to aneuploidy, a characteristic of many tumors (Wong et al. 2006; Stockum et al. 2018; Babu et al. 2003; Jeganathan et al. 2006; Funasaka et al. 2011). Therefore, in terms of maintaining genomic stability, RAE1 exerts tumor-suppressive effects. However, other studies have shown that RAE1 promotes cancer progression by regulating peroxisome proliferator-activated receptor alpha (PPARα)-mediated lipid metabolism (He et al. 2023) and regulating EMT signals (Oh et al. 2017, 2019). During viral infection, the matrix protein of vesicular stomatitis virus inhibits nuclear export of host cell mRNAs by binding to the mRNA export factor RAE1 (Quan et al. 2014). Similarly, ORF6, one of the proteins encoded by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), reduced interactions with NUP98-RAE1 and consequently impaired immune evasion (Kehrer et al. 2023) (Fig. 3, Table 2).
Inner Ring Complex
In yeast, a core feature of IR is the Z-shaped Nup188-Nup192 complex located in the middle layer, which is flanked by two roughly parallel rhomboidal structures in the inner and outer layers (Li et al. 2022). Nup188, Nup192, and Nic96 are pivotal for linking all subunits within the inner ring (IR) monomer, contributing to its stability in yeast (Li et al. 2022). At the heart of the IR subunit, a homodimer of Nup205 is central, with a pair of Nup188 molecules situated on either side of Xenopus laevis (Huang et al. 2022). This arrangement is further stabilized by four Nup93 molecules, each inserting an extended helix into the axial groove of either Nup205 or Nup188, forming part of the central scaffold of NPCs (Huang et al. 2022). Channel Nups comprising a heterotrimer of Nup62/58/54 are anchored to this central scaffold, which is critical for the function of NPC in nuclear transport in Xenopus laevis (Huang et al. 2022). In addition, six Nup155 molecules interacted with the central scaffold (Huang et al. 2022). Together with NDC1-ALADIN heterodimers, they serve to anchor the IR subunit to both the NE and outer rings of the NPCs (Huang et al. 2022).
NUP205
The physiological roles of human NUP205 include regulating cell division by modulating mitotic timing (Bao et al. 2019), enhancing viral gene expression and replication (Lu et al. 2014; Pashkov et al. 2022), and contributing to podocyte homeostasis through the localization and activity of YAP and TAZ proteins (Ester et al. 2023). Additionally, NUP205 facilitates proper cilia function (Marquez et al. 2021; Zhang et al. 2020a), and its expression is predominantly found in the testis and cancer cells, suggesting that it may be an oncogenic marker (Fujitomo et al. 2012). Specifications: c.5984T>C (p. P1995S) disrupts the NUP205-NUP93 interaction, reducing the presence of NUP205 in NPCs (Braun et al. 2016). Together with NUP93 and XPO5, NUP205 modulates mothers against decapentaplegic homolog 4 (SMAD4), which is central to the TGF-β signaling pathway implicated in podocyte damage and SRNS (Ester et al. 2023; Braun et al. 2016; Song et al. 2022; Rahbar Saadat et al. 2020). The nuclear import of the crucial TFs YAP and TAZ mediated by NUP205 in podocytes highlights its potential involvement in the pathogenesis of SRNS through the Hippo pathway (Ester et al. 2023).
In gliomass, elevated NUP205 expression is correlated with reduced patient survival (Liang et al. 2023). In CRC, NUP93, NUP188 and NUP205, are enriched at HOXA promoters, and their overexpression is associated with leukemia proliferation, invasion, and metastasis (Paço et al. 2020; Abuhantash et al. 2021; Labade et al. 2016). Furthermore, NUP205 has been shown to promote cell growth in lung cancer through its interaction with transmembrane protein 209 (TMEM209), which stabilizes NUP205 and regulates c-Myc nuclear entry (Fujitomo et al. 2012). Additional studies have implicated NUP205 in the progression and oncogenesis of nasopharyngeal carcinoma (Yue et al. 2020), AML (Bao et al. 2019), and papillary thyroid carcinoma (Xia et al. 2020) (Fig. 4, Table 3).

Summary of the physiological and pathological effects of each component of the inner ring complex and the central channel in humans
NUP188
Research on human NUP188 has revealed its multifaceted roles in cellular function and development, with implications for the understanding of human diseases. In Drosophila models, knockout of Nup188 has been shown to cause motor deficits and seizure susceptibility, partially mirroring the neurological symptoms observed in humans with NUP188-related conditions (Muir et al. 2020). In addition, this phenotype is further characterized by abnormal dendrite tiling, highlighting its potential role for NUP188 in the development of dendritic structures (Muir et al. 2020). Interestingly, another study revealed that knockdown of human NUP188 or its binding partner NUP93 results in the loss of cilia during embryonic development, while NPC function remains largely unaffected (Viso et al. 2016). Additionally, NUP188 has been implicated in centriole duplication, a key event in cell division. NUP188 surrounds centrioles at cilia bases, functions as a component of the pericentriolar material, and is necessary for the proper duplication of centrioles, which has implications for understanding the mechanisms underlying congenital heart diseases (Viso et al. 2016; Vishnoi et al. 2020). The direct interaction between NUP188 and the centrosomal protein 152 kDa (Cep152), as well as its role in functioning at or upstream of the spikele assembly abnormal protein 6 homolog (Sas6), underscores its essential role in cell division (Vishnoi et al. 2020). NUP188 also contributes to mitotic chromosome alignment by promoting K-fiber formation and recruiting NuMA to the spindle poles, which is crucial for accurate chromosome segregation (Itoh et al. 2013). While investigating the Nup93 complex in Xenopus laevis, which includes Nup188 and Nup205, researchers found that these Nups are not essential for NPC formation (Theerthagiri et al. 2010). However, nuclei lacking Nup188 grew several-fold in size compared to those of the wild type (Theerthagiri et al. 2010). This dramatic change is due to the accelerated translocation of integral membrane proteins through NPCs, suggesting that Nup188 restricts the passage of these proteins, thereby maintaining NE homeostasis (Theerthagiri et al. 2010). Clinically, severe developmental syndromes are characterized by hypotonia, congenital cataracts, microcephaly, abnormal brain imaging, and early death due to respiratory failure, usually within the first year after birth, and are caused by homozygous nonsense mutations in the human NUP188 gene (Sandestig et al. 2020) (Fig. 4, Table 3).
NUP155
In Xenopus, Nup155 is involved in the fusion and formation of NEs by promoting the accumulation of Nups at the nuclear periphery (Franz et al. 2005). Additionally, proper formation of NPCs requires a self-inhibitory interaction with Nup155 in Xenopus (Magistris et al. 2018). The function of Nup155 relies on its structure, which includes the β-propeller domain that anchors the protein to the NPC and the α-solenoid region, which is essential for the correct localization of INM proteins such as the lamin-B receptor and otefin in Drosophila (Busayavalasa et al. 2012). Additionally, the α-solenoid of Nup155 exhibits chromatin-binding activity, which intensifies at the end of mitosis (Busayavalasa et al. 2012). Another study revealed that the depletion of Xenopus Nup155 in vivo resulted in the failure of nuclear lamina formation and defects in chromosome segregation at anaphase (Franz et al. 2005). Furthermore, human NUP155 may play a role in mental and developmental retardation associated with hemizygous deletions of the 5p13 region, given the genomic location of NUP155 (Zhang et al. 1999).
Mutations in human NUP155 have been linked to specific phenotypes including atrial fibrillation (AF), a common cardiac arrhythmia (Zhang et al. 2008; Preston et al. 2018). A homozygous mutation, R391H, in NUP155 co-segregates with AF and affects the nuclear localization of NUP155, reducing NE permeability (Zhang et al. 2008). Interestingly, another study demonstrated that the lamin A/C mutation p. R399C weakens the interaction between lamin A/C and NUP155, leading to AF (Han et al. 2019). This finding suggests that NUP155 functions upstream of AF at the molecular level during the pathogenesis of AF. Additionally, NUP155 is a p53 repression target that regulates p21 mRNA translation in HCC (Holzer et al. 2019) (Fig. 4 and Table 3).
NUP93
In the assembly and function of NPCs, Nup93 functions by non-selectively binding to various Nup62-containing heterotrimers (Madheshiya et al. 2022). In addition, the conserved amino-terminal domain of Nup93 is crucial for anchoring the central channel to NPCs (Sonawane et al. 2020). In humans, the interaction of NUP93 with CCCTC-binding factor (CTCF) influences the spatial and temporal dynamics of the HOXA locus during cellular differentiation, indicating its role in gene regulation (Labade et al. 2021). Notably, the repression of HOXA by NUP93 was aided by its interacting partners NUP188 and NUP205 (Labade et al. 2016). Additionally, another study revealed that NUP93 directly activates genes with high levels of RNA polymerase II loading and transcriptional elongation by recruiting BRD4 to their active enhancers (Zhu et al. 2022). In cardiovascular research, NUP93 deficiency has been linked to impaired endothelial NPC transport, leading to nuclear retention of YAP and subsequent exacerbation of YAP transcription, while exacerbating hypoxia-induced cardiomyocyte death and inflammation (Nguyen et al. 2024; Pan et al. 2023). In addition, Nup93 can indirectly promote IRF3 nuclear translocation by enhancing serine/threonine-protein kinase TBK1 activity and regulating antiviral innate immunity in mice (Monwan et al. 2020).
In the context of BC and cervical cancer, overexpression of NUP93 has been shown to enhance cancer growth by stimulating cell proliferation and remodeling of the actin cytoskeleton and Ras homology (Rho) family proteins (Bersini et al. 2020; Ouyang et al. 2019). Pathological mutations in NUP93 have been implicated in early-onset, severe kidney diseases, such as SRNS and focal segmental glomerulosclerosis (FSGS), with studies revealing its essential role in kidney cell function (Braun et al. 2016; Bierzynska et al. 2022; Dhanorkar et al. 2023; Hashimoto et al. 2019; Wasilewska et al. 2023). Mechanistically, NUP93 mutations abrogated the interaction with SMAD4, causing aberrant SMAD signaling in SRNS (Braun et al. 2016). At the cellular level, NUP93 is ubiquitously expressed in various cell types within the human kidney, and its altered expression due to mutations could elucidate the pathogenic mechanisms of SRNS and FSGS (Hashimoto et al. 2019). Interestingly, NUP93 intronic variants (c.2137-18G>A) cause SRNS by leading to aberrant splicing and protein mislocalization, thereby expanding the clinical understanding of rare genetic variants underlying the disease (Rossanti et al. 2019) (Fig. 4, Table 3).
NUP53
Nup53, also known as Nup35, is a pivotal component of NPCs and exhibits a high degree of evolutionary conservation across species (Chen et al. 2018b). Structurally, Nup53 is a highly conserved RNA-recognition motif (RRM) domain in mice, which is implicated in its ability to form homodimers (Handa et al. 2006). This dimerization is not only a structural feature but is also critical for membrane interactions and its functional role in NPC assembly (Vollmer et al. 2012). Intriguingly, a point mutation within the RRM domain of mouse Nup53 (p.F192L) leads to severe megacolon in mice, indicating its significant role in the pathology of degenerative myopathy (Parish et al. 2016). In addition to its structural roles, human NUP53 has been shown to selectively regulates intracellular pH homeostasis in cardiomyocytes by post-transcriptionally controlling the expression of sodium-hydrogen exchanger 1 (NHE1) (Xu et al. 2015b). This regulation underscores a novel functional dimension of NUP53 that connects NPC components to the regulation of cellular physiological processes.
Nup53 plays a critical role in maintaining NPC structure and nuclear integrity during the interphase in vertebrate cells (Eisenhardt et al. 2014). During mouse cell meiosis, Nup53 functions as a novel microtubule-associated protein that is important for the architecture of the oocyte meiotic spindle (Chen et al. 2018b). Its role is particularly crucial during rapid cellular divisions that occur during early embryogenesis, emphasizing its fundamental contribution to NE formation and function (Ródenas et al. 2009). The involvement of Nup53 in mitotic regulation is further underscored by its phosphorylation by two mitotic kinases, Cdk1p and Hrr25p, in yeast (Lusk et al. 2007). Furthermore, the interaction of Xenopus Nup53 with the transmembrane protein Ndc1 is vital for NPC assembly (Eisenhardt et al. 2014). Ndc1 and Nup53 also cooperatively regulate NPC density and nuclear size in early C. elegans embryos (Mauro et al. 2022) (Fig. 4 and Table 3).
Central channel
FG repeats of the human NUP62 complex, which consists of NUP62, NUP58, and NUP54, are located on the IR plane of the central channel (Beck and Hurt 2017). During entry into mitosis, the human NUP62 complex dissolves through acetylation of NUP62 at K432 by the histone acetyltransferase TIP60, orchestrating the correct spindle orientation (Akbar et al. 2022). Additionally, the NUP62 complex anchors PLK1 to NE, facilitating NE breakdown (Martino et al. 2017). The FG repeat domains of FG-NUPs transiently bind to nuclear transport receptors via low-affinity interactions, allowing them to pass through NPCs alone or with bound cargo (Rexach and Blobel 1995). Specifically, the NUP62 complex mainly mediates the nuclear import of cargo with unconventional nuclear localization sequences (Chen et al. 2023). Genetically, the promoters of Nup54, Nup58, and Nup62 exhibit rapid accumulation of insertions/deletions (indexes) in Drosophila (McQuarrie et al. 2023), suggesting that they are vital for safeguarding genomic stability. Coincidently, another study showed that depletion of NUP54, NUP62, and NUP58 increased cell sensitivity to ionizing radiation (Rodriguez-Berriguete et al. 2018).
NUP58
Human NUP58 originates from the nucleoporin-like protein 1 (NUPL1) gene and results from alternative mRNA splicing, which differs only in unstructured regions (Hu and Gerace 1998; Ishikawa et al. 1997). Physiologically, NUP58 contributes to the maintenance of proper mitosis (Hartono et al. 2019; Li et al. 2014) and amyloid formation (Danilov et al. 2021, 2023). Moreover, one study reported that, in the initial response to the reduced functionality of NUP58, cells exhibit physiological adaptation without genetic changes (Targa et al. 2021). However, the cells appeared to develop genetic adaptations that specifically addressed the primary impairment via focal amplification of NUP58 and restoration of mutant protein expression (Targa et al. 2021) (Fig. 4).
In cancers, NUP58 regulates the glycogen synthase kinase 3 beta/zinc finger protein Snai1 (GSK-3β/Snail) signaling pathway (Shi et al. 2019) and bipolar spindle formation (Li et al. 2014), thereby exerting its carcinogenic effect. Additionally, NUP58 may be a potential target for proximal tubular injury (Tang et al. 2023). NUP58 is closely associated with amyloid formation (Danilov et al. 2021, 2023). Amyloid deposits are associated with different diseases, among which the most prominent are Aβ (Alzheimer's disease), α-synuclein (Parkinson's disease), amylin (type 2 diabetes), and huntingtin with polyQ expansion (Huntington's disease) (Iadanza et al. 2018). Indeed, a previous study demonstrated that NUP58 interacts with huntingtin fragments in a yeast 2-hybrid assay; thus, the underlying mechanism should be studied in the future (Kaltenbach et al. 2007) (Fig. 4, Table 3).
NUP54
To date, the physiological roles of human NUP54 include regulating nuclear translocation (Wang et al. 2022), maintaining genome integrity (Rodriguez-Berriguete et al. 2018), and promoting the proliferation of pulmonary arterial smooth muscle cells (Yang et al. 2009). Additionally, Nup54 is specifically expressed in previtellogenic oocytes in zebrafish, which may be related to its differentiation effects (Gautier et al. 2011). Furthermore, NUP54 and other central channel NUPs exhibited high expression levels in the brain, which is consistent with the critical role of NPCs in neurogenesis and neural maintenance (Thul and Lindskog 2018; Guglielmi et al. 2020) (Fig. 4).
Diseases associated with NUP54 include early onset dystonia (Harrer et al. 2023), cancers (Huang et al. 2010; Wang et al. 2022), psoriasis (Li et al. 2015), and ALS (Shi et al. 2017; Khosravi et al. 2017). Dystonia-associated patients had homozygous or compound-heterozygous missense and in-frame deletion variants in NUP54, all of which were located near the C-terminal end of protein (Harrer et al. 2023). Thus, these mutations in NUP54 (c.1073T > G, p.I358S; c.1126A > G, p.K376E; c.1410_1412del, p.Q471del; c.1414G > A, p.E472K; c.1420C > T, p.L474F) may lead to destabilization of the interactions between functionally related NUPs and disassembly of the central channel (Harrer et al. 2023). In cancers, NUP54 induces nuclear import of coactivator-associated arginine methyltransferase 1 (CARM1) and, consequently, transcriptional activation and neurogenic locus notch homolog protein 2 (Notch2) methylation, thereby accelerating GC cell proliferation and tumorigenesis (Wang et al. 2022). However, NUP54-depleted cells also exhibited increased formation of chromosomal aberrations arising from replicated DNA (Rodriguez-Berriguete et al. 2018), suggesting that Nup54 may also have a cancer suppressive effect by maintaining gene stability. Consistently, a previous study demonstrated that NUP54 mediates downstream critical protein phosphorylation through recruitment and direct interaction with PLK1, leading to NE disassembly and consequently, proper chromosome segregation (Martino et al. 2017). In addition, loss of heterozygosity in NUP54 is associated with tumor size in patients with HCC (Huang et al. 2010). In ALS, proline:arginine (PRn) poly-dipeptides, caused by bidirectional transcription and ATG-independent translation of the expanded (GGGGCC)n repeat, bind to NUP54 and inhibit the movement of macromolecules into and out of the nucleus (Shi et al. 2017). Interestingly, the PRn poly-dipeptide did not bind to the unstructured monomeric form of FG repeats, but did bind to labile, amyloid-like FG polymers (Shi et al. 2017), indicating that the amyloid-like fibers formed by NUP54 enhance the toxicity of the PRn poly-dipeptide. However, a previous study showed that NUP54 can restore the proper nuclear localization of TDP-43 by alleviating the negative nuclear transport effects caused by another poly-dipeptide protein, poly-GA (Khosravi et al. 2017), suggesting that NUP54 has paradoxical pathological mechanisms through interactions with multiple poly-dipeptides in ALS (Fig. 4, Table 3).
Nuclear basket
The human nuclear basket extends into the nucleoplasm, a substance within the nucleus. The main components of the nuclear basket include NUP153, TPR, NUP50, and ZC3HC1 (Beck and Hurt 2017; Appen et al. 2015; Gunkel et al. 2021). These proteins form distinct subcomplexes and create a lattice-like structure, similar to an actual basket, hanging from the nuclear side of NPCs (Nakielny et al. 1999). Functionally, the nuclear basket plays a pivotal role in the nuclear transport process, affecting both the import and export stages in human (Nakielny et al. 1999; Schertzer et al. 2023; Buffone et al. 2018). Moreover, the nuclear basket is implicated in chromatin organization and gene regulation in humans (Lelek et al. 2015; Kadota et al. 2020), suggesting that its influence extends beyond transport, and it may also participate in the spatial organization of the RNA processing machinery, thus playing a role in the quality control and sorting of RNAs destined for export to the cytoplasm (Nakielny et al. 1999; Ball et al. 2007). Notably, the nuclear basket is connected to the NPC central framework and can interact with the nuclear lamina and chromatin (Smythe et al. 2000; Cobb et al. 2016), further underscoring its multifunctional nature.
NUP153
In addition to being an important component of NPCs, human NUP153 is necessary for the assembly and anchoring of NPCs (Walther et al. 2001), and also acts as a docking site for the import of karyopherin (Moroianu et al. 1997). Structurally, it contains three distinct domains: an N-terminal region containing RNA-binding and pore-targeting domains (Ball et al. 2004), a central region containing multiple zinc finger motifs (Fahrenkrog et al. 2002), and a C-terminal region containing multiple FG repeats (Fahrenkrog et al. 2002). Human Nup153 has also been shown to directly interact with sentrin-specific protease 1/2 (SENP1/2) (Hang and Dasso 2002; Chow et al. 2012), importin α (Ogawa et al. 2012), importin β (Shah et al. 1998), TPR (Hase and Cordes 2003), and regulator of telomere elongation helicase 1 (RTEL1) (Schertzer et al. 2023) (Fig. 5).

Summary of physiological and pathological effects of each component of the transmembrane nucleoporin nuclear basket in humans
Previous studies on human NUP153 have focused on nuclear transport (Shen et al. 2023a), human immunodeficiency virus (HIV) host integration (Bester et al. 2020; Xue et al. 2023), and the maintenance of genome integrity (Moudry et al. 2012; Mackay et al. 2017). Recently, growing evidence has indicated that NUP153 is closely associated with tumorigenesis. NUP153 is aberrantly expressed in various cancers, including CRC (Wu et al. 2019), HCC (Gan et al. 2022), BC (Zhou and Panté 2010), PCa (Re et al. 2018), and thyroid cancer (Ma et al. 2021). NUP153 exerts a carcinogenic effect by promoting cell motility (Zhou and Panté 2010), regulating the c-Myc/P15 axis (Gan et al. 2022), and facilitating the nuclear translocation of nitric oxide synthase 3 (eNOS) and estrogen receptor beta (ERβ) (Re et al. 2018). However, the overexpression of NUP153 negatively regulates Wnt/β-catenin signaling and suppresses the proliferation of CRC cells (Wu et al. 2019). Moreover, a previous study revealed that enhancer-specific chromatin structure and organization regulation by mammalian NUP153 is another underlying mechanism of cancer (Kadota et al. 2020). During viral infection, NUP153 interacts with the HIV-1 capsid protein, contributing to the entry of the intact capsid into the nucleus (Buffone et al. 2018; Shen et al. 2023a). However, in influenza viruses and herpesviruses, loss of function of NUP153 facilitates the nuclear import of viral proteins for viral DNA replication and assembly (Mühlbauer et al. 2015; Chang et al. 2012, 2015) (Fig. 5).
TPR
Human nucleoporin TPR is a large protein of ~ 267 kDa that contains a coiled-coil N-terminal domain capable of forming homodimers and a highly post-translationally modified C-terminus (Hase et al. 2001). The N-terminal domain of TPR mediates intranuclear attachment to NUP153 (Hase and Cordes 2003; McCloskey et al. 2018), whereas its C-terminal domain facilitates nuclear import (Snow et al. 2013). Nucleoporin TPR forms the nuclear basket of NPCs and is vital for the enrichment of open chromatin around NPCs (Uhlířová et al. 2021). TPR plays multiple roles in the nucleus, including regulating the TREX-2-dependent mRNA export pathway (Aksenova et al. 2020; Lee et al. 2020), export of unspliced RNA (Rajanala and Nandicoori 2012), intranuclear transport of poly(A)+ RNA (Shibata et al. 2002), nuclear export sequence-dependent nuclear export of proteins (David-Watine 2011), and provision of a scaffold for extracellular signal-regulated kinase 2 (ERK2) (Vomastek et al. 2008) and c-Myc (Su et al. 2018). In addition, it participates in establishing nuclear-peripheral chromatin compartmentalization (Krull et al. 2010) and mitotic spindle checkpoint signaling during mitosis (Liu et al. 2023a; Lee et al. 2008) (Fig. 5).
Pathologically, TPR has been implicated in cancer through several types of abnormalities, including chromosomal translocations that generate fusion proteins with several receptor tyrosine kinases (Greco et al. 1997; Choi et al. 2014; Lee et al. 2021), regulation of nuclear translocation of RNAs (Kosar et al. 2021; Chen et al. 2021), and point mutations found in several types of cancer (Forbes et al. 2017; Moon et al. 2021); however, the underlying mechanism is unclear. One well-studied example is the TPR-MET oncogene, which arises from a chromosomal rearrangement that fuses the TPR gene with the MET proto-oncogene (Cooper et al. 1984; Soman et al. 1990; Yu et al. 2000; Liang et al. 1996), leading to the constitutive activation of the tyrosine kinase domain of MET and consequently activating the c-Jun N-terminal kinase (JNK) pathway, driving uncontrolled cell growth, and contributing to oncogenesis (Pal et al. 2017; Rodrigues et al. 1997). Similarly, other studies have reported the fusion of TPR with kinases, including tropomyosin receptor kinase (TRK) (Greco et al. 1992), neurotrophic tyrosine receptor kinase 1 (NTRK1) (Greco et al. 1997; Russell et al. 2000; Créancier et al. 2015; Rekhi et al. 2021; Roccato et al. 2005), fibroblast growth factor receptor 1 (FGFR1) (Kim et al. 2014; Malli et al. 2016; Qiu et al. 2017; Li et al. 2012), and ALK (Choi et al. 2014). TPR promotes transfer RNA (tRNA) nuclear export and protein synthesis in lung cancer cells (Chen et al. 2021), suggesting an oncogenic role in this context. In CC, TPR further supports oncogenesis by promoting mitotic processes and cell proliferation through its interaction with GSK3β (Dewi et al. 2018). TPR mutations have been identified in microsatellite instability CC, with specific examples (c.6381delT, p. F2127LfsX65 and c.6381dupT, p. D2128X), which may contribute to the development of cancer (Moon et al. 2021), indicating that these mutations could be function-enhancing. However, loss of heterozygosity and homozygous deletion of the TPR locus have been observed in human GC, which suggests that loss of function contributes to tumorigenesis in this context (Cunningham et al. 1997). Interestingly, TPR interacts with Mad 1/2 and dynein light chain to promote proper chromosome segregation during mitosis, highlighting its role in genomic stability (Lee et al. 2008; Nakano et al. 2010). Moreover, TPR depletion has been shown to induce mitotic catastrophe and increase the rate of tetraploidy and polyploidy, likely due to disruption of its interaction with Aurora A kinase (Kobayashi et al. 2015). These findings show that depending on the maintenance of genetic stability, TPR may also play a role in cancer inhibition. The TPR also plays a role in the response to cellular stress and senescence. Silencing of TPR elicits a senescent-like phenotype in cancer cells (David-Watine 2011). During oncogene-induced senescence, TPR is essential for both the formation and maintenance of internal senescence-associated heterochromatin foci, and this process is controlled by nuclear pore density (Boumendil et al. 2019). Intriguingly, another study showed that TPR depletion affected the level and function of the SUMO protease SENP2, thus affecting SUMOylation regulation at the nuclear pore and overall SUMOylation in the cell, which has broad implications for nuclear transport and cell regulation (David-Watine 2011). Furthermore, TPR protects cells from RNA-mediated replication stress by regulating heat-shock transcription factor 1 (HSF1) mRNA trafficking, maintaining mTORC1 activity to phosphorylate Unc-51-like kinase 1 (ULK1), and preventing macroautophagy/autophagy induction in ependymoma (Kosar et al. 2021; Dewi et al. 2021). In addition to its cellular functions, TPR has been implicated in viral infection, as it is required for viral mRNA export, distinct from other basket NUPs, such as NUP153 and NUP50 (Bhat et al. 2023) (Fig. 5, Table 4).
NUP50
The human nucleoporin NUP50, also known as Npap60, is a key component of NPCs (Lindsay et al. 2002a). NUP50 is characterized by a motif with FG repeats and is known for its interaction with importin α/β, which is essential for the import of proteins containing a nuclear localization signal (NLS) (Park et al. 2016). Located on the nuclear side of NPCs, NUP50 interacts with the nuclear transporter importin α/β (Lindsay et al. 2002b). The function of NUP50 extends beyond merely chaperoning the import complex through NPC. It is also critical for disassembling the importin α/β-cargo complex and recycling importin (Ogawa et al. 2010a; Pumroy et al. 2012). The N-terminal region of NUP50 is particularly crucial for its function in NPC assembly, where it binds to the regulator of chromosome condensation 1 (RCC1) and stimulates guanine nucleotide exchange factor (GEF) activity toward Ran. This interaction is proposed to increase local concentrations of RanGTP, which could stimulate mitotic NPC assembly (Lindsay et al. 2002b; Ogawa et al. 2010b; Matsuura and Stewart 2005; Moore 2003) (483). Interestingly, the function of NUP50 in NPC assembly is sufficiently robust that excess RCC1 can compensate for NUP50 depletion, indicating that NUP50, while essential, is not required for NPC formation (Holzer et al. 2021). In addition to its roles in nucleocytoplasmic transport and NPC assembly, Nup50 impacts various cellular processes, including cell cycle control and DNA repair. NUP50 has been identified as an interactor of CDKN1B, and disruption of this interaction can lead to cytosolic accumulation of CDKN1B and defects in cell proliferation (Müller et al. 2000). In terms of development, NUP50 is weakly expressed in most tissues but is highly expressed in the developing neural tube and adult testes (Smitherman et al. 2000; Park et al. 2016). Its deficiency can cause embryonic lethality, neural tube abnormalities, exencephaly, and intrauterine growth retardation in mice, indicating its importance in embryonic development (Smitherman et al. 2000; Park et al. 2016) (Fig. 5).
NUP50 is strongly associated with ALS pathogenesis in neurological diseases. The expansion of GGGGCC repeats in the non-coding region of C9orf72 is the most common cause of sporadic and familial forms of ALS and frontotemporal dementia (Megat et al. 2023). Loss of Nup50 enhances the toxicity of GGGGCC repeats in Drosophila (Freibaum et al. 2015). In addition, another study demonstrated that mutations in NUP50 associated with ALS can decrease the expression of NUP50 and disrupt its normal function, linking NUP50 and ALS (Megat et al. 2023). Previous studies have shown that NUP50 knockdown can cause cytoplasmic inclusions of nuclear pore components and RanGAP1 (Zhang et al. 2015; Chou et al. 2018), impairing the nuclear pore function. RanGAP1 is a key protein that regulates nucleocytoplasmic shuttling, and its aberrant localization and expression have been observed in ALS patients (Gasset-Rosa et al. 2019; Li et al. 2017). In addition, alterations to the nuclear pore are sufficient to cause dysfunction of TDP-43 in both human neurons and ALS patients (Coyne et al. 2021; Lek et al. 2016). In bladder cancer, NUP50 promotes the nuclear translocation of the SWI/SNF complex subunit SMARCC1, facilitating cell migration and proliferation (Wei et al. 2022). However, NUP50 can promote the recruitment of DNA double-strand break repair factor 53BP1 to DNA repair foci by antagonizing BRCA1-dependent events (Mackay et al. 2017) (Fig. 5, Table 4).
ZC3HC1
ZC3HC1, also known as the nuclear-interacting partner of ALK (NIPA), is a protein that is encoded by the ZC3HC1 gene on chr 7 in humans (Zhang et al. 2000). Recent findings have shown that ZC3HC1 constitutes a novel and intrinsic element of the nuclear basket ubiquitously present at the NE in a diverse array of cells, encompassing both proliferating and non-dividing terminally differentiated cell types of various morphogenetic lineages (Gunkel et al. 2021; Gunkel and Cordes 2022). In the context of its molecular function, ZC3HC1 is instrumental in the assembly of the SCF (NIPA) E3 ubiquitin ligase complex (Bassermann et al. 2005). This complex is pivotal for the temporal regulation of the cell cycle, specifically orchestrating the transition to mitosis by mediating ubiquitination and the subsequent proteasomal degradation of Cyclin B1 (Bassermann et al. 2005). The regulatory influence of ZC3HC1 extends to the maintenance of NE integrity (Gunkel et al. 2021) and is thought to contribute to broader nuclear activities (Gunkel et al. 2021; Gunkel and Cordes 2022), underscoring its versatility within the cellular environment. Pathologically, ZC3HC1 is associated with an increased risk of coronary artery disease by regulating Cyclin B1 stability (Mega et al. 2015; Jones et al. 2016; Jafaripour et al. 2019). Additionally, a single nucleotide polymorphism in ZC3HC1 is associated with increased carotid intima-media thickness in patients with rheumatoid arthritis (López-Mejías et al. 2013; Linseman et al. 2017). Furthermore, certain ZC3HC1 polymorphisms are involved in the incidence of hypertension (Ma et al. 2019a; Kunnas and Nikkari 2015), suggesting that this gene has a far-reaching impact on vascular health. Overall, the emerging portrait of ZC3HC1 is that of a multifaceted protein integral to cell cycle progression and nuclear architecture, and is potentially implicated in various disease states, meriting further investigation to fully delineate its biological roles and therapeutic potential (Fig. 5, Table 4).
Transmembrane nucleoporins
Human transmembrane nucleoporins, including POM121, GP210, and NDC1, play important roles in the selective transport of macromolecules across the NE through NPCs and carry the onus of anchoring the NPCs at the NE and ER (Eisenhardt et al. 2014; Bindra and Mishra 2021; Mansfeld et al. 2006). In addition to their transport function, human transmembrane nucleoporins are involved in multiple cell processes, including the regulating of gene expression (Franks et al. 2016), cell differentiation (Gomez-Cavazos and Hetzer 2015; Sharma et al. 2014), and NE assembly (Güttinger et al. 2009).
POM121
POM121 is encoded by the tandem gene locus POM121A/C on human chromosome 7q11.23 (Funakoshi et al. 2007). Notably, this chr region is associated with the inherited developmental disease Williams–Beuren syndrome (Osborne and Mervis 2007). Structurally, POM121 contains a single-pass N-terminal membrane-spanning domain that directly binds to the β-propeller regions of NUP155 and NUP160, an NLS that interacts with importin α/β, and a C-terminal cytoplasmic/nucleoplasmic domain with FG repeats that aid cargo carriage (Mitchell et al. 2010; Funakoshi et al. 2011). Notably, specific regions within POM121, specifically the FG repeats and an adjacent α-helix structure, are critical for its role in mediating the nuclear import of HIV-1 pre-integration complex (Guo et al. 2018). In terms of transport, POM121 is known to facilitate the nucleocytoplasmic transport of several TFs, including E2F1, c-Myc, the endothelial transcription factor GATA2, proliferator-activated receptor alpha gamma (PPARγ), and AR (Rodriguez-Bravo et al. 2018; Yu et al. 2024). Interestingly, a recent study revealed that POM121 is not only a structural component of the nuclear pore, but also has a chaperone-like capacity to facilitate the signaling required for transcription factor EB (TFEB)-mediated autophagy (Wang et al. 2023c) (Figure 5).
In various cancer types, including CRC (Wang et al. 2020), lung cancer (Guan et al. 2021; Zhang et al. 2020b), laryngocarcinoma (Zhao et al. 2020), PCa (Becker et al. 2022), GC (Kang et al. 2023), and oral SCC (Ma et al. 2019b), the oncoprotein POM121 is universally overexpressed and plays a role in modulating key signaling pathways. In lung cancer (Guan et al. 2021; Zhang et al. 2020b) and GC (Kang et al. 2023), POM121 overexpression has been associated with abnormal activation of important signaling pathways, such as the TGF-β/SMAD and PI3K/AKT pathways. In addition, it promotes PCa tumor growth by enhancing the nuclear transport of oncogenic TFs, such as E2F1 and c-Myc, as well as PCa-specific factors, such as AR and GATA2 (Rodriguez-Bravo et al. 2018). Furthermore, POM121 expression increases with tumor progression, suggesting that POM121 may be a target molecule for patients with advanced PCa (Becker et al. 2022). In triple-negative breast cancer (TNBC), overexpressed POM121 facilitates cell drug resistance (Tang et al. 2022c). Regarding the immune response, POM121 has been shown to suppress macrophage inflammatory responses by selectively inhibiting the nuclear accumulation of NF-κB (Ge et al. 2019) (Fig. 5, Table 4).
In C9orf72-associated ALS, there is a reduction in the levels of POM121 and consequently other NUPs in induced pluripotent stem cell-derived neurons (Coyne et al. 2020). The reduction in POM121 was believed to result from expanded C9orf72 amyotrophic lateral sclerosis overlapping with frontotemporal dementia (ALS/FTD) repeat RNA alone (Coyne et al. 2020). This reduction appears to initiate a cascade of events leading to cellular toxicity, which may contribute to neurodegeneration observed in ALS. In addition to the aberrant expression of POM121, other studies have shown that the normal function of POM121 in the transport of TFEB (Wang et al. 2023c; Lin et al. 2024), which is vital for the autophagy-lysosome pathway (Brady et al. 2018), is impaired in C9orf72-ALS. Autophagy is a cellular process for degrading and recycling cellular components, and its dysfunction is a feature of several neurodegenerative diseases, including ALS (Chua et al. 2022). Additionally, the sigma-1 receptor (SIGMAR1) facilitates the nuclear transport of TFEB by interacting with POM121 (Wang et al. 2023c). However, pathological hexanucleotide repeat expansion disrupts this interaction in C9orf72-ALS models, leading to impaired autophagy (Wang et al. 2023c). Interventions that enhance SIGMAR1 activity or directly increase POM121 function, such as the use of SIGMAR1 agonists such as pridopidine or fluvoxamine, have shown promise in restoring normal nucleocytoplasmic transport and autophagy, and may provide therapeutic benefits for patients with ALS (Wang et al. 2023c). In summary, POM121 has been implicated in a wide range of cellular processes, from viral infection to cancer progression and neurodegeneration (Fig. 5, Table 4).
GP210
Human GP210, a transmembrane protein, was the first nucleoporin identified and has been described as having tissue-specific expression, particularly within epithelial cells (Wozniak and Blobel 1992; Olsson et al. 1999). Its expression is not static; gp210 is dynamically regulated during cellular differentiation, as evidenced in mouse myoblasts (Sakuma et al. 2022). Although initially not expressed in these cells, gp210 expression markedly increases during the mouse myogenic differentiation process, highlighting its critical role in the proper function and maintenance of skeletal muscle integrity (Sakuma et al. 2022). Consistently, the N-terminal domain of gp210, which projects into the perinuclear space, has been identified as a mediator of mouse muscle cell differentiation in vitro, and is believed to be vital for NE and ER homeostasis (Gomez-Cavazos and Hetzer 2015). In addition to its role in muscle differentiation, GP210 is involved in fundamental processes of nuclear pore formation and dilation (Cohen et al. 2003). Additionally, gp210 is implicated in NPC disassembly and NE breakdown during embryonic cell division (Galy et al. 2008), which is pivotal for ensuring proper cell cycle progression. Interestingly, the presence of Drosophila gp210 in an abundant non-nuclear form during embryogenesis suggests that it may play a key role in facilitating the swift assembly of nuclei, which is critical during the rapid cellular division and differentiation stages of development (Berrios et al. 1995). Moreover, gp210 is dispensable for the incorporation of other Nups, such as Pom121 or Nup107, and for maintaining the stability of NPCs in mouse cell (Eriksson et al. 2004) (Fig. 5).
The relevance of human GP210 extends to its clinical applications, particularly in the context of autoimmune diseases. It has been implicated in endometriosis (Cipollini et al. 2019) and more notably, in primary biliary cirrhosis (PBC) (Nakamura et al. 2005, 2006; Wang et al. 2023b), a chronic liver condition. The detection of antibodies against GP210 is a strong indicator of PBC, and antibody titers against its C-terminal peptide serve as a clinical parameter for monitoring the disease (Nakamura et al. 2005; Wang et al. 2023b). Moreover, the heightened expression of GP210 on the NE of biliary epithelial cells in PBC patients might be associated with inflammatory damage that contributes to the autoimmune response, potentially leading to the progression of liver failure in this disease (Nakamura et al. 2006) (Fig. 5, Table 4). These insights into the functions of GP210 and its association with disease states underscore its potential as a biomarker and target for therapeutic intervention.
NDC1
Human NDC1 is a multifunctional protein that plays a crucial role in the structural and functional organization of NE. NDC1, as a component of NPCs, plays a key role in assembly (Stavru et al. 2006) and anchoring of NPCs to the NE via interactions with NUP53 and ALADIN (Huang et al. 2022; Mansfeld et al. 2006). In addition, Ndc1 acts within the NE to mediate the insertion of the nascent spindle pole body in yeast, which is critical during cell division (Winey et al. 1993; Chen et al. 2014). Aberrations in the functions of Ndc1, such as overexpression, have been shown to cause spindle pole body duplication defects in yeast, highlighting its importance in maintaining cellular integrity (Chial et al. 1999). Moreover, loss-of-function mutations in Ndc1 can lead to severe developmental consequences, such as high mortality rates in organisms such as Caenorhabditis elegans, due to NPC defects (Stavru et al. 2006). In the broader context of cellular architecture, the role of Ndc1 extends to regulating NPC density and nuclear size in the elegans embryo, acting in concert with the Nup53 and membrane biogenesis pathways (Mauro et al. 2022). In humans, SEPT12-NDC1 complexes have been implicated in spermiogenesis in mammals (Lai et al. 2016), emphasizing the versatility of NDC1. The reach of the protein also extends to vesicle trafficking, where it has been suggested that ARRDC5 could influence the transport and localization of mouse Ndc1, alongside other proteins vital for the formation of the sperm head–tail coupling apparatus (Liu et al. 2023c). Furthermore, human NDC1's direct interaction with ALADIN is not only a structural requirement, but also critical in disease, as mutations in ALADIN that disrupt this interaction are implicated in the pathogenesis of triple-A syndrome (Yamazumi et al. 2009; Kind et al. 2009) (Fig. 5).
The relevance of NDC1 is also evident in the context of oncology. It is implicated in the promotion of tumorigenesis in HCC by enhancing the PI3K/AKT signaling pathway in cooperation with B-cell receptor-associated protein 31 (BCAP31) (Liu et al. 2024). Moreover, compared with those in normal tissues, the expression levels of NDC1 in CC tissues were significantly elevated, with higher expression levels correlating with increased patient survival (Liu et al. 2021b). Conversely, elevated NDC1 is a marker of poor prognosis in HCC (Liu et al. 2023b, 2024) and pancreatic cancer (Shen et al. 2023b), suggesting its dual role in cancer biology. In summary, NDC1 is a key player in the maintenance of cellular structure and function, with roles ranging from cell division to spermiogenesis, and from vesicle trafficking to disease pathogenesis (Fig. 5, Table 4). The diverse roles of this protein in various cancers further illustrate its importance as a focal point for both the understanding and treatment of these diseases.
Conclusion and perspective
Human NPCs serve as critical gateways that regulate the flow of molecules between the nucleus and cytoplasm, and play a pivotal role in numerous cellular processes. Their importance extends far beyond their barrier function, as they are actively involved in gene regulation, RNA processing, and cell cycle progression. The intricate structure and function of NPCs are essential for maintaining cellular homeostasis, and disruptions in their components can lead to a range of human diseases. Cancers, for instance, have been linked to NPC dysfunction, where alterations in nucleocytoplasmic transport may contribute to the aberrant growth and division of cells (Gan et al. 2022; Nofrini et al. 2016; Fujitomo et al. 2012). Neurodegenerative conditions such as ALS/FTD have also been associated with dysregulation of human NUPs (Coyne et al. 2020; Gasset-Rosa et al. 2019; Khalil et al. 2022), indicating a broader impact of NPC integrity on neuronal health. Furthermore, pathogens, such as viruses, exploit NPCs to propagate and disrupt cellular integrity (Sadasivan et al. 2022; Quan et al. 2014). Moreover, inherited conditions such as triple-A syndrome underscore the critical nature of NPC components in human biology (Yamazumi et al. 2009; Kind et al. 2009; Osborne and Mervis 2007). The insights gained into NPCs could revolutionize our approach for the diagnosis and treatment of diseases. In addition, new NPC components, such as the newly discovered ZC3HC1, still need further discovery (Gunkel et al. 2021; Gunkel and Cordes 2022). This provides a new direction for fully understanding NPC functions. Research is increasingly focused on unraveling the complex molecular mechanisms underlying NPC functions. This could pave the way for identifying therapeutic targets within NPC assembly and transport pathways. The potential of NPCs as biomarkers is also significant, as changes in NPC function could signal the early onset of the disease or track its progression.
Currently, there are no drugs that specifically target NPC components. However, certain drugs that interfere with transport factors dependent on NPCs have been identified. One such example is importazole, a 2,4-diamino quinazoline compound that disrupts the interaction between importin β and NPCs (Soderholm et al. 2011). This disruption effectively inhibits the nuclear import of cargo proteins while leaving transportin-mediated nuclear import or CRM1-mediated nuclear export unaffected (Soderholm et al. 2011). Another compound, leptomycin B, is a natural compound that selectively binds to the nuclear export signal-binding region of CRM1 (Rahmani and Dean 2017). Leptomycin B decreases the nuclear availability of CRM1 by promoting the redistribution of CRM1 from the nucleus to the cytoplasm (Rahmani and Dean 2017). Additionally, a previous study demonstrated that 5-fluorouracil disrupts nuclear export and NPC permeability in a calcium-dependent manner, which has been associated with drug resistance (Cristoferi et al. Mar 2021). Therefore, further investigations of drugs targeting specific components of NPCs are promising for refining treatment strategies.
Availability of data and materials
Not applicable.
References
Abuhantash M, Collins EM, Thompson A (2021) Role of the HOXA cluster in HSC emergence and blood cancer. Biochem Soc Trans 49(4):1817–1827. https://doi.org/10.1042/BST20210234
Addetia A, Lieberman NAP, Phung Q et al (2021) SARS-CoV-2 ORF6 disrupts bidirectional nucleocytoplasmic transport through interactions with Rae1 and Nup98. Mbio. https://doi.org/10.1128/mBio.00065-21
Aditi, Folkmann AW, Wente SR (2015) Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell 26(8):1476–1490. https://doi.org/10.1091/mbc.E14-11-1523
Aditi, Mason AC, Sharma M, Dawson TR, Wente SR (2019) MAPK- and glycogen synthase kinase 3-mediated phosphorylation regulates the DEAD-box protein modulator Gle1 for control of stress granule dynamics. J Biol Chem 294(2):559–575. https://doi.org/10.1074/jbc.RA118.005749
Ageberg M, Drott K, Olofsson T, Gullberg U, Lindmark A (2008) Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47(4):276–287. https://doi.org/10.1002/gcc.20531
Akbar H, Cao J, Wang D et al (2022) Acetylation of Nup62 by TIP60 ensures accurate chromosome segregation in mitosis. J Mol Cell Biol. https://doi.org/10.1093/jmcb/mjac056
Aksenova V, Smith A, Lee H et al (2020) Nucleoporin TPR is an integral component of the TREX-2 mRNA export pathway. Nat Commun 11(1):4577. https://doi.org/10.1038/s41467-020-18266-2
Alanee S, Delfino K, Wilber A, Robinson K, Brard L, Semaan A (2017) Single nucleotide variant in Nucleoporin 107 may be predictive of sensitivity to chemotherapy in patients with ovarian cancer. Pharmacogenet Genom 27(7):264–269. https://doi.org/10.1097/fpc.0000000000000288
Alcázar-Román AR, Bolger TA, Wente SR (2010) Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. J Biol Chem 285(22):16683–16692. https://doi.org/10.1074/jbc.M109.082370
Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM (2003) Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 160(3):341–353
Ball JR, Dimaano C, Ullman KS (2004) The RNA binding domain within the nucleoporin Nup153 associates preferentially with single-stranded RNA. RNA 10(1):19–27
Ball JR, Dimaano C, Bilak A, Kurchan E, Zundel MT, Ullman KS (2007) Sequence preference in RNA recognition by the nucleoporin Nup153. J Biol Chem 282(12):8734–8740
Banerjee HN, Gibbs J, Jordan T, Blackshear M (2010) Depletion of a single nucleoporin, Nup107, induces apoptosis in eukaryotic cells. Mol Cell Biochem 343(1–2):21–25. https://doi.org/10.1007/s11010-010-0494-6
Bao XL, Zhang L, Song WP (2019) LncRNA SNHG1 overexpression regulates the proliferation of acute myeloid leukemia cells through miR-488-5p/NUP205 axis. Eur Rev Med Pharmacol Sci 23(13):5896–5903. https://doi.org/10.26355/eurrev_201907_18334
Barr FA, Silljé HHW, Nigg EA (2004) Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5(6):429–440
Basel-Vanagaite L, Muncher L, Straussberg R et al (2006) Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol 60(2):214–222
Bashiri FA, Al Johani S, Hamad MH et al (2020) Acute necrotizing encephalopathy of childhood: a multicenter experience in Saudi Arabia. Front Pediatr 8:526. https://doi.org/10.3389/fped.2020.00526
Bassermann F, von Klitzing C, Münch S et al (2005) NIPA defines an SCF-type mammalian E3 ligase that regulates mitotic entry. Cell 122(1):45–57
Batliwalla FM, Li W, Ritchlin CT et al (2005) Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol Med 11(1–12):21–29
Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18(2):73–89. https://doi.org/10.1038/nrm.2016.147
Becker F, Offermann A, Roesch MC et al (2022) Up-regulation of POM121 is linked to prostate cancer aggressiveness and serves as a prognostic biomarker. Urol Oncol 40(8):380.e11-380.e18. https://doi.org/10.1016/j.urolonc.2022.05.019
Bernad R, Engelsma D, Sanderson H, Pickersgill H, Fornerod M (2006) Nup214-Nup88 nucleoporin subcomplex is required for CRM1-mediated 60 S preribosomal nuclear export. J Biol Chem 281(28):19378–19386
Berrios M, Meller VH, McConnell M, Fisher PA (1995) Drosophila gp210, an invertebrate nuclear pore complex glycoprotein. Eur J Cell Biol 67(1):1–7
Bersini S, Lytle NK, Schulte R, Huang L, Wahl GM, Hetzer MW (2020) Nup93 regulates breast tumor growth by modulating cell proliferation and actin cytoskeleton remodeling. Life Sci Alliance. https://doi.org/10.26508/lsa.201900623
Besse B, Candé C, Spano JP et al (2004) Nuclear localization of apoptosis protease activating factor-1 predicts survival after tumor resection in early-stage non-small cell lung cancer. Clin Cancer Res 10(17):5665–5669. https://doi.org/10.1158/1078-0432.Ccr-04-0415
Bester SM, Wei G, Zhao H et al (2020) Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 370(6514):360–364. https://doi.org/10.1126/science.abb4808
Bhat P, Aksenova V, Gazzara M et al (2023) Influenza virus mRNAs encode determinants for nuclear export via the cellular TREX-2 complex. Nat Commun 14(1):2304. https://doi.org/10.1038/s41467-023-37911-0
Bierzynska A, Bull K, Miellet S et al (2022) Exploring the relevance of NUP93 variants in steroid-resistant nephrotic syndrome using next generation sequencing and a fly kidney model. Pediatr Nephrol 37(11):2643–2656. https://doi.org/10.1007/s00467-022-05440-5
Bilokapic S, Schwartz TU (2012) Molecular basis for Nup37 and ELY5/ELYS recruitment to the nuclear pore complex. Proc Natl Acad Sci USA 109(38):15241–15246
Bindra D, Mishra RK (2021) In pursuit of distinctiveness: transmembrane nucleoporins and their disease associations. Front Oncol 11:784319. https://doi.org/10.3389/fonc.2021.784319
Bley CJ, Nie S, Mobbs GW et al (2022) Architecture of the cytoplasmic face of the nuclear pore. Science 376(6598):eabm9129. https://doi.org/10.1126/science.abm9129
Blower MD, Nachury M, Heald R, Weis K (2005) A Rae1-containing ribonucleoprotein complex is required for mitotic spindle assembly. Cell 121(2):223–234
Boer J, Bonten-Surtel J, Grosveld G (1998) Overexpression of the nucleoporin CAN/NUP214 induces growth arrest, nucleocytoplasmic transport defects, and apoptosis. Mol Cell Biol 18(3):1236–1247
Bonnin E, Cabochette P, Filosa A et al (2018) Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet 14(12):e1007845. https://doi.org/10.1371/journal.pgen.1007845
Borlido J, D’Angelo MA (2018) Nup62-mediated nuclear import of p63 in squamous cell carcinoma. EMBO Rep 19(1):3–4. https://doi.org/10.15252/embr.201745497
Borrow J, Shearman AM, Stanton VP et al (1996) The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 12(2):159–167
Boumendil C, Hari P, Olsen KCF, Acosta JC, Bickmore WA (2019) Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev 33(3–4):144–149. https://doi.org/10.1101/gad.321117.118
Brackenbury WJ (2012) Voltage-gated sodium channels and metastatic disease. Channels (austin) 6(5):352–361. https://doi.org/10.4161/chan.21910
Brady OA, Martina JA, Puertollano R (2018) Emerging roles for TFEB in the immune response and inflammation. Autophagy 14(2):181–189. https://doi.org/10.1080/15548627.2017.1313943
Braun DA, Sadowski CE, Kohl S et al (2016) Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48(4):457–465. https://doi.org/10.1038/ng.3512
Braun DA, Lovric S, Schapiro D et al (2018a) Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J Clin Investig 128(10):4313–4328. https://doi.org/10.1172/JCI98688
Braun DA, Lovric S, Schapiro D et al (2018b) Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J Clin Investig 128(10):4313–4328. https://doi.org/10.1172/jci98688
Buffone C, Martinez-Lopez A, Fricke T et al (2018) Nup153 unlocks the nuclear pore complex for HIV-1 nuclear translocation in nondividing cells. J Virol. https://doi.org/10.1128/JVI.00648-18
Bui KH, von Appen A, DiGuilio AL et al (2013) Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155(6):1233–1243. https://doi.org/10.1016/j.cell.2013.10.055
Busayavalasa K, Chen X, Farrants A-KÖ, Wagner N, Sabri N (2012) The Nup155-mediated organisation of inner nuclear membrane proteins is independent of Nup155 anchoring to the metazoan nuclear pore complex. J Cell Sci 125(18):4214–4218. https://doi.org/10.1242/jcs.105809
Capitanio JS, Montpetit B, Wozniak RW (2017) Human Nup98 regulates the localization and activity of DExH/D-box helicase DHX9. Elife. https://doi.org/10.7554/eLife.18825
Carlon-Andres I, Lagadec F, Pied N et al (2020) Nup358 and transportin 1 cooperate in adenoviral genome import. J Virol. https://doi.org/10.1128/JVI.00164-20
Cassany A, Ragues J, Guan T et al (2015) Nuclear import of adenovirus DNA involves direct interaction of hexon with an N-terminal domain of the nucleoporin Nup214. J Virol 89(3):1719–1730. https://doi.org/10.1128/JVI.02639-14
Castagnet P, Mavlyutov T, Cai Y, Zhong F, Ferreira P (2003) RPGRIP1s with distinct neuronal localization and biochemical properties associate selectively with RanBP2 in amacrine neurons. Hum Mol Genet 12(15):1847–1863
Chakraborty P, Wang Y, Wei J-H et al (2008) Nucleoporin levels regulate cell cycle progression and phase-specific gene expression. Dev Cell 15(5):657–667. https://doi.org/10.1016/j.devcel.2008.08.020
Chandra B, Michmerhuizen NL, Shirnekhi HK et al (2022) Phase separation mediates NUP98 fusion oncoprotein leukemic transformation. Cancer Discov 12(4):1152–1169. https://doi.org/10.1158/2159-8290.CD-21-0674
Chang C-W, Lee C-P, Huang Y-H, Yang P-W, Wang J-T, Chen M-R (2012) Epstein–Barr virus protein kinase BGLF4 targets the nucleus through interaction with nucleoporins. J Virol 86(15):8072–8085. https://doi.org/10.1128/JVI.01058-12
Chang C-W, Lee C-P, Su M-T, Tsai C-H, Chen M-R (2015) BGLF4 kinase modulates the structure and transport preference of the nuclear pore complex to facilitate nuclear import of Epstein–Barr virus lytic proteins. J Virol 89(3):1703–1718. https://doi.org/10.1128/JVI.02880-14
Chatel G, Desai SH, Mattheyses AL, Powers MA, Fahrenkrog B (2012) Domain topology of nucleoporin Nup98 within the nuclear pore complex. J Struct Biol 177(1):81–89. https://doi.org/10.1016/j.jsb.2011.11.004
Chen J, Smoyer CJ, Slaughter BD, Unruh JR, Jaspersen SL (2014) The SUN protein Mps3 controls Ndc1 distribution and function on the nuclear membrane. J Cell Biol 204(4):523–539. https://doi.org/10.1083/jcb.201307043
Chen T, Wang D, Xie T, Xu L-G (2018a) Sec13 is a positive regulator of VISA-mediated antiviral signaling. Virus Genes 54(4):514–526. https://doi.org/10.1007/s11262-018-1581-0
Chen F, Jiao X-F, Zhang J-Y et al (2018b) Nucleoporin35 is a novel microtubule associated protein functioning in oocyte meiotic spindle architecture. Exp Cell Res 371(2):435–443. https://doi.org/10.1016/j.yexcr.2018.09.004
Chen J, Wo D, Ma E et al (2019) Deletion of low-density lipoprotein-related receptor 5 inhibits liver cancer cell proliferation via destabilizing nucleoporin 37. Cell Commun Signal 17(1):174. https://doi.org/10.1186/s12964-019-0495-3
Chen M, Long Q, Borrie MS et al (2021) Nucleoporin TPR promotes tRNA nuclear export and protein synthesis in lung cancer cells. PLoS Genet 17(11):e1009899. https://doi.org/10.1371/journal.pgen.1009899
Chen M, Choi S, Wen T et al (2022) A p53-phosphoinositide signalosome regulates nuclear AKT activation. Nat Cell Biol 24(7):1099–1113. https://doi.org/10.1038/s41556-022-00949-1
Chen G, Xu D, Liu Q et al (2023) Regulation of FLC nuclear import by coordinated action of the NUP62-subcomplex and importin β SAD2. J Integr Plant Biol 65(9):2086–2106. https://doi.org/10.1111/jipb.13540
Chew HB, Ngu LH (2020) RANBP2 susceptibility to infection-induced encephalopathy: clinicoradiologic and molecular description in a Malaysian family. Mol Genet Metab Rep 24:100627. https://doi.org/10.1016/j.ymgmr.2020.100627
Chial HJ, Giddings TH, Siewert EA, Hoyt MA, Winey M (1999) Altered dosage of the Saccharomyces cerevisiae spindle pole body duplication gene, NDC1, leads to aneuploidy and polyploidy. Proc Natl Acad Sci USA 96(18):10200–10205
Chien M-L, Lai J-H, Lin T-F, Yang W-S, Juang Y-L (2020) NUP62 is required for the maintenance of the spindle assembly checkpoint and chromosomal stability. Int J Biochem Cell Biol 128:105843. https://doi.org/10.1016/j.biocel.2020.105843
Choi Y-L, Lira ME, Hong M et al (2014) A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol 9(4):563–566. https://doi.org/10.1097/JTO.0000000000000093
Chou C-C, Zhang Y, Umoh ME et al (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21(2):228–239. https://doi.org/10.1038/s41593-017-0047-3
Chow K-H, Elgort S, Dasso M, Ullman KS (2012) Two distinct sites in Nup153 mediate interaction with the SUMO proteases SENP1 and SENP2. Nucleus 3(4):349–358
Chua JP, De Calbiac H, Kabashi E, Barmada SJ (2022) Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 18(2):254–282. https://doi.org/10.1080/15548627.2021.1926656
Cibulka J, Bisaccia F, Radisavljević K, Gudino Carrillo RM, Köhler A (2022) Assembly principle of a membrane-anchored nuclear pore basket scaffold. Sci Adv 8(6):eabl6863. https://doi.org/10.1126/sciadv.abl6863
Cigdem S, Saito S, Nishikata D, Nagata K, Okuwaki M (2021) SET-NUP214 and MLL cooperatively regulate the promoter activity of the HoxA10 gene. Genes Cells 26(10):830–837. https://doi.org/10.1111/gtc.12886
Cipollini M, Luisi S, Piomboni P et al (2019) Functional polymorphism within NUP210 encoding for nucleoporin GP210 is associated with the risk of endometriosis. Fertil Steril. https://doi.org/10.1016/j.fertnstert.2019.04.011
Cobb AM, Larrieu D, Warren DT et al (2016) Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell 15(6):1039–1050. https://doi.org/10.1111/acel.12506
Cohen M, Feinstein N, Wilson KL, Gruenbaum Y (2003) Nuclear pore protein gp210 is essential for viability in HeLa cells and Caenorhabditis elegans. Mol Biol Cell 14(10):4230–4237
Cooper CS, Park M, Blair DG et al (1984) Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311(5981):29–33
Cooper HJ, Tatham MH, Jaffray E et al (2005) Fourier transform ion cyclotron resonance mass spectrometry for the analysis of small ubiquitin-like modifier (SUMO) modification: identification of lysines in RanBP2 and SUMO targeted for modification during the E3 autoSUMOylation reaction. Anal Chem 77(19):6310–6319
Cordes VC, Reidenbach S, Franke WW (1996) Cytoplasmic annulate lamellae in cultured cells: composition, distribution, and mitotic behavior. Cell Tissue Res 284(2):177–191
Coyne AN, Zaepfel BL, Hayes L et al (2020) G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron. https://doi.org/10.1016/j.neuron.2020.06.027
Coyne AN, Baskerville V, Zaepfel BL et al (2021) Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abe1923
Créancier L, Vandenberghe I, Gomes B et al (2015) Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett 365(1):107–111. https://doi.org/10.1016/j.canlet.2015.05.013
Cristoferi L, Gerussi A, Invernizzi P (2021) Anti-gp210 and other anti-nuclear pore complex autoantibodies in primary biliary cholangitis: what we know and what we should know. Liver Int 41(3):432–435. https://doi.org/10.1111/liv.14791
Cross MK, Powers MA (2011) Nup98 regulates bipolar spindle assembly through association with microtubules and opposition of MCAK. Mol Biol Cell 22(5):661–672. https://doi.org/10.1091/mbc.E10-06-0478
Culjkovic-Kraljacic B, Baguet A, Volpon L, Amri A, Borden KLB (2012) The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep 2(2):207–215. https://doi.org/10.1016/j.celrep.2012.07.007
Cunningham JD, Schwartz GK, Karpeh M et al (1997) Loss of heterozygosity and homozygous deletion of the tpr locus in human gastric cancer. Am J Surg 173(6):521–522
Danilov LG, Moskalenko SE, Matveenko AG et al (2021) The human NUP58 nucleoporin can form amyloids in vitro and in vivo. Biomedicines. https://doi.org/10.3390/biomedicines9101451
Danilov LG, Sukhanova XV, Rogoza TM et al (2023) Identification of new FG-repeat nucleoporins with amyloid properties. Int J Mol Sci. https://doi.org/10.3390/ijms24108571
David-Watine B (2011) Silencing nuclear pore protein Tpr elicits a senescent-like phenotype in cancer cells. PLoS ONE 6(7):e22423. https://doi.org/10.1371/journal.pone.0022423
Dawlaty MM, Malureanu L, Jeganathan KB et al (2008) Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 133(1):103–115. https://doi.org/10.1016/j.cell.2008.01.045
De Magistris P, Tatarek-Nossol M, Dewor M, Antonin W (2018) A self-inhibitory interaction within Nup155 and membrane binding are required for nuclear pore complex formation. J Cell Sci. https://doi.org/10.1242/jcs.208538
Del Viso F, Huang F, Myers J et al (2016) Congenital heart disease genetics uncovers context-dependent organization and function of nucleoporins at cilia. Dev Cell 38(5):478–492. https://doi.org/10.1016/j.devcel.2016.08.002
Deshmukh P, Singh A, Khuperkar D, Joseph J (2021) Acute necrotizing encephalopathy-linked mutations in Nup358 impair interaction of Nup358 with TNRC6/GW182 and miRNA function. Biochem Biophys Res Commun 559:230–237. https://doi.org/10.1016/j.bbrc.2021.04.027
Dewi FRP, Domoto T, Hazawa M et al (2018) Colorectal cancer cells require glycogen synthase kinase-3β for sustaining mitosis via translocated promoter region (TPR)-dynein interaction. Oncotarget 9(17):13337–13352. https://doi.org/10.18632/oncotarget.24344
Dewi FRP, Jiapaer S, Kobayashi A et al (2021) Nucleoporin TPR (translocated promoter region, nuclear basket protein) upregulation alters MTOR-HSF1 trails and suppresses autophagy induction in ependymoma. Autophagy 17(4):1001–1012. https://doi.org/10.1080/15548627.2020.1741318
Dey G, Baum B (2021) Nuclear envelope remodeling during mitosis. Curr Opin Cell Biol 70:67–74. https://doi.org/10.1016/j.ceb.2020.12.004
Dhanorkar MA, Rao SN, Malhotra KP, Ansari M, Chandra A (2023) Novel NUP93 gene variant and diffuse mesangial sclerosis in steroid resistant nephrotic syndrome. Indian J Pediatr. https://doi.org/10.1007/s12098-023-04887-0
Di Nunzio F, Fricke T, Miccio A et al (2013) Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology. https://doi.org/10.1016/j.virol.2013.02.008
Ding B, Sepehrimanesh M (2021) Nucleocytoplasmic transport: regulatory mechanisms and the implications in neurodegeneration. Int J Mol Sci. https://doi.org/10.3390/ijms22084165
Eisenhardt N, Redolfi J, Antonin W (2014) Interaction of Nup53 with Ndc1 and Nup155 is required for nuclear pore complex assembly. J Cell Sci 127(Pt 4):908–921. https://doi.org/10.1242/jcs.141739
Endlich N, Kress KR, Reiser J et al (2001) Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol 12(3):413–422. https://doi.org/10.1681/asn.V123413
Engelsma D, Bernad R, Calafat J, Fornerod M (2004) Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J 23(18):3643–3652
Enninga J, Levy DE, Blobel G, Fontoura BMA (2002) Role of nucleoporin induction in releasing an mRNA nuclear export block. Science 295(5559):1523–1525
Enninga J, Levay A, Fontoura BMA (2003) Sec13 shuttles between the nucleus and the cytoplasm and stably interacts with Nup96 at the nuclear pore complex. Mol Cell Biol 23(20):7271–7284
Eriksson C, Rustum C, Hallberg E (2004) Dynamic properties of nuclear pore complex proteins in gp210 deficient cells. FEBS Lett 572(1–3):261–265
Esaki K, Yoshinaga S, Tsuji T et al (2014) Structural basis for the binding of the membrane-proximal C-terminal region of chemokine receptor CCR2 with the cytosolic regulator FROUNT. FEBS J 281(24):5552–5566. https://doi.org/10.1111/febs.13096
Ester L, Cabrita I, Ventzke M et al (2023) The role of the FSGS disease gene product and nuclear pore protein NUP205 in regulating nuclear localization and activity of transcriptional regulators YAP and TAZ. Hum Mol Genet 32(22):3153–3165. https://doi.org/10.1093/hmg/ddad135
Fahrenkrog B, Maco B, Fager AM et al (2002) Domain-specific antibodies reveal multiple-site topology of Nup153 within the nuclear pore complex. J Struct Biol 140(1–3):254–267
Faria AMC, Levay A, Wang Y et al (2006) The nucleoporin Nup96 is required for proper expression of interferon-regulated proteins and functions. Immunity 24(3):295–304
Fedorchak GR, Kaminski A, Lammerding J (2014) Cellular mechanosensing: getting to the nucleus of it all. Prog Biophys Mol Biol 115(2–3):76–92. https://doi.org/10.1016/j.pbiomolbio.2014.06.009
Fellenberg J, Sähr H, Kunz P et al (2016) Restoration of miR-127-3p and miR-376a-3p counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells by targeting COA1, GLE1 and PDIA6. Cancer Lett 371(1):134–141. https://doi.org/10.1016/j.canlet.2015.10.039
Fichtman B, Harel T, Biran N et al (2019) Pathogenic variants in NUP214 cause “plugged” nuclear pore channels and acute febrile encephalopathy. Am J Hum Genet 105(1):48–64. https://doi.org/10.1016/j.ajhg.2019.05.003
Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR (2013) Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 155(3):582–593. https://doi.org/10.1016/j.cell.2013.09.023
Fontana P, Dong Y, Pi X et al (2022) Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold. Science 376(6598):eabm9326. https://doi.org/10.1126/science.abm9326
Fontoura BM, Blobel G, Matunis MJ (1999) A conserved biogenesis pathway for nucleoporins: proteolytic processing of a 186-kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J Cell Biol 144(6):1097–1112
Forbes SA, Beare D, Boutselakis H et al (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45(D1):D777–D783. https://doi.org/10.1093/nar/gkw1121
Forler D, Rabut G, Ciccarelli FD et al (2004) RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol Cell Biol 24(3):1155–1167
Fornerod M, van Deursen J, van Baal S et al (1997) The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J 16(4):807–816
Franks TM, Benner C, Narvaiza I et al (2016) Evolution of a transcriptional regulator from a transmembrane nucleoporin. Genes Dev 30(10):1155–1171. https://doi.org/10.1101/gad.280941.116
Franz C, Askjaer P, Antonin W et al (2005) Nup155 regulates nuclear envelope and nuclear pore complex formation in nematodes and vertebrates. EMBO J 24(20):3519–3531
Freibaum BD, Lu Y, Lopez-Gonzalez R et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525(7567):129–133. https://doi.org/10.1038/nature14974
Frey S, Richter RP, Görlich D (2006) FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314(5800):815–817
Frohnert C, Hutten S, Wälde S, Nath A, Kehlenbach RH (2014) Importin 7 and Nup358 promote nuclear import of the protein component of human telomerase. PLoS ONE 9(2):e88887. https://doi.org/10.1371/journal.pone.0088887
Fujita A, Tsukaguchi H, Koshimizu E et al (2018) Homozygous splicing mutation in NUP133 causes Galloway-Mowat syndrome. Ann Neurol 84(6):814–828. https://doi.org/10.1002/ana.25370
Fujitomo T, Daigo Y, Matsuda K, Ueda K, Nakamura Y (2012) Critical function for nuclear envelope protein TMEM209 in human pulmonary carcinogenesis. Cancer Res 72(16):4110–4118. https://doi.org/10.1158/0008-5472.CAN-12-0159
Fukuhara T, Sakaguchi N, Katahira J, Yoneda Y, Ogino K, Tachibana T (2006) Functional analysis of nuclear pore complex protein Nup62/p62 using monoclonal antibodies. Hybridoma (larchmt) 25(2):51–59
Funakoshi T, Maeshima K, Yahata K, Sugano S, Imamoto F, Imamoto N (2007) Two distinct human POM121 genes: requirement for the formation of nuclear pore complexes. FEBS Lett 581(25):4910–4916
Funakoshi T, Clever M, Watanabe A, Imamoto N (2011) Localization of Pom121 to the inner nuclear membrane is required for an early step of interphase nuclear pore complex assembly. Mol Biol Cell 22(7):1058–1069. https://doi.org/10.1091/mbc.E10-07-0641
Funasaka T, Nakano H, Wu Y et al (2011) RNA export factor RAE1 contributes to NUP98-HOXA9-mediated leukemogenesis. Cell Cycle 10(9):1456–1467
Funasaka T, Balan V, Raz A, Wong RW (2013) Nucleoporin Nup98 mediates galectin-3 nuclear-cytoplasmic trafficking. Biochem Biophys Res Commun 434(1):155–161. https://doi.org/10.1016/j.bbrc.2013.03.052
Gaik M, Flemming D, von Appen A et al (2015) Structural basis for assembly and function of the Nup82 complex in the nuclear pore scaffold. J Cell Biol 208(3):283–297. https://doi.org/10.1083/jcb.201411003
Galganski L, Urbanek MO, Krzyzosiak WJ (2017) Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 45(18):10350–10368. https://doi.org/10.1093/nar/gkx759
Galy V, Antonin W, Jaedicke A et al (2008) A role for gp210 in mitotic nuclear-envelope breakdown. J Cell Sci 121(Pt 3):317–328. https://doi.org/10.1242/jcs.022525
Gan C, Zhou K, Li M, Shang J, Liu L, Zhao Q (2022) NUP153 promotes HCC cells proliferation via c-Myc-mediated downregulation of P15INK4b. Dig Liver Dis 54(12):1706–1715. https://doi.org/10.1016/j.dld.2022.02.008
Gasset-Rosa F, Lu S, Yu H et al (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron. https://doi.org/10.1016/j.neuron.2019.02.038
Gautier A, Le Gac F, Lareyre J-J (2011) The gsdf gene locus harbors evolutionary conserved and clustered genes preferentially expressed in fish previtellogenic oocytes. Gene. https://doi.org/10.1016/j.gene.2010.10.014
Ge W, Yue Y, Xiong S (2019) POM121 inhibits the macrophage inflammatory response by impacting NF-κB P65 nuclear accumulation. Exp Cell Res 377(1–2):17–23. https://doi.org/10.1016/j.yexcr.2019.02.021
Gibson JM, Cui H, Ali MY et al (2022) Coil-to-α-helix transition at the Nup358–BicD2 interface activates BicD2 for dynein recruitment. Elife. https://doi.org/10.7554/eLife.74714
Gibson JM, Zhao X, Ali MY, Solmaz SR, Wang C (2023) A structural model for the core Nup358–BicD2 interface. Biomolecules. https://doi.org/10.3390/biom13101445
Gleixner AM, Verdone BM, Otte CG et al (2022) NUP62 localizes to ALS/FTLD pathological assemblies and contributes to TDP-43 insolubility. Nat Commun 13(1):3380. https://doi.org/10.1038/s41467-022-31098-6
Gloerich M, Vliem MJ, Prummel E et al (2011) The nucleoporin RanBP2 tethers the cAMP effector Epac1 and inhibits its catalytic activity. J Cell Biol 193(6):1009–1020. https://doi.org/10.1083/jcb.201011126
Gomez-Cavazos JS, Hetzer MW (2015) The nucleoporin gp210/Nup210 controls muscle differentiation by regulating nuclear envelope/ER homeostasis. J Cell Biol 208(6):671–681. https://doi.org/10.1083/jcb.201410047
Gong Z, Zhu J, Chen J et al (2023) CircRREB1 mediates lipid metabolism related senescent phenotypes in chondrocytes through FASN post-translational modifications. Nat Commun 14(1):5242. https://doi.org/10.1038/s41467-023-40975-7
Gonzalez-Estevez A, Verrico A, Orniacki C, Reina-San-Martin B, Doye V (2021) Integrity of the short arm of the nuclear pore Y-complex is required for mouse embryonic stem cell growth and differentiation. J Cell Sci. https://doi.org/10.1242/jcs.258340
Gorello P, La Starza R, Di Giacomo D et al (2010) SQSTM1-NUP214: a new gene fusion in adult T-cell acute lymphoblastic leukemia. Haematologica 95(12):2161–2163. https://doi.org/10.3324/haematol.2010.029769
Gray S, Cao W, Montpetit B, De La Cruz EM (2022) The nucleoporin Gle1 activates DEAD-box protein 5 (Dbp5) by promoting ATP binding and accelerating rate limiting phosphate release. Nucleic Acids Res 50(7):3998–4011. https://doi.org/10.1093/nar/gkac164
Greco A, Pierotti MA, Bongarzone I, Pagliardini S, Lanzi C, Della PG (1992) TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene 7(2):237–242
Greco A, Miranda C, Pagliardini S, Fusetti L, Bongarzone I, Pierotti MA (1997) Chromosome 1 rearrangements involving the genes TPR and NTRK1 produce structurally different thyroid-specific TRK oncogenes. Genes Chromosomes Cancer 19(2):112–123
Grill B, Chen L, Tulgren ED et al (2012) RAE-1, a novel PHR binding protein, is required for axon termination and synapse formation in Caenorhabditis elegans. J Neurosci 32(8):2628–2636. https://doi.org/10.1523/JNEUROSCI.2901-11.2012
Guan Y, Gao X, Tang Q et al (2019) Nucleoporin 107 facilitates the nuclear export of Scn5a mRNA to regulate cardiac bioelectricity. J Cell Mol Med 23(2):1448–1457. https://doi.org/10.1111/jcmm.14051
Guan L, Zhang L, Wang T et al (2021) POM121 promotes proliferation and metastasis in non-small-cell lung cancer through TGF-β/SMAD and PI3K/AKT pathways. Cancer Biomark 32(3):293–302. https://doi.org/10.3233/CBM-210001
Guglielmi V, Sakuma S, D’Angelo MA (2020) Nuclear pore complexes in development and tissue homeostasis. Development. https://doi.org/10.1242/dev.183442
Gunkel P, Cordes VC (2022) ZC3HC1 is a structural element of the nuclear basket effecting interlinkage of TPR polypeptides. Mol Biol Cell 33(9):ar82. https://doi.org/10.1091/mbc.E22-02-0037
Gunkel P, Iino H, Krull S, Cordes VC (2021) ZC3HC1 is a novel inherent component of the nuclear basket, resident in a state of reciprocal dependence with TPR. Cells. https://doi.org/10.3390/cells10081937
Guo J, Liu X, Wu C et al (2018) The transmembrane nucleoporin Pom121 ensures efficient HIV-1 pre-integration complex nuclear import. Virology 521:169–174. https://doi.org/10.1016/j.virol.2018.06.008
Guo Q, Liu Q, Wang N et al (2022) The function of Nucleoporin 37 on mouse oocyte maturation and preimplantation embryo development. J Assist Reprod Genet 39(1):107–116. https://doi.org/10.1007/s10815-021-02330-x
Gupta S, Kumar M, Chaudhuri S, Kumar A (2022) The non-canonical nuclear functions of key players of the PI3K-AKT-MTOR pathway. J Cell Physiol 237(8):3181–3204. https://doi.org/10.1002/jcp.30782
Güttinger S, Laurell E, Kutay U (2009) Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 10(3):178–191. https://doi.org/10.1038/nrm2641
Hamada M, Haeger A, Jeganathan KB et al (2011) Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J Cell Biol 194(4):597–612. https://doi.org/10.1083/jcb.201102018
Han M, Zhao M, Cheng C et al (2019) Lamin A mutation impairs interaction with nucleoporin NUP155 and disrupts nucleocytoplasmic transport in atrial fibrillation. Hum Mutat 40(3):310–325. https://doi.org/10.1002/humu.23691
Handa N, Kukimoto-Niino M, Akasaka R et al (2006) The crystal structure of mouse Nup35 reveals atypical RNP motifs and novel homodimerization of the RRM domain. J Mol Biol 363(1):114–124
Hang J, Dasso M (2002) Association of the human SUMO-1 protease SENP2 with the nuclear pore. J Biol Chem 277(22):19961–19966
Harrer P, Schalk A, Shimura M et al (2023) Recessive NUP54 variants underlie early-onset dystonia with striatal lesions. Ann Neurol 93(2):330–335. https://doi.org/10.1002/ana.26544
Hartono, Hazawa M, Lim KS, Dewi FRP, Kobayashi A, Wong RW (2019) Nucleoporin Nup58 localizes to centrosomes and mid-bodies during mitosis. Cell Div 14:7. https://doi.org/10.1186/s13008-019-0050-z
Hase ME, Cordes VC (2003) Direct interaction with nup153 mediates binding of Tpr to the periphery of the nuclear pore complex. Mol Biol Cell 14(5):1923–1940
Hase ME, Kuznetsov NV, Cordes VC (2001) Amino acid substitutions of coiled-coil protein Tpr abrogate anchorage to the nuclear pore complex but not parallel, in-register homodimerization. Mol Biol Cell 12(8):2433–2452
Hashimoto T, Harita Y, Takizawa K et al (2019) In vivo expression of NUP93 and its alteration by NUP93 mutations causing focal segmental glomerulosclerosis. Kidney Int Rep 4(9):1312–1322. https://doi.org/10.1016/j.ekir.2019.05.1157
Hashizume C, Nakano H, Yoshida K, Wong RW (2010) Characterization of the role of the tumor marker Nup88 in mitosis. Mol Cancer 9:119. https://doi.org/10.1186/1476-4598-9-119
Hashizume C, Moyori A, Kobayashi A, Yamakoshi N, Endo A, Wong RW (2013) Nucleoporin Nup62 maintains centrosome homeostasis. Cell Cycle 12(24):3804–3816. https://doi.org/10.4161/cc.26671
Hatano Y, Miura I, Nakamura T, Yamazaki Y, Takahashi N, Miura AB (1999) Molecular heterogeneity of the NUP98/HOXA9 fusion transcript in myelodysplastic syndromes associated with t(7;11)(p15;p15). Br J Haematol 107(3):600–604
Hazawa M, Lin D-C, Kobayashi A et al (2018) ROCK-dependent phosphorylation of NUP62 regulates p63 nuclear transport and squamous cell carcinoma proliferation. EMBO Rep 19(1):73–88. https://doi.org/10.15252/embr.201744523
He Y, Li J, Shen L et al (2022) Pan-cancer analysis reveals NUP37 as a prognostic biomarker correlated with the immunosuppressive microenvironment in glioma. Aging (albany N y) 14(2):1033–1047. https://doi.org/10.18632/aging.203862
He L, Zhou X, Liu J et al (2023) RAE1 promotes nitrosamine-induced malignant transformation of human esophageal epithelial cells through PPARα-mediated lipid metabolism. Ecotoxicol Environ Saf 265:115513. https://doi.org/10.1016/j.ecoenv.2023.115513
Hergott CB, Dal Cin P, Hornick JL, Winer ES, Carrasco RD, Kim AS (2023) Characteristic nuclear membrane ALK reactivity in chronic myelomonocytic leukemia with RANBP2-ALK fusion. Am J Hematol 98(2):365–367. https://doi.org/10.1002/ajh.26107
Holzer K, Ori A, Cooke A et al (2019) Nucleoporin Nup155 is part of the p53 network in liver cancer. Nat Commun 10(1):2147. https://doi.org/10.1038/s41467-019-10133-z
Holzer G, De Magistris P, Gramminger C et al (2021) The nucleoporin Nup50 activates the Ran guanine nucleotide exchange factor RCC1 to promote NPC assembly at the end of mitosis. EMBO J 40(23):e108788. https://doi.org/10.15252/embj.2021108788
Hu T, Gerace L (1998) cDNA cloning and analysis of the expression of nucleoporin p45. Gene 221(2):245–253
Huang G-L, Li B-K, Zhang M-Y et al (2010) LOH analysis of genes around D4S2964 identifies ARD1B as a prognostic predictor of hepatocellular carcinoma. World J Gastroenterol 16(16):2046–2054
Huang G, Zhang Y, Zhu X et al (2020) Structure of the cytoplasmic ring of the Xenopus laevis nuclear pore complex by cryo-electron microscopy single particle analysis. Cell Res 30(6):520–531. https://doi.org/10.1038/s41422-020-0319-4
Huang G, Zhan X, Zeng C et al (2022) Cryo-EM structure of the inner ring from the Xenopus laevis nuclear pore complex. Cell Res 32(5):451–460. https://doi.org/10.1038/s41422-022-00633-x
Hussein NA, Rashad MM, Abdou AS, Hussein AM, Mohamed HM (2022) Gene profiling of SEC13, SMAD7, GHRL, long non-coding RNA GHRLOS, HIF-1α in gastric cancer patients. Sci Rep 12(1):6555. https://doi.org/10.1038/s41598-022-10402-w
Hutten S, Flotho A, Melchior F, Kehlenbach RH (2008) The Nup358–RanGAP complex is required for efficient importin alpha/beta-dependent nuclear import. Mol Biol Cell 19(5):2300–2310
Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE (2018) A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol 19(12):755–773. https://doi.org/10.1038/s41580-018-0060-8
Ikliptikawati DK, Hirai N, Makiyama K et al (2023) Nuclear transport surveillance of p53 by nuclear pores in glioblastoma. Cell Rep 42(8):112882. https://doi.org/10.1016/j.celrep.2023.112882
Ishikawa K, Nagase T, Nakajima D et al (1997) Prediction of the coding sequences of unidentified human genes. VIII. 78 new cDNA clones from brain which code for large proteins in vitro. DNA Res 4(5):307–313
Itoh G, Sugino S, Ikeda M et al (2013) Nucleoporin Nup188 is required for chromosome alignment in mitosis. Cancer Sci 104(7):871–879. https://doi.org/10.1111/cas.12159
Jafaripour S, Sasanejad P, Dadgarmoghaddam M, Sadr-Nabavi A (2019) ADAMTS7 and ZC3HC1 share genetic predisposition to coronary artery disease and large artery ischemic stroke. Crit Rev Eukaryot Gene Expr 29(4):351–361. https://doi.org/10.1615/CritRevEukaryotGeneExpr.2019028209
Jagot-Lacoussiere L, Faye A, Bruzzoni-Giovanelli H, Villoutreix BO, Rain JC, Poyet JL (2015) DNA damage-induced nuclear translocation of Apaf-1 is mediated by nucleoporin Nup107. Cell Cycle 14(8):1242–1251. https://doi.org/10.1080/15384101.2015.1014148
Jahanshahi M, Hsiao K, Jenny A, Pfleger CM (2016) The hippo pathway targets Rae1 to regulate mitosis and organ size and to feed back to regulate upstream components merlin, hippo, and warts. PLoS Genet 12(8):e1006198. https://doi.org/10.1371/journal.pgen.1006198
Jeganathan KB, Baker DJ, van Deursen JM (2006) Securin associates with APCCdh1 in prometaphase but its destruction is delayed by Rae1 and Nup98 until the metaphase/anaphase transition. Cell Cycle 5(4):366–370
Jiang F, Wu C, Wang M, Wei K, Wang J (2021) Identification of novel cell glycolysis related gene signature predicting survival in patients with breast cancer. Sci Rep 11(1):3986. https://doi.org/10.1038/s41598-021-83628-9
Jiang J, Wang YE, Palazzo AF, Shen Q (2022) Roles of nucleoporin RanBP2/Nup358 in acute necrotizing encephalopathy type 1 (ANE1) and viral infection. Int J Mol Sci. https://doi.org/10.3390/ijms23073548
Jones PD, Kaiser MA, Ghaderi Najafabadi M et al (2016) The coronary artery disease-associated coding variant in zinc finger C3HC-type containing 1 (ZC3HC1) affects cell cycle regulation. J Biol Chem 291(31):16318–16327. https://doi.org/10.1074/jbc.M116.734020
Joseph J, Liu S-T, Jablonski SA, Yen TJ, Dasso M (2004) The RanGAP1-RanBP2 complex is essential for microtubule–kinetochore interactions in vivo. Curr Biol 14(7):611–617
Kadota S, Ou J, Shi Y, Lee JT, Sun J, Yildirim E (2020) Nucleoporin 153 links nuclear pore complex to chromatin architecture by mediating CTCF and cohesin binding. Nat Commun 11(1):2606. https://doi.org/10.1038/s41467-020-16394-3
Kaltenbach LS, Romero E, Becklin RR et al (2007) Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet 3(5):e82
Kaneb HM, Folkmann AW, Belzil VV et al (2015) Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet 24(5):1363–1373. https://doi.org/10.1093/hmg/ddu545
Kang C, Jia L, Hao L, Zhang N, Liu Y, Zhang L (2023) POM121 promotes the proliferation and metastasis of gastric cancer via PI3K/AKT/MYC pathway. Am J Cancer Res 13(2):485–497
Kapphahn RJ, Bigelow EJ, Ferrington DA (2007) Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp Eye Res 84(4):646–654
Ke H, Han M, Kim J, Gustin KE, Yoo D (2019) Porcine reproductive and respiratory syndrome virus nonstructural protein 1 beta interacts with nucleoporin 62 to promote viral replication and immune evasion. J Virol. https://doi.org/10.1128/JVI.00469-19
Kehrer T, Cupic A, Ye C et al (2023) Impact of SARS-CoV-2 ORF6 and its variant polymorphisms on host responses and viral pathogenesis. Cell Host Microbe. https://doi.org/10.1016/j.chom.2023.08.003
Kelley K, Knockenhauer KE, Kabachinski G, Schwartz TU (2015) Atomic structure of the Y complex of the nuclear pore. Nat Struct Mol Biol 22(5):425–431. https://doi.org/10.1038/nsmb.2998
Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR (2003) An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol 160(7):1029–1040
Kendirgi F, Rexer DJ, Alcázar-Román AR, Onishko HM, Wente SR (2005) Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell 16(9):4304–4315
Kessoku T, Kobayashi T, Imajo K et al (2021) Endotoxins and non-alcoholic fatty liver disease. Front Endocrinol (lausanne) 12:770986. https://doi.org/10.3389/fendo.2021.770986
Khalaf B, Roncador A, Pischedda F et al (2019) Ankyrin-G induces nucleoporin Nup358 to associate with the axon initial segment of neurons. J Cell Sci. https://doi.org/10.1242/jcs.222802
Khalil B, Chhangani D, Wren MC et al (2022) Nuclear import receptors are recruited by FG-nucleoporins to rescue hallmarks of TDP-43 proteinopathy. Mol Neurodegener 17(1):80. https://doi.org/10.1186/s13024-022-00585-1
Khosravi B, Hartmann H, May S et al (2017) Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum Mol Genet 26(4):790–800. https://doi.org/10.1093/hmg/ddw432
Kim SY, Kim J-E, Park S, Kim HK (2014) Molecular identification of a TPR-FGFR1 fusion transcript in an adult with myeloproliferative neoplasm, T-lymphoblastic lymphoma, and a t(1;8)(q25;p11.2). Cancer Genet 207(6):258–262. https://doi.org/10.1016/j.cancergen.2014.05.011
Kim D-H, Kwon S, Byun S et al (2016) Critical role of RanBP2-mediated SUMOylation of Small Heterodimer Partner in maintaining bile acid homeostasis. Nat Commun 7:12179. https://doi.org/10.1038/ncomms12179
Kind B, Koehler K, Lorenz M, Huebner A (2009) The nuclear pore complex protein ALADIN is anchored via NDC1 but not via POM121 and GP210 in the nuclear envelope. Biochem Biophys Res Commun 390(2):205–210. https://doi.org/10.1016/j.bbrc.2009.09.080
Kindermann B, Valkova C, Krämer A et al (2019) The nuclear pore proteins Nup88/214 and T-cell acute lymphatic leukemia-associated NUP214 fusion proteins regulate Notch signaling. J Biol Chem 294(31):11741–11750. https://doi.org/10.1074/jbc.RA118.006357
Kinoshita Y, Hunter RG, Gray JD et al (2014) Role for NUP62 depletion and PYK2 redistribution in dendritic retraction resulting from chronic stress. Proc Natl Acad Sci USA 111(45):16130–16135. https://doi.org/10.1073/pnas.1418896111
Kleppe M, Soulier J, Asnafi V et al (2011) PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood 117(26):7090–7098. https://doi.org/10.1182/blood-2010-10-314286
Knockenhauer KE, Schwartz TU (2016) The nuclear pore complex as a flexible and dynamic gate. Cell 164(6):1162–1171. https://doi.org/10.1016/j.cell.2016.01.034
Kobayashi A, Hashizume C, Dowaki T, Wong RW (2015) Therapeutic potential of mitotic interaction between the nucleoporin Tpr and aurora kinase A. Cell Cycle 14(9):1447–1458. https://doi.org/10.1080/15384101.2015.1021518
Kosar M, Giannattasio M, Piccini D et al (2021) The human nucleoporin Tpr protects cells from RNA-mediated replication stress. Nat Commun 12(1):3937. https://doi.org/10.1038/s41467-021-24224-3
Kosinski J, Mosalaganti S, von Appen A et al (2016) Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352(6283):363–365. https://doi.org/10.1126/science.aaf0643
Kraemer D, Wozniak RW, Blobel G, Radu A (1994) The human CAN protein, a putative oncogene product associated with myeloid leukemogenesis, is a nuclear pore complex protein that faces the cytoplasm. Proc Natl Acad Sci USA 91(4):1519–1523
Krull S, Dörries J, Boysen B et al (2010) Protein Tpr is required for establishing nuclear pore-associated zones of heterochromatin exclusion. EMBO J 29(10):1659–1673. https://doi.org/10.1038/emboj.2010.54
Kunnas T, Nikkari ST (2015) Association of zinc finger, C3HC-type containing 1 (ZC3HC1) rs11556924 genetic variant with hypertension in a Finnish population, the TAMRISK study. Medicine (baltimore) 94(32):e1221. https://doi.org/10.1097/MD.0000000000001221
Labade AS, Karmodiya K, Sengupta K (2016) HOXA repression is mediated by nucleoporin Nup93 assisted by its interactors Nup188 and Nup205. Epigenet Chromatin 9:54
Labade AS, Salvi A, Kar S, Karmodiya K, Sengupta K (2021) Nup93 and CTCF modulate spatiotemporal dynamics and function of the HOXA gene locus during differentiation. J Cell Sci. https://doi.org/10.1242/jcs.259307
Lai T-H, Wu Y-Y, Wang Y-Y, et al. SEPT12-NDC1 Complexes Are Required for Mammalian Spermiogenesis. Int J Mol Sci. 2016;17(11):1911
Lavau CP, Aumann WK, Sze S-GK et al (2020) The SQSTM1-NUP214 fusion protein interacts with Crm1, activates Hoxa and Meis1 genes, and drives leukemogenesis in mice. PLoS ONE 15(4):e0232036. https://doi.org/10.1371/journal.pone.0232036
Lee SH, Sterling H, Burlingame A, McCormick F (2008) Tpr directly binds to Mad1 and Mad2 and is important for the Mad1–Mad2-mediated mitotic spindle checkpoint. Genes Dev 22(21):2926–2931. https://doi.org/10.1101/gad.1677208
Lee J-C, Li C-F, Huang H-Y et al (2017) ALK oncoproteins in atypical inflammatory myofibroblastic tumours: novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol 241(3):316–323. https://doi.org/10.1002/path.4836
Lee ES, Wolf EJ, Ihn SSJ, Smith HW, Emili A, Palazzo AF (2020) TPR is required for the efficient nuclear export of mRNAs and lncRNAs from short and intron-poor genes. Nucleic Acids Res 48(20):11645–11663. https://doi.org/10.1093/nar/gkaa919
Lee YA, Lee H, Im S-W et al (2021) NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J Clin Investig. https://doi.org/10.1172/JCI144847
Lek M, Karczewski KJ, Minikel EV et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. https://doi.org/10.1038/nature19057
Lelek M, Casartelli N, Pellin D et al (2015) Chromatin organization at the nuclear pore favours HIV replication. Nat Commun 6:6483. https://doi.org/10.1038/ncomms7483
Lemke EA (2016) The multiple faces of disordered nucleoporins. J Mol Biol 428(10 Pt A):2011–2024. https://doi.org/10.1016/j.jmb.2016.01.002
Li K, Liu T (2021) Evaluation of oncogene NUP37 as a potential novel biomarker in breast cancer. Front Oncol 11:669655. https://doi.org/10.3389/fonc.2021.669655
Li F, Zhai Y-P, Tang Y-M, Wang L-P, Wan P-J (2012) Identification of a novel partner gene, TPR, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 51(9):890–897. https://doi.org/10.1002/gcc.21973
Li Y, Xu J, Xiong H et al (2014) Cancer driver candidate genes AVL9, DENND5A and NUPL1 contribute to MDCK cystogenesis. Oncoscience 1(12):854–865
Li J, Li X, Hou R et al (2015) Psoriatic T cells reduce epidermal turnover time and affect cell proliferation contributed from differential gene expression. J Dermatol 42(9):874–880. https://doi.org/10.1111/1346-8138.12961
Li Q, Ohta Y, Yamashita T et al (2017) Dynamic mislocalizations of nuclear pore complex proteins after focal cerebral ischemia in rat. J Neurosci Res 95(9):1745–1759. https://doi.org/10.1002/jnr.24005
Li W, Duan J, Shi W, Lei L, Lv P (2020) Long non-coding RNA NCK1-AS1 serves an oncogenic role in gastric cancer by regulating miR-137/NUP43 axis. Onco Targets Ther 13:9929–9939. https://doi.org/10.2147/OTT.S259336
Li Z, Chen S, Zhao L et al (2022) Near-atomic structure of the inner ring of the Saccharomyces cerevisiae nuclear pore complex. Cell Res 32(5):437–450. https://doi.org/10.1038/s41422-022-00632-y
Liang TJ, Reid AE, Xavier R, Cardiff RD, Wang TC (1996) Transgenic expression of tpr-met oncogene leads to development of mammary hyperplasia and tumors. J Clin Investig 97(12):2872–2877
Liang W, Hu C, Zhu Q et al (2023) Exploring the relationship between abnormally high expression of NUP205 and the clinicopathological characteristics, immune microenvironment, and prognostic value of lower-grade glioma. Front Oncol 13:1007198. https://doi.org/10.3389/fonc.2023.1007198
Lin DH, Hoelz A (2019) The structure of the nuclear pore complex (an update). Annu Rev Biochem 88:725–783. https://doi.org/10.1146/annurev-biochem-062917-011901
Lin C-Y, Wu H-E, Weng EF-J, Wu H-C, Su T-P, Wang S-M (2024) Fluvoxamine exerts sigma-1R to rescue autophagy via Pom121-mediated nucleocytoplasmic transport of TFEB. Mol Neurobiol. https://doi.org/10.1007/s12035-023-03885-9
Lindsay ME, Plafker K, Smith AE, Clurman BE, Macara IG (2002a) Npap60/Nup50 is a tri-stable switch that stimulates importin-alpha:beta-mediated nuclear protein import. Cell 110(3):349–360
Lindsay ME, Plafker K, Smith AE, Clurman BE, Macara IG (2002b) Npap60/Nup50 is a tri-stable switch that stimulates importin-alpha:beta-mediated nuclear protein import. Cell 110(3):349–360. https://doi.org/10.1016/s0092-8674(02)00836-x
Linseman T, Soubeyrand S, Martinuk A, Nikpay M, Lau P, McPherson R (2017) Functional validation of a common nonsynonymous coding variant in ZC3HC1 associated with protection from coronary artery disease. Circ Cardiovasc Genet. https://doi.org/10.1161/CIRCGENETICS.116.001498
Liu H-L, De Souza CPC, Osmani AH, Osmani SA (2009) The three fungal transmembrane nuclear pore complex proteins of Aspergillus nidulans are dispensable in the presence of an intact An-Nup84-120 complex. Mol Biol Cell 20(2):616–630
Liu YY, Jiao WY, Li T, Bao YY (2019) MiRNA-409-5p dysregulation promotes imatinib resistance and disease progression in children with chronic myeloid leukemia. Eur Rev Med Pharmacol Sci 23(19):8468–8475. https://doi.org/10.26355/eurrev_201910_19159
Liu X, Liu J, Xiao W et al (2020) SIRT1 regulates N6-methyladenosine RNA modification in hepatocarcinogenesis by inducing RANBP2-dependent FTO SUMOylation. Hepatology 72(6):2029–2050. https://doi.org/10.1002/hep.31222
Liu Z, Wang H, Jia Y et al (2021a) Significantly high expression of NUP37 leads to poor prognosis of glioma patients by promoting the proliferation of glioma cells. Cancer Med 10(15):5218–5234. https://doi.org/10.1002/cam4.3954
Liu M, Yuan R, Liu S, Xue Y, Wang X (2021b) NDC1 is a prognostic biomarker and associated with immune infiltrates in colon cancer. Int J Gen Med 14:8811–8817. https://doi.org/10.2147/IJGM.S325720
Liu X, Chen X, Xiao M et al (2021c) RANBP2 activates O-GlcNAcylation through inducing CEBPα-dependent OGA downregulation to promote hepatocellular carcinoma malignant phenotypes. Cancers (Basel). https://doi.org/10.3390/cancers13143475
Liu Z, Yan M, Lei W et al (2022) Sec13 promotes oligodendrocyte differentiation and myelin repair through autocrine pleiotrophin signaling. J Clin Investig. https://doi.org/10.1172/JCI155096
Liu G, Wu X, Chen J (2022) Identification and validation of a glycolysis-related gene signature for depicting clinical characteristics and its relationship with tumor immunity in patients with colon cancer. Aging (albany N y) 14(21):8700–8718. https://doi.org/10.18632/aging.204226
Liu J, Xue H, Li C et al (2023a) MTA1 localizes to the mitotic spindle apparatus and interacts with TPR in spindle assembly checkpoint regulation. Biochem Biophys Res Commun 675:106–112. https://doi.org/10.1016/j.bbrc.2023.07.021
Liu Q, Gu L, Qiu J, Qian J (2023b) Elevated NDC1 expression predicts poor prognosis and correlates with immunity in hepatocellular carcinoma. J Gastrointest Oncol 14(1):245–264. https://doi.org/10.21037/jgo-22-1166
Liu R, Qu R, Li Q et al (2023c) ARRDC5 deficiency impairs spermatogenesis by affecting SUN5 and NDC1. Development. https://doi.org/10.1242/dev.201959
Liu Y-P, Guo G, Ren M et al (2024) NDC1 promotes hepatocellular carcinoma tumorigenesis by targeting BCAP31 to activate PI3K/AKT signaling. J Biochem Mol Toxicol 38(2):e23647. https://doi.org/10.1002/jbt.23647
Loïodice I, Alves A, Rabut G et al (2004) The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell 15(7):3333–3344
López-Mejías R, Genre F, García-Bermúdez M et al (2013) The ZC3HC1 rs11556924 polymorphism is associated with increased carotid intima-media thickness in patients with rheumatoid arthritis. Arthritis Res Ther 15(5):R152. https://doi.org/10.1186/ar4335
Lu Y, Kucharski TJ, Gamache I, Blanchette P, Branton PE, Teodoro JG (2014) Interaction of adenovirus type 5 E4orf4 with the nuclear pore subunit Nup205 is required for proper viral gene expression. J Virol 88(22):13249–13259. https://doi.org/10.1128/JVI.00933-14
Luiz AP, Wood JN (2016) Sodium channels in pain and cancer: new therapeutic opportunities. Adv Pharmacol (san Diego, Calif) 75:153–178. https://doi.org/10.1016/bs.apha.2015.12.006
Luo X, Liu Y, Feng W et al (2017) NUP37, a positive regulator of YAP/TEAD signaling, promotes the progression of hepatocellular carcinoma. Oncotarget 8(58):98004–98013. https://doi.org/10.18632/oncotarget.20336
Lupu F, Alves A, Anderson K, Doye V, Lacy E (2008) Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev Cell 14(6):831–842. https://doi.org/10.1016/j.devcel.2008.03.011
Lusk CP, Waller DD, Makhnevych T et al (2007) Nup53p is a target of two mitotic kinases, Cdk1p and Hrr25p. Traffic 8(6):647–660
Ma H, He Y, Bai M et al (2019a) The genetic polymorphisms of ZC3HC1 and SMARCA4 are associated with hypertension risk. Mol Genet Genomic Med 7(11):e942. https://doi.org/10.1002/mgg3.942
Ma H, Li L, Jia L et al (2019b) POM121 is identified as a novel prognostic marker of oral squamous cell carcinoma. J Cancer 10(19):4473–4480. https://doi.org/10.7150/jca.33368
Ma Y, Yin S, Liu X-F et al (2021) Comprehensive analysis of the functions and prognostic value of RNA-binding proteins in thyroid cancer. Front Oncol 11:625007. https://doi.org/10.3389/fonc.2021.625007
Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;37(1):98–105
Mackay DR, Howa AC, Werner TL, Ullman KS (2017) Nup153 and Nup50 promote recruitment of 53BP1 to DNA repair foci by antagonizing BRCA1-dependent events. J Cell Sci 130(19):3347–3359. https://doi.org/10.1242/jcs.203513
Madheshiya PK, Shukla E, Singh J, Bawaria S, Ansari MY, Chauhan R (2022) Insights into the role of Nup62 and Nup93 in assembling cytoplasmic ring and central transport channel of the nuclear pore complex. Mol Biol Cell 33(14):ar139. https://doi.org/10.1091/mbc.E22-01-0027
Maehara K, Murata T, Aoyama N, Matsuno K, Sawamura K. Genetic dissection of Nucleoporin 160 (Nup160), a gene involved in multiple phenotypes of reproductive isolation in Drosophila. Genes Genet Syst. 2012;87(2):99–106
Makise M, Nakamura H, Kuniyasu A (2018) The role of vimentin in the tumor marker Nup88-dependent multinucleated phenotype. BMC Cancer 18(1):519. https://doi.org/10.1186/s12885-018-4454-y
Makise M, Uchimura R, Higashi K et al (2021) Overexpression of the nucleoporin Nup88 stimulates migration and invasion of HeLa cells. Histochem Cell Biol 156(5):409–421. https://doi.org/10.1007/s00418-021-02020-w
Malhas A, Goulbourne C, Vaux DJ (2011) The nucleoplasmic reticulum: form and function. Trends Cell Biol 21(6):362–373. https://doi.org/10.1016/j.tcb.2011.03.008
Malli T, Buxhofer-Ausch V, Rammer M et al (2016) Functional characterization, localization, and inhibitor sensitivity of the TPR-FGFR1 fusion in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 55(1):60–68. https://doi.org/10.1002/gcc.22311
Mancini MA, He D, Ouspenski II, Brinkley BR (1996) Dynamic continuity of nuclear and mitotic matrix proteins in the cell cycle. J Cell Biochem 62(2):158–164
Mansfeld J, Güttinger S, Hawryluk-Gara LA, et al. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol Cell. 2006;22(1):93–103
Mariño-Enríquez A, Wang WL, Roy A et al (2011) Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol 35(1):135–144. https://doi.org/10.1097/PAS.0b013e318200cfd5
Marquez J, Bhattacharya D, Lusk CP, Khokha MK (2021) Nucleoporin NUP205 plays a critical role in cilia and congenital disease. Dev Biol 469:46–53. https://doi.org/10.1016/j.ydbio.2020.10.001
Martínez N, Alonso A, Moragues MD, Pontón J, Schneider J (1999) The nuclear pore complex protein Nup88 is overexpressed in tumor cells. Cancer Res 59(21):5408–5411
Martino L, Morchoisne-Bolhy S, Cheerambathur DK et al (2017) Channel nucleoporins recruit PLK-1 to nuclear pore complexes to direct nuclear envelope breakdown in C. elegans. Dev Cell. https://doi.org/10.1016/j.devcel.2017.09.019
Mason AC, Wente SR (2020) Functions of Gle1 are governed by two distinct modes of self-association. J Biol Chem 295(49):16813–16825. https://doi.org/10.1074/jbc.RA120.015715
Matsuura Y, Stewart M (2005) Nup50/Npap60 function in nuclear protein import complex disassembly and importin recycling. Embo J 24(21):3681–3689. https://doi.org/10.1038/sj.emboj.7600843
Mauro MS, Celma G, Zimyanin V et al (2022) Ndc1 drives nuclear pore complex assembly independent of membrane biogenesis to promote nuclear formation and growth. Elife. https://doi.org/10.7554/eLife.75513
McCloskey A, Ibarra A, Hetzer MW (2018) Tpr regulates the total number of nuclear pore complexes per cell nucleus. Genes Dev 32(19–20):1321–1331. https://doi.org/10.1101/gad.315523.118
McNeer NA, Philip J, Geiger H et al (2019) Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33(8):1934–1943. https://doi.org/10.1038/s41375-019-0402-3
McQuarrie DWJ, Read AM, Stephens FHS, Civetta A, Soller M (2023) Indel driven rapid evolution of core nuclear pore protein gene promoters. Sci Rep 13(1):8035. https://doi.org/10.1038/s41598-023-34985-0
Mega JL, Stitziel NO, Smith JG et al (2015) Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385(9984):2264–2271. https://doi.org/10.1016/s0140-6736(14)61730-x
Megat S, Mora N, Sanogo J et al (2023) Integrative genetic analysis illuminates ALS heritability and identifies risk genes. Nat Commun 14(1):342. https://doi.org/10.1038/s41467-022-35724-1
Mendes A, Fahrenkrog B (2019) NUP214 in leukemia: it’s more than transport. Cells. https://doi.org/10.3390/cells8010076
Michmerhuizen NL, Klco JM, Mullighan CG (2020) Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood 136(20):2275–2289. https://doi.org/10.1182/blood.2020007093
Miorin L, Kehrer T, Sanchez-Aparicio MT et al (2020) SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA 117(45):28344–28354. https://doi.org/10.1073/pnas.2016650117
Mitchell JM, Mansfeld J, Capitanio J, Kutay U, Wozniak RW (2010) Pom121 links two essential subcomplexes of the nuclear pore complex core to the membrane. J Cell Biol 191(3):505–521. https://doi.org/10.1083/jcb.201007098
Miyake N, Tsukaguchi H, Koshimizu E et al (2015) Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am J Hum Genet 97(4):555–566. https://doi.org/10.1016/j.ajhg.2015.08.013
Monwan W, Kawasaki T, Hasan MZ, Ori D, Kawai T (2020) Identification of nucleoporin 93 (Nup93) that mediates antiviral innate immune responses. Biochem Biophys Res Commun 521(4):1077–1082. https://doi.org/10.1016/j.bbrc.2019.11.035
Moon SW, Mo HY, Choi EJ, Yoo NJ, Lee SH (2021) Cancer-related SRCAP and TPR mutations in colon cancers. Pathol Res Pract 217:153292. https://doi.org/10.1016/j.prp.2020.153292
Moore MS (2003) Npap60: a new player in nuclear protein import. Trends Cell Biol 13(2):61–64. https://doi.org/10.1016/s0962-8924(02)00044-2
Moreno-Oñate M, Herrero-Ruiz AM, García-Dominguez M, Cortés-Ledesma F, Ruiz JF (2020) RanBP2-mediated SUMOylation promotes human DNA polymerase lambda nuclear localization and DNA repair. J Mol Biol 432(13):3965–3979. https://doi.org/10.1016/j.jmb.2020.03.020
Moroianu J, Blobel G, Radu A (1997) RanGTP-mediated nuclear export of karyopherin alpha involves its interaction with the nucleoporin Nup153. Proc Natl Acad Sci USA 94(18):9699–9704
Moudry P, Lukas C, Macurek L et al (2012) Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ 19(5):798–807. https://doi.org/10.1038/cdd.2011.150
Mühlbauer D, Dzieciolowski J, Hardt M et al (2015) Influenza virus-induced caspase-dependent enlargement of nuclear pores promotes nuclear export of viral ribonucleoprotein complexes. J Virol 89(11):6009–6021. https://doi.org/10.1128/JVI.03531-14
Muir AM, Cohen JL, Sheppard SE et al (2020) Bi-allelic loss-of-function variants in NUP188 cause a recognizable syndrome characterized by neurologic, ocular, and cardiac abnormalities. Am J Hum Genet 106(5):623–631. https://doi.org/10.1016/j.ajhg.2020.03.009
Müller D, Thieke K, Bürgin A, Dickmanns A, Eilers M (2000) Cyclin E-mediated elimination of p27 requires its interaction with the nuclear pore-associated protein mNPAP60. EMBO J 19(10):2168–2180
Munafò M, Lawless VR, Passera A et al (2021) Channel nuclear pore complex subunits are required for transposon silencing in Drosophila. Elife. https://doi.org/10.7554/eLife.66321
Murakami N, Okuno Y, Yoshida K et al (2018) Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131(14):1576–1586. https://doi.org/10.1182/blood-2017-07-798157
Nakamura T, Largaespada DA, Lee MP et al (1996) Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet 12(2):154–158
Nakamura M, Shimizu-Yoshida Y, Takii Y et al (2005) Antibody titer to gp210-C terminal peptide as a clinical parameter for monitoring primary biliary cirrhosis. J Hepatol 42(3):386–392
Nakamura M, Takii Y, Ito M et al (2006) Increased expression of nuclear envelope gp210 antigen in small bile ducts in primary biliary cirrhosis. J Autoimmun 26(2):138–145
Nakano H, Funasaka T, Hashizume C, Wong RW (2010) Nucleoporin translocated promoter region (Tpr) associates with dynein complex, preventing chromosome lagging formation during mitosis. J Biol Chem 285(14):10841–10849. https://doi.org/10.1074/jbc.M110.105890
Nakielny S, Shaikh S, Burke B, Dreyfuss G (1999) Nup153 is an M9-containing mobile nucleoporin with a novel Ran-binding domain. EMBO J 18(7):1982–1995
Naylor RM, Jeganathan KB, Cao X, van Deursen JM (2016) Nuclear pore protein NUP88 activates anaphase-promoting complex to promote aneuploidy. J Clin Investig 126(2):543–559. https://doi.org/10.1172/JCI82277
Neely AE, Blumensaadt LA, Ho PJ et al (2023) NUP98 and RAE1 sustain progenitor function through HDAC-dependent chromatin targeting to escape from nucleolar localization. Commun Biol 6(1):664. https://doi.org/10.1038/s42003-023-05043-2
Neilson DE, Adams MD, Orr CMD et al (2009) Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet 84(1):44–51. https://doi.org/10.1016/j.ajhg.2008.12.009
Nguyen TD, Rao MK, Dhyani SP et al (2024) Nucleoporin93 limits Yap activity to prevent endothelial cell senescence. Aging Cell. https://doi.org/10.1111/acel.14095
Nofrini V, Di Giacomo D, Mecucci C (2016) Nucleoporin genes in human diseases. Eur J Hum Genet 24(10):1388–1395. https://doi.org/10.1038/ejhg.2016.25
Nong JS, Zhou X, Liu JQ et al (2023) Nucleoporin 107 is a prognostic biomarker in hepatocellular carcinoma associated with immune infiltration. Cancer Med 12(9):10990–11009. https://doi.org/10.1002/cam4.5807
Nousiainen HO, Kestilä M, Pakkasjärvi N et al (2008) Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet 40(2):155–157. https://doi.org/10.1038/ng.2007.65
Ogawa Y, Miyamoto Y, Asally M, Oka M, Yasuda Y, Yoneda Y (2010a) Two isoforms of Npap60 (Nup50) differentially regulate nuclear protein import. Mol Biol Cell 21(4):630–638
Ogawa Y, Miyamoto Y, Asally M, Oka M, Yasuda Y, Yoneda Y (2010b) Two isoforms of Npap60 (Nup50) differentially regulate nuclear protein import. Mol Biol Cell 21(4):630–638. https://doi.org/10.1091/mbc.e09-05-0374
Ogawa Y, Miyamoto Y, Oka M, Yoneda Y (2012) The interaction between importin-α and Nup153 promotes importin-α/β-mediated nuclear import. Traffic 13(7):934–946. https://doi.org/10.1111/j.1600-0854.2012.01367.x
Oh JH, Hur H, Lee J-Y, Kim Y, Seo Y, Kim MH (2017) The mitotic checkpoint regulator RAE1 induces aggressive breast cancer cell phenotypes by mediating epithelial-mesenchymal transition. Sci Rep 7:42256. https://doi.org/10.1038/srep42256
Oh JH, Lee J-Y, Yu S et al (2019) RAE1 mediated ZEB1 expression promotes epithelial-mesenchymal transition in breast cancer. Sci Rep 9(1):2977. https://doi.org/10.1038/s41598-019-39574-8
Oka M, Mura S, Otani M et al (2019) Chromatin-bound CRM1 recruits SET-Nup214 and NPM1c onto HOX clusters causing aberrant HOX expression in leukemia cells. Elife. https://doi.org/10.7554/eLife.46667
Oka M, Otani M, Miyamoto Y et al (2023) Phase-separated nuclear bodies of nucleoporin fusions promote condensation of MLL1/CRM1 and rearrangement of 3D genome structure. Cell Rep 42(8):112884. https://doi.org/10.1016/j.celrep.2023.112884
Okazaki R, Yamazoe K, Inoue YH (2020) Nuclear export of cyclin B mediated by the Nup62 complex is required for meiotic initiation in Drosophila males. Cells. https://doi.org/10.3390/cells9020270
Olsson M, Ekblom M, Fecker L, Kurkinen M, Ekblom P (1999) cDNA cloning and embryonic expression of mouse nuclear pore membrane glycoprotein 210 mRNA. Kidney Int 56(3):827–838
Orniacki C, Verrico A, Pelletier S et al (2023) Y-complex nucleoporins independently contribute to nuclear pore assembly and gene regulation in neuronal progenitors. J Cell Sci. https://doi.org/10.1242/jcs.261151
Osborne LR, Mervis CB. Rearrangements of the Williams-Beuren syndrome locus: molecular basis and implications for speech and language development. Expert Rev Mol Med. 2007;9(15):1–16
Ouyang X, Hao X, Liu S, Hu J, Hu L (2019) Expression of Nup93 is associated with the proliferation, migration and invasion capacity of cervical cancer cells. Acta Biochim Biophys Sin (shanghai) 51(12):1276–1285. https://doi.org/10.1093/abbs/gmz131
Paakkola T, Vuopala K, Kokkonen H et al (2018) A homozygous I684T in GLE1 as a novel cause of arthrogryposis and motor neuron loss. Clin Genet 93(1):173–177. https://doi.org/10.1111/cge.13086
Paço A, Aparecida de Bessa Garcia S, Leitão Castro J, Costa-Pinto AR, Freitas R (2020) Roles of the HOX proteins in cancer invasion and metastasis. Cancers (basel). https://doi.org/10.3390/cancers13010010
Pal K, Bandyopadhyay A, Zhou XE et al (2017) Structural basis of TPR-mediated oligomerization and activation of oncogenic fusion kinases. Structure. https://doi.org/10.1016/j.str.2017.04.015
Pan L, Song X-W, Song J-C et al (2023) Downregulation of NUP93 aggravates hypoxia-induced death of cardiomyocytes in vitro through abnormal regulation of gene transcription. Acta Pharmacol Sin 44(5):969–983. https://doi.org/10.1038/s41401-022-01036-9
Parish IA, Stamp LA, Lorenzo AMD et al (2016) A novel mutation in nucleoporin 35 causes murine degenerative colonic smooth muscle myopathy. Am J Pathol 186(9):2254–2261. https://doi.org/10.1016/j.ajpath.2016.04.016
Park E, Lee B, Clurman BE, Lee K (2016) NUP50 is necessary for the survival of primordial germ cells in mouse embryos. Reproduction 151(1):51–58. https://doi.org/10.1530/REP-14-0649
Park E, Ahn YH, Kang HG, Miyake N, Tsukaguchi H, Cheong HI (2017) NUP107 mutations in children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 32(6):1013–1017. https://doi.org/10.1093/ndt/gfw103
Pashkov E, Korchevaya E, Faizuloev E et al (2022) Knockdown of FLT4, Nup98, and Nup205 Cellular Genes Effectively Suppresses the Reproduction of Influenza Virus Strain A/WSN/1933 (H1N1) In vitro. Infect Disord Drug Targets 22(5):e250322202629. https://doi.org/10.2174/1871526522666220325121403
Patil H, Yoon D, Bhowmick R, Cai Y, Cho K-I, Ferreira PA (2019) Impairments in age-dependent ubiquitin proteostasis and structural integrity of selective neurons by uncoupling Ran GTPase from the Ran-binding domain 3 of Ranbp2 and identification of novel mitochondrial isoforms of ubiquitin-conjugating enzyme E2I (ubc9) and Ranbp2. Small GTPases 10(2):146–161. https://doi.org/10.1080/21541248.2017.1356432
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23(1):27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Peng Y, Shen J, Gao Y et al (2022) Nucleoporin37 may play a role in early embryo development in human and mice. Mol Hum Reprod. https://doi.org/10.1093/molehr/gaac017
Petermann AT, Hiromura K, Blonski M et al (2002) Mechanical stress reduces podocyte proliferation in vitro. Kidney Int 61(1):40–50. https://doi.org/10.1046/j.1523-1755.2002.00102.x
Pichler A, Gast A, Seeler JS, Dejean A, Melchior F (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108(1):109–120
Platani M, Santarella-Mellwig R, Posch M, Walczak R, Swedlow JR, Mattaj IW (2009) The Nup107-160 nucleoporin complex promotes mitotic events via control of the localization state of the chromosome passenger complex. Mol Biol Cell 20(24):5260–5275
Platani M, Samejima I, Samejima K, Kanemaki MT, Earnshaw WC (2018) Seh1 targets GATOR2 and Nup153 to mitotic chromosomes. J Cell Sci. https://doi.org/10.1242/jcs.213140
Popivanova BK, Kostadinova FI, Furuichi K et al (2009) Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res 69(19):7884–7892. https://doi.org/10.1158/0008-5472.CAN-09-1451
Port SA, Monecke T, Dickmanns A et al (2015) Structural and functional characterization of CRM1–Nup214 interactions reveals multiple FG-binding sites involved in nuclear export. Cell Rep 13(4):690–702. https://doi.org/10.1016/j.celrep.2015.09.042
Preston CC, Wyles SP, Reyes S, Storm EC, Eckloff BW, Faustino RS (2018) NUP155 insufficiency recalibrates a pluripotent transcriptome with network remodeling of a cardiogenic signaling module. BMC Syst Biol 12(1):62. https://doi.org/10.1186/s12918-018-0590-x
Pritchard CE, Fornerod M, Kasper LH, van Deursen JM (1999) RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J Cell Biol 145(2):237–254
Prunuske AJ, Liu J, Elgort S, Joseph J, Dasso M, Ullman KS (2006) Nuclear envelope breakdown is coordinated by both Nup358/RanBP2 and Nup153, two nucleoporins with zinc finger modules. Mol Biol Cell 17(2):760–769
Pulianmackal AJ, Kanakousaki K, Flegel K et al (2022) Misregulation of Nucleoporins 98 and 96 leads to defects in protein synthesis that promote hallmarks of tumorigenesis. Dis Model Mech. https://doi.org/10.1242/dmm.049234
Pumroy RA, Nardozzi JD, Hart DJ, Root MJ, Cingolani G (2012) Nucleoporin Nup50 stabilizes closed conformation of armadillo repeat 10 in importin α5. J Biol Chem 287(3):2022–2031. https://doi.org/10.1074/jbc.M111.315838
Qiu X-H, Li F, Cao H-Q et al (2017) Activity of fibroblast growth factor receptor inhibitors TKI258, ponatinib and AZD4547 against TPR-FGFR1 fusion. Mol Med Rep 15(3):1024–1030. https://doi.org/10.3892/mmr.2017.6140
Quan B, Seo H-S, Blobel G, Ren Y (2014) Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1 • Nup98). Proc Natl Acad Sci USA 111(25):9127–9132. https://doi.org/10.1073/pnas.1409076111
Quentmeier H, Schneider B, Röhrs S et al (2009) SET-NUP214 fusion in acute myeloid leukemia- and T-cell acute lymphoblastic leukemia-derived cell lines. J Hematol Oncol 2:3. https://doi.org/10.1186/1756-8722-2-3
Quintás-Cardama A, Tong W, Manshouri T et al (2008) Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22(6):1117–1124. https://doi.org/10.1038/leu.2008.80
Rahbar Saadat Y, Hejazian SM, Nariman-Saleh-Fam Z et al (2020) Glucocorticoid receptors and their upstream epigenetic regulators in adults with steroid-resistant nephrotic syndrome. BioFactors. https://doi.org/10.1002/biof.1680
Rahmani K, Dean DA (2017) Leptomycin B alters the subcellular distribution of CRM1 (Exportin 1). Biochem Biophys Res Commun 488(2):253–258. https://doi.org/10.1016/j.bbrc.2017.04.042
Rajanala K, Nandicoori VK (2012) Localization of nucleoporin Tpr to the nuclear pore complex is essential for Tpr mediated regulation of the export of unspliced RNA. PLoS ONE 7(1):e29921. https://doi.org/10.1371/journal.pone.0029921
Ravindran E, Jühlen R, Vieira-Vieira CH et al (2021) Expanding the phenotype of NUP85 mutations beyond nephrotic syndrome to primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders. Hum Mol Genet 30(22):2068–2081. https://doi.org/10.1093/hmg/ddab160
Ravindran E, Lesca G, Januel L et al (2023) Case report: compound heterozygous NUP85 variants cause autosomal recessive primary microcephaly. Front Neurol 14:1124886. https://doi.org/10.3389/fneur.2023.1124886
Rayala HJ, Kendirgi F, Barry DM, Majerus PW, Wente SR (2004) The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol Cell Proteom 3(2):145–155
Re A, Colussi C, Nanni S et al (2018) Nucleoporin 153 regulates estrogen-dependent nuclear translocation of endothelial nitric oxide synthase and estrogen receptor beta in prostate cancer. Oncotarget 9(46):27985–27997. https://doi.org/10.18632/oncotarget.25462
Rekhi B, Shetty O, Bapat P, Gurav M, Qureshi S (2021) A case of Inv(1)(q23q31) TPR-NTRK1 fusion-positive spindle cell neoplasm in an infant-uncovered by next-generation sequencing: diagnostic challenge, review, and therapeutic implications. Int J Surg Pathol 29(1):102–108. https://doi.org/10.1177/1066896920927467
Ren Y, Seo H-S, Blobel G, Hoelz A (2010) Structural and functional analysis of the interaction between the nucleoporin Nup98 and the mRNA export factor Rae1. Proc Natl Acad Sci USA 107(23):10406–10411. https://doi.org/10.1073/pnas.1005389107
Ren Y, Diao F, Katari S et al (2018) Functional study of a novel missense single-nucleotide variant of NUP107 in two daughters of Mexican origin with premature ovarian insufficiency. Mol Genet Genom Med 6(2):276–281. https://doi.org/10.1002/mgg3.345
Ren X, Cui H, Wu J et al (2022) Identification of a combined apoptosis and hypoxia gene signature for predicting prognosis and immune infiltration in breast cancer. Cancer Med 11(20):3886–3901. https://doi.org/10.1002/cam4.4755
Rexach M, Blobel G (1995) Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell 83(5):683–692
Roccato E, Bressan P, Sabatella G et al (2005) Proximity of TPR and NTRK1 rearranging loci in human thyrocytes. Cancer Res 65(7):2572–2576
Ródenas E, Klerkx EPF, Ayuso C, Audhya A, Askjaer P (2009) Early embryonic requirement for nucleoporin Nup35/NPP-19 in nuclear assembly. Dev Biol 327(2):399–409. https://doi.org/10.1016/j.ydbio.2008.12.024
Rodrigues GA, Park M, Schlessinger J (1997) Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J 16(10):2634–2645
Rodriguez-Berriguete G, Granata G, Puliyadi R et al (2018) Nucleoporin 54 contributes to homologous recombination repair and post-replicative DNA integrity. Nucleic Acids Res 46(15):7731–7746. https://doi.org/10.1093/nar/gky569
Rodriguez-Bravo V, Pippa R, Song W-M et al (2018) Nuclear pores promote lethal prostate cancer by increasing POM121-driven E2F1, MYC, and AR nuclear import. Cell. https://doi.org/10.1016/j.cell.2018.07.015
Roepman R, Letteboer SJF, Arts HH et al (2005) Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc Natl Acad Sci USA 102(51):18520–18525
Rogg M, Maier JI, Ehle M et al (2022) NUP133 controls nuclear pore assembly, transcriptome composition, and cytoskeleton regulation in podocytes. Cells. https://doi.org/10.3390/cells11081259
Rossanti R, Shono A, Miura K et al (2019) Molecular assay for an intronic variant in NUP93 that causes steroid resistant nephrotic syndrome. J Hum Genet 64(7):673–679. https://doi.org/10.1038/s10038-019-0606-4
Rosti RO, Sotak BN, Bielas SL et al (2017) Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway–Mowat syndrome. J Med Genet 54(6):399–403. https://doi.org/10.1136/jmedgenet-2016-104237
Röttgers S, Gombert M, Teigler-Schlegel A et al (2010) ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia 24(6):1197–1200. https://doi.org/10.1038/leu.2010.18
Russell JP, Powell DJ, Cunnane M et al (2000) The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene 19(50):5729–5735
Sadasivan J, Vlok M, Wang X, Nayak A, Andino R, Jan E (2022) Targeting Nup358/RanBP2 by a viral protein disrupts stress granule formation. PLoS Pathog 18(12):e1010598. https://doi.org/10.1371/journal.ppat.1010598
Sahoo MR, Gaikwad S, Khuperkar D et al (2017) Nup358 binds to AGO proteins through its SUMO-interacting motifs and promotes the association of target mRNA with miRISC. EMBO Rep 18(2):241–263. https://doi.org/10.15252/embr.201642386
Saito S, Cigdem S, Okuwaki M, Nagata K (2016) Leukemia-associated Nup214 fusion proteins disturb the XPO1-mediated nuclear-cytoplasmic transport pathway and thereby the NF-κB signaling pathway. Mol Cell Biol 36(13):1820–1835. https://doi.org/10.1128/MCB.00158-16
Sakiyama Y, Mazur A, Kapinos LE, Lim RYH (2016) Spatiotemporal dynamics of the nuclear pore complex transport barrier resolved by high-speed atomic force microscopy. Nat Nanotechnol 11(8):719–723. https://doi.org/10.1038/nnano.2016.62
Sakiyama Y, Panatala R, Lim RYH (2017) Structural dynamics of the nuclear pore complex. Semin Cell Dev Biol 68:27–33. https://doi.org/10.1016/j.semcdb.2017.05.021
Sakuma S, Zhu EY, Raices M, Zhang P, Murad R, D’Angelo MA (2022) Loss of Nup210 results in muscle repair delays and age-associated alterations in muscle integrity. Life Sci Alliance. https://doi.org/10.2650/lsa.202101216
Sandén C, Gullberg U (2015) The DEK oncoprotein and its emerging roles in gene regulation. Leukemia 29(8):1632–1636. https://doi.org/10.1038/leu.2015.72
Sandestig A, Engström K, Pepler A et al (2020) NUP188 biallelic loss of function may underlie a new syndrome: nucleoporin 188 insufficiency syndrome? Mol Syndromol 10(6):313–319. https://doi.org/10.1159/000504818
Sanford DE, Belt BA, Panni RZ et al (2013) Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 19(13):3404–3415. https://doi.org/10.1158/1078-0432.CCR-13-0525
Satow R, Shitashige M, Jigami T et al (2012) β-Catenin inhibits promyelocytic leukemia protein tumor suppressor function in colorectal cancer cells. Gastroenterology 142(3):572–581. https://doi.org/10.1053/j.gastro.2011.11.041
Schertzer M, Jullien L, Pinto AL, Calado RT, Revy P, Londoño-Vallejo A (2023) Human RTEL1 interacts with KPNB1 (importin β) and NUP153 and connects nuclear import to nuclear envelope stability in S-phase. Cells. https://doi.org/10.3390/cells12242798
Schwarz DS, Blower MD (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73(1):79–94. https://doi.org/10.1007/s00018-015-2052-6
Senécal JL, Isabelle C, Fritzler MJ et al (2014) An autoimmune myositis-overlap syndrome associated with autoantibodies to nuclear pore complexes: description and long-term follow-up of the anti-Nup syndrome. Medicine (baltimore) 93(24):383–394. https://doi.org/10.1097/md.0000000000000223
Seytanoglu A, Alsomali NI, Valori CF et al (2016) Deficiency in the mRNA export mediator Gle1 impairs Schwann cell development in the zebrafish embryo. Neuroscience 322:287–297. https://doi.org/10.1016/j.neuroscience.2016.02.039
Shah S, Tugendreich S, Forbes D (1998) Major binding sites for the nuclear import receptor are the internal nucleoporin Nup153 and the adjacent nuclear filament protein Tpr. J Cell Biol 141(1):31–49
Sharma M, Wente SR (2020) Nucleocytoplasmic shuttling of Gle1 impacts DDX1 at transcription termination sites. Mol Biol Cell 31(21):2398–2408. https://doi.org/10.1091/mbc.E20-03-0215
Sharma M, Johnson M, Brocardo M, Jamieson C, Henderson BR (2014) Wnt signaling proteins associate with the nuclear pore complex: implications for cancer. Adv Exp Med Biol 773:353–372. https://doi.org/10.1007/978-1-4899-8032-8_16
Shen Q, Wang YE, Truong M et al (2021) RanBP2/Nup358 enhances miRNA activity by sumoylating Argonautes. PLoS Genet 17(2):e1009378. https://doi.org/10.1371/journal.pgen.1009378
Shen W, Gong B, Xing C et al (2022) Comprehensive maturity of nuclear pore complexes regulates zygotic genome activation. Cell. https://doi.org/10.1016/j.cell.2022.11.011
Shen Q, Kumari S, Xu C et al (2023a) The capsid lattice engages a bipartite NUP153 motif to mediate nuclear entry of HIV-1 cores. Proc Natl Acad Sci USA 120(13):e2202815120. https://doi.org/10.1073/pnas.2202815120
Shen Q, Li J, Zhang C et al (2023b) Pan-cancer analysis and experimental validation identify ndc1 as a potential immunological, prognostic and therapeutic biomarker in pancreatic cancer. Aging (albany N y) 15(18):9779–9796. https://doi.org/10.18632/aging.205048
Shi KY, Mori E, Nizami ZF et al (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci USA 114(7):E1111–E1117. https://doi.org/10.1073/pnas.1620293114
Shi J, Li C, Wang H, Xiao B, Qiu W (2019) NUP58 facilitates metastasis and epithelial-mesenchymal transition of lung adenocarcinoma via the GSK-3β/Snail signaling pathway. Am J Transl Res 11(1):393–405
Shi R, Xu L, Huang L, Cheng J-X (2020) Nucleoporin 107 promotes the survival of tumor cells in cervical cancers. Gynecol Obstet Invest 85(1):41–52. https://doi.org/10.1159/000502788
Shibata S, Matsuoka Y, Yoneda Y (2002) Nucleocytoplasmic transport of proteins and poly(A)+ RNA in reconstituted Tpr-less nuclei in living mammalian cells. Genes Cells 7(4):421–434
Shima Y, Yumoto M, Katsumoto T, Kitabayashi I (2017) MLL is essential for NUP98-HOXA9-induced leukemia. Leukemia 31(10):2200–2210. https://doi.org/10.1038/leu.2017.62
Shimozono N, Jinnin M, Masuzawa M et al (2015) NUP160-SLC43A3 is a novel recurrent fusion oncogene in angiosarcoma. Cancer Res 75(21):4458–4465. https://doi.org/10.1158/0008-5472.CAN-15-0418
Sihn C-R, Suh E-J, Lee K-H, Kim SH (2005) Sec13 induces genomic instability in U2OS cells. Exp Mol Med 37(3):255–260
Simioni C, Ultimo S, Martelli AM et al (2016) Synergistic effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway or NUP214-ABL1 fusion protein in human acute lymphoblastic leukemia. Oncotarget 7(48):79842–79853. https://doi.org/10.18632/oncotarget.13035
Singer S, Zhao R, Barsotti AM et al (2012) Nuclear pore component Nup98 is a potential tumor suppressor and regulates posttranscriptional expression of select p53 target genes. Mol Cell 48(5):799–810. https://doi.org/10.1016/j.molcel.2012.09.020
Singh U, Bindra D, Samaiya A, Mishra RK (2023) Overexpressed Nup88 stabilized through interaction with Nup62 promotes NF-κB dependent pathways in cancer. Front Oncol 13:1095046. https://doi.org/10.3389/fonc.2023.1095046
Smitherman M, Lee K, Swanger J, Kapur R, Clurman BE (2000) Characterization and targeted disruption of murine Nup50, a p27(Kip1)-interacting component of the nuclear pore complex. Mol Cell Biol 20(15):5631–5642
Smythe C, Jenkins HE, Hutchison CJ (2000) Incorporation of the nuclear pore basket protein nup153 into nuclear pore structures is dependent upon lamina assembly: evidence from cell-free extracts of Xenopus eggs. EMBO J 19(15):3918–3931
Snow CJ, Dar A, Dutta A, Kehlenbach RH, Paschal BM (2013) Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. J Cell Biol 201(4):541–557. https://doi.org/10.1083/jcb.201212117
Soderholm JF, Bird SL, Kalab P et al (2011) Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem Biol 6(7):700–708. https://doi.org/10.1021/cb2000296
Soman NR, Wogan GN, Rhim JS (1990) TPR-MET oncogenic rearrangement: detection by polymerase chain reaction amplification of the transcript and expression in human tumor cell lines. Proc Natl Acad Sci USA 87(2):738–742
Sonawane PJ, Dewangan SP, Madheshiya PK et al (2020) Molecular and structural analysis of central transport channel in complex with Nup93 of nuclear pore complex. Protein Sci 29(12):2510–2527. https://doi.org/10.1002/pro.3983
Song S, Shi C, Bian Y et al (2022) Sestrin2 remedies podocyte injury via orchestrating TSP-1/TGF-β1/Smad3 axis in diabetic kidney disease. Cell Death Dis 13(7):663. https://doi.org/10.1038/s41419-022-05120-0
Souquet B, Freed E, Berto A et al (2018) Nup133 is required for proper nuclear pore basket assembly and dynamics in embryonic stem cells. Cell Rep 23(8):2443–2454. https://doi.org/10.1016/j.celrep.2018.04.070
Stavru F, Hülsmann BB, Spang A, Hartmann E, Cordes VC, Görlich D (2006) NDC1: a crucial membrane-integral nucleoporin of metazoan nuclear pore complexes. J Cell Biol 173(4):509–519
Stockum A, Snijders AP, Maertens GN (2018) USP11 deubiquitinates RAE1 and plays a key role in bipolar spindle formation. PLoS ONE 13(1):e0190513. https://doi.org/10.1371/journal.pone.0190513
Struski S, Lagarde S, Bories P et al (2017) NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31(3):565–572. https://doi.org/10.1038/leu.2016.267
Su Y, Pelz C, Huang T et al (2018) Post-translational modification localizes MYC to the nuclear pore basket to regulate a subset of target genes involved in cellular responses to environmental signals. Genes Dev 32(21–22):1398–1419. https://doi.org/10.1101/gad.314377.118
Sump B, Brickner JH (2017) Nup98 regulation of histone methylation promotes normal gene expression and may drive leukemogenesis. Genes Dev 31(22):2201–2203. https://doi.org/10.1101/gad.310359.117
Sun Y, Guo H-C (2008) Structural constraints on autoprocessing of the human nucleoporin Nup98. Protein Sci 17(3):494–505. https://doi.org/10.1110/ps.073311808
Suntharalingam M, Alcázar-Román AR, Wente SR (2004) Nuclear export of the yeast mRNA-binding protein Nab2 is linked to a direct interaction with Gfd1 and to Gle1 function. J Biol Chem 279(34):35384–35391
Swift J, Discher DE (2014) The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J Cell Sci 127(Pt 14):3005–3015. https://doi.org/10.1242/jcs.149203
Szwed A, Kim E, Jacinto E (2021) Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev 101(3):1371–1426. https://doi.org/10.1152/physrev.00026.2020
Takahashi N, van Kilsdonk JWJ, Ostendorf B et al (2008) Tumor marker nucleoporin 88 kDa regulates nucleocytoplasmic transport of NF-kappaB. Biochem Biophys Res Commun 374(3):424–430. https://doi.org/10.1016/j.bbrc.2008.06.128
Tan QKG, McConkie-Rosell A, Juusola J et al (2017) The importance of managing the patient and not the gene: expanded phenotype of GLE1-associated arthrogryposis. Cold Spring Harb Mol Case Stud. https://doi.org/10.1101/mcs.a002063
Tang Z, Yang Y, Chen W, Li E, Liang T (2022a) Demethylation at enhancer upregulates MCM2 and NUP37 expression predicting poor survival in hepatocellular carcinoma patients. J Transl Med 20(1):49. https://doi.org/10.1186/s12967-022-03249-2
Tang Z, Yang Y, Chen W, Li E, Liang T (2022b) Correction: Demethylation at enhancer upregulates MCM2 and NUP37 expression predicting poor survival in hepatocellular carcinoma patients. J Transl Med 20(1):594. https://doi.org/10.1186/s12967-022-03766-0
Tang Q, Zhou F, Yang C, Dai J, Li J, He Y (2022c) CircINTS4 facilitates chemoresistance of TNBC by competitively binding miR-129-5p/POM121 axis. J Oncol 2022:2630864. https://doi.org/10.1155/2022/2630864
Tang R, Lin W, Shen C et al (2023) Single-cell transcriptomics uncover hub genes and cell–cell crosstalk in patients with hypertensive nephropathy. Int Immunopharmacol 125(Pt A):111104. https://doi.org/10.1016/j.intimp.2023.111104
Targa A, Larrimore KE, Wong CK et al (2021) Non-genetic and genetic rewiring underlie adaptation to hypomorphic alleles of an essential gene. EMBO J 40(21):e107839. https://doi.org/10.15252/embj.2021107839
Terashima Y, Toda E, Itakura M et al (2020) Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties. Nat Commun 11(1):609. https://doi.org/10.1038/s41467-020-14338-5
Theerthagiri G, Eisenhardt N, Schwarz H, Antonin W (2010) The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. J Cell Biol 189(7):1129–1142. https://doi.org/10.1083/jcb.200912045
Thompson ME (2010) BRCA1 16 years later: nuclear import and export processes. FEBS J 277(15):3072–3078. https://doi.org/10.1111/j.1742-4658.2010.07733.x
Thul PJ, Lindskog C (2018) The human protein atlas: a spatial map of the human proteome. Protein Sci 27(1):233–244. https://doi.org/10.1002/pro.3307
Tian X, Li J, Valakh V, DiAntonio A, Wu C (2011) Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat Neurosci 14(10):1267–1275. https://doi.org/10.1038/nn.2922
Tian C, Zhou S, Yi C (2018) High NUP43 expression might independently predict poor overall survival in luminal A and in HER2+ breast cancer. Future Oncol 14(15):1431–1442. https://doi.org/10.2217/fon-2017-0690
Toda E, Terashima Y, Esaki K et al (2014) Identification of a binding element for the cytoplasmic regulator FROUNT in the membrane-proximal C-terminal region of chemokine receptors CCR2 and CCR5. Biochem J 457(2):313–322. https://doi.org/10.1042/BJ20130827
Uhlířová J, Šebestová L, Fišer K, Sieger T, Fišerová J, Hozák P (2021) Nucleoporin TPR affects C2C12 myogenic differentiation via regulation of Myh4 expression. Cells. https://doi.org/10.3390/cells10061271
Valenstein ML, Rogala KB, Lalgudi PV et al (2022) Structure of the nutrient-sensing hub GATOR2. Nature 607(7919):610–616. https://doi.org/10.1038/s41586-022-04939-z
Van Vlierberghe P, van Grotel M, Tchinda J et al (2008) The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 111(9):4668–4680. https://doi.org/10.1182/blood-2007-09-111872
Vasu S, Shah S, Orjalo A, Park M, Fischer WH, Forbes DJ (2001) Novel vertebrate nucleoporins Nup133 and Nup160 play a role in mRNA export. J Cell Biol 155(3):339–354
Vishnoi N, Dhanasekeran K, Chalfant M, Surovstev I, Khokha MK, Lusk CP (2020) Differential turnover of Nup188 controls its levels at centrosomes and role in centriole duplication. J Cell Biol. https://doi.org/10.1083/jcb.201906031
Vollmer B, Schooley A, Sachdev R et al (2012) Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J 31(20):4072–4084. https://doi.org/10.1038/emboj.2012.256
Vomastek T, Iwanicki MP, Burack WR et al (2008) Extracellular signal-regulated kinase 2 (ERK2) phosphorylation sites and docking domain on the nuclear pore complex protein Tpr cooperatively regulate ERK2-Tpr interaction. Mol Cell Biol 28(22):6954–6966. https://doi.org/10.1128/MCB.00925-08
von Appen A, Kosinski J, Sparks L et al (2015) In situ structural analysis of the human nuclear pore complex. Nature 526(7571):140–143. https://doi.org/10.1038/nature15381
Vuopala K, Herva R (1994) Lethal congenital contracture syndrome: further delineation and genetic aspects. J Med Genet 31(7):521–527
Vyas P, Singh A, Murawala P, Joseph J (2013) Nup358 interacts with Dishevelled and aPKC to regulate neuronal polarity. Biol Open 2(11):1270–1278. https://doi.org/10.1242/bio.20135363
Wälde S, Thakar K, Hutten S et al (2012) The nucleoporin Nup358/RanBP2 promotes nuclear import in a cargo- and transport receptor-specific manner. Traffic 13(2):218–233. https://doi.org/10.1111/j.1600-0854.2011.01302.x
Walther TC, Fornerod M, Pickersgill H, Goldberg M, Allen TD, Mattaj IW (2001) The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J 20(20):5703–5714
Wang J, Zhuo Z, Chen M et al (2018) RAN/RANBP2 polymorphisms and neuroblastoma risk in Chinese children: a three-center case-control study. Aging (albany NY) 10(4):808–818. https://doi.org/10.18632/aging.101429
Wang T, Sun H, Bao Y et al (2020) POM121 overexpression is related to a poor prognosis in colorectal cancer. Expert Rev Mol Diagn 20(3):345–353. https://doi.org/10.1080/14737159.2020.1707670
Wang H, Lin Y, Jin J, Shen H, Dai C (2021a) Nuclear pore complex 62 promotes metastasis of gastric cancer by regulating Wnt/β-catenin and TGF-β signaling pathways. J Environ Pathol Toxicol Oncol 40(2):81–87. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2021037136
Wang H, Luo Q, Kang J et al (2021b) YTHDF1 aggravates the progression of cervical cancer through m6A-mediated up-regulation of RANBP2. Front Oncol 11:650383. https://doi.org/10.3389/fonc.2021.650383
Wang F, Zhang J, Tang H et al (2022) Nup54-induced CARM1 nuclear importation promotes gastric cancer cell proliferation and tumorigenesis through transcriptional activation and methylation of Notch2. Oncogene 41(2):246–259. https://doi.org/10.1038/s41388-021-02078-9
Wang Q, Gu R, Li F-W, Gu W-Y, Zhang J-J (2023a) Steroid-resistant nephrotic syndrome caused by nuclear pore gene NUP133 variation. Clin Genet 104(2):272–274. https://doi.org/10.1111/cge.14339
Wang X, Yang Z, Ran Y, Li L, Wang B, Zhou L (2023b) Anti-gp210-positive primary biliary cholangitis: the dilemma of clinical treatment and emerging mechanisms. Ann Hepatol 28(5):101121. https://doi.org/10.1016/j.aohep.2023.101121
Wang S-M, Wu H-E, Yasui Y et al (2023c) Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy 19(1):126–151. https://doi.org/10.1080/15548627.2022.2063003
Wasilewska A, Rybi-Szuminska A, Dubiela P (2023) Steroid-resistant nephrotic syndrome caused by NUP93 pathogenic variants. J Clin Med. https://doi.org/10.3390/jcm12185810
Wei Z, Xu J, Li W et al (2022) SMARCC1 enters the nucleus via KPNA2 and plays an oncogenic role in bladder cancer. Front Mol Biosci 9:902220. https://doi.org/10.3389/fmolb.2022.902220
Weinberg-Shukron A, Renbaum P, Kalifa R et al (2015) A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis. J Clin Investig 125(11):4295–4304. https://doi.org/10.1172/jci83553
Whittle J, Johnson A, Dobbs MB, Gurnett CA (2021) Models of distal arthrogryposis and lethal congenital contracture syndrome. Genes (basel). https://doi.org/10.3390/genes12060943
Winey M, Hoyt MA, Chan C, Goetsch L, Botstein D, Byers B (1993) NDC1: a nuclear periphery component required for yeast spindle pole body duplication. J Cell Biol 122(4):743–751
Wong RW, Blobel G (2008) Cohesin subunit SMC1 associates with mitotic microtubules at the spindle pole. Proc Natl Acad Sci USA 105(40):15441–15445. https://doi.org/10.1073/pnas.0807660105
Wong RW, Blobel G, Coutavas E (2006) Rae1 interaction with NuMA is required for bipolar spindle formation. Proc Natl Acad Sci USA 103(52):19783–19787
Wozniak RW, Blobel G (1992) The single transmembrane segment of gp210 is sufficient for sorting to the pore membrane domain of the nuclear envelope. J Cell Biol 119(6):1441–1449
Wu J, Matunis MJ, Kraemer D, Blobel G, Coutavas E (1995) Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J Biol Chem 270(23):14209–14213
Wu Y, Fang G, Wang X et al (2019) NUP153 overexpression suppresses the proliferation of colorectal cancer by negatively regulating Wnt/β-catenin signaling pathway and predicts good prognosis. Cancer Biomark 24(1):61–70. https://doi.org/10.3233/CBM-181703
Xia F, Xia W, Yu X (2020) LncRNA HOTAIR influences the growth, migration, and invasion of papillary thyroid carcinoma via affection on the miR-488-5p/NUP205 axis. Technol Cancer Res Treat 19:1533033820962125. https://doi.org/10.1177/1533033820962125
Xie P, Peng Z, Chen Y et al (2021a) Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res 31(3):291–311. https://doi.org/10.1038/s41422-020-00443-z
Xie J, Yuan Y, Yao G, Chen Z, Yu W, Zhu Q (2021b) Nucleoporin 160 (NUP160) inhibition alleviates diabetic nephropathy by activating autophagy. Bioengineered 12(1):6390–6402. https://doi.org/10.1080/21655979.2021.1968777
Xiong H, Li Y, Liu M (2023) DEPDC1B is involved in the proliferation, metastasis, cell cycle arrest and apoptosis of colon cancer cells by regulating NUP37. Mol Med Rep. https://doi.org/10.3892/mmr.2023.13013
Xu C, Li Z, He H et al (2015a) Crystal structure of human nuclear pore complex component NUP43. FEBS Lett 589(21):3247–3253. https://doi.org/10.1016/j.febslet.2015.09.008
Xu L, Pan L, Li J et al (2015b) Nucleoporin 35 regulates cardiomyocyte pH homeostasis by controlling Na+-H+ exchanger-1 expression. J Mol Cell Biol 7(5):476–485. https://doi.org/10.1093/jmcb/mjv054
Xue G, Yu HJ, Buffone C et al (2023) The HIV-1 capsid core is an opportunistic nuclear import receptor. Nat Commun 14(1):3782. https://doi.org/10.1038/s41467-023-39146-5
Xylourgidis N, Roth P, Sabri N, Tsarouhas V, Samakovlis C (2006) The nucleoporin Nup214 sequesters CRM1 at the nuclear rim and modulates NFkappaB activation in Drosophila. J Cell Sci 119(Pt 21):4409–4419
Yamazumi Y, Kamiya A, Nishida A et al (2009) The transmembrane nucleoporin NDC1 is required for targeting of ALADIN to nuclear pore complexes. Biochem Biophys Res Commun 389(1):100–104. https://doi.org/10.1016/j.bbrc.2009.08.096
Yang M-X, Liu Z-X, Zhang S et al (2009) Proteomic analysis of the effect of iptakalim on human pulmonary arterial smooth muscle cell proliferation. Acta Pharmacol Sin 30(2):175–183. https://doi.org/10.1038/aps.2008.30
Yang J, Liu Y, Wang B et al (2017) Sumoylation in p27kip1 via RanBP2 promotes cancer cell growth in cholangiocarcinoma cell line QBC939. BMC Mol Biol 18(1):23. https://doi.org/10.1186/s12867-017-0100-5
Yang Y, Chen Z, Zhou L et al (2022) In silico development and validation of a novel glucose and lipid metabolism-related gene signature in gastric cancer. Transl Cancer Res 11(7):1977–1993. https://doi.org/10.21037/tcr-22-168
Yi H, Friedman JL, Ferreira PA (2007) The cyclophilin-like domain of Ran-binding protein-2 modulates selectively the activity of the ubiquitin-proteasome system and protein biogenesis. J Biol Chem 282(48):34770–34778
Yu J, Miehlke S, Ebert MP et al (2000) Frequency of TPR-MET rearrangement in patients with gastric carcinoma and in first-degree relatives. Cancer 88(8):1801–1806
Yu Y, Farooq MS, Eberhart Meessen S et al (2024) Nuclear pore protein POM121 regulates subcellular localization and transcriptional activity of PPARγ. Cell Death Dis 15(1):7. https://doi.org/10.1038/s41419-023-06371-1
Yue WJ, Wang Y, Li WY, Wang ZD (2020) LINC00887 regulates the proliferation of nasopharyngeal carcinoma via targeting miRNA-203b-3p to upregulate NUP205. Eur Rev Med Pharmacol Sci 24(17):8863–8870. https://doi.org/10.26355/eurrev_202009_22826
Zermati Y, Mouhamad S, Stergiou L et al (2007) Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol Cell 28(4):624–637. https://doi.org/10.1016/j.molcel.2007.09.030
Zhang X, Yang H, Corydon MJ et al (1999) Localization of a human nucleoporin 155 gene (NUP155) to the 5p13 region and cloning of its cDNA. Genomics 57(1):144–151
Zhang QH, Ye M, Wu XY et al (2000) Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells. Genome Res 10(10):1546–1560
Zhang X, Chen S, Yoo S et al (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135(6):1017–1027. https://doi.org/10.1016/j.cell.2008.10.022
Zhang X-W, Qin X, Qin CY, Yin Y-L, Chen Y, Zhu H-L (2013) Expression of monocyte chemoattractant protein-1 and CC chemokine receptor 2 in non-small cell lung cancer and its significance. Cancer Immunol Immunother 62(3):563–570. https://doi.org/10.1007/s00262-012-1361-y
Zhang K, Donnelly CJ, Haeusler AR et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525(7567):56–61. https://doi.org/10.1038/nature14973
Zhang XT, Dong SH, Zhang JY, Shan B (2019) MicroRNA-577 promotes the sensitivity of chronic myeloid leukemia cells to imatinib by targeting NUP160. Eur Rev Med Pharmacol Sci 23(16):7008–7015. https://doi.org/10.26355/eurrev_201908_18741
Zhang Y, Chen W, Zeng W, Lu Z, Zhou X (2020a) Biallelic loss of function NEK3 mutations deacetylate α-tubulin and downregulate NUP205 that predispose individuals to cilia-related abnormal cardiac left-right patterning. Cell Death Dis 11(11):1005. https://doi.org/10.1038/s41419-020-03214-1
Zhang S, Zheng C, Li D et al (2020b) Clinical significance of POM121 expression in lung cancer. Genet Test Mol Biomarkers 24(12):819–824. https://doi.org/10.1089/gtmb.2020.0053
Zhang J, Lv W, Liu Y et al (2021a) Nucleoporin 37 promotes the cell proliferation, migration, and invasion of gastric cancer through activating the PI3K/AKT/mTOR signaling pathway. In Vitro Cell Dev Biol Anim 57(10):987–997. https://doi.org/10.1007/s11626-021-00627-w
Zhang X, Wang J, Zhuang J et al (2021b) A novel glycolysis-related four-mRNA signature for predicting the survival of patients with breast cancer. Front Genet 12:606937. https://doi.org/10.3389/fgene.2021.606937
Zhang M, Tian SY, Ma SY et al (2023) Deficient chaperone-mediated autophagy in macrophage aggravates inflammation of nonalcoholic steatohepatitis by targeting Nup85. Liver Int 43(5):1021–1034. https://doi.org/10.1111/liv.15547
Zhao Z-R, Zhang Z-Y, He X-Q et al (2010) Nup88 mRNA overexpression in colorectal cancers and relationship with p53. Cancer Biomark 8(2):73–80. https://doi.org/10.3233/DMA-2011-0842
Zhao F, Zhu J-Y, Richman A et al (2019) Mutations in NUP160 are implicated in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 30(5):840–853. https://doi.org/10.1681/ASN.2018080786
Zhao R, Tang G, Wang T et al (2020) POM121 is a novel marker for predicting the prognosis of laryngeal cancer. Histol Histopathol 35(11):1285–1293. https://doi.org/10.14670/HH-18-267
Zheng X, Zhao X, Zhang Y et al (2019) RAE1 promotes BMAL1 shuttling and regulates degradation and activity of CLOCK: BMAL1 heterodimer. Cell Death Dis 10(2):62. https://doi.org/10.1038/s41419-019-1346-2
Zhou L, Panté N (2010) The nucleoporin Nup153 maintains nuclear envelope architecture and is required for cell migration in tumor cells. FEBS Lett 584(14):3013–3020. https://doi.org/10.1016/j.febslet.2010.05.038
Zhu X, Huang G, Zeng C et al (2022) Structure of the cytoplasmic ring of the Xenopus laevis nuclear pore complex. Science 376(6598):eabl8280. https://doi.org/10.1126/science.abl8280
Zhu X, Qi C, Wang R et al (2022) Acute depletion of human core nucleoporin reveals direct roles in transcription control but dispensability for 3D genome organization. Cell Rep 41(5):111576. https://doi.org/10.1016/j.celrep.2022.111576
Zuccolo M, Alves A, Galy V et al (2007) The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J 26(7):1853–1864
Funding
This work was supported by the Youth Science and technology innovation leader of Ningbo (no. 2023QL052), the Natural Science Foundation of Ningbo (no. 2021J294), the National Natural Science Foundation of China (no. 32270821), the National Natural Science Foundation of Zhejiang (no. LY24C050001), the General Surgery Clinical Key Specialty Construction Project of Zhejiang Province (no. 2023-SZZ), the Project of Zhejiang Medical and Health Platform Plan (no. 2022KY1079), the Ningbo Public Welfare Science and Technology Major Project (no. 2021S106), the Ningbo Major Research and Development Plan Project (no. 2024Z215), and the K.C. Wong Magna Fund in Ningbo University.
Author information
Authors and Affiliations
Contributions
Yuxuan Li: conceptualization, writing the original draft, writing review and editing, and visualization. Jie Zhu: conceptualization, writing review and editing. Fengguang Zhai: conceptualization, writing review and editing. Lili Kong: conceptualization, writing review, and editing. Hong Li: methodology, writing review and editing. Xiaofeng Jin: methodology, writing review and editing, supervision, funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Zhu, J., Zhai, F. et al. Advances in the understanding of nuclear pore complexes in human diseases. J Cancer Res Clin Oncol 150, 374 (2024). https://doi.org/10.1007/s00432-024-05881-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-024-05881-5