
Abstract
Background
Uveal melanoma (UM) is an aggressive intraocular malignant tumor. The present study aimed to identify the key genes associated with UM metastasis and established a gene signature to analyze the relationship between the signature and prognosis and immune cell infiltration. Later, a predictive model combined with clinical variables was developed and validated.
Methods
Two UM gene expression profile chip datasets were downloaded from TCGA and GEO databases. Immune-related genes (IRGs) were obtained from IMPORT database. First, these mRNAs were intersected with IRGs, and weighted gene co-expression network analysis (WGCNA) was used to identify the co-expression of genes primarily associated with metastasis of UM. Univariate Cox regression analysis screened the genes related to prognosis. LASSO-Cox established a risk score to distinguish high-risk group and low-risk group. Then the GSEA enrichment pathway and immune cell infiltration of the two groups were compared. And combined with clinical variables, a predictive model was constructed. The time-dependent receiver operating characteristic (ROC) curve, calibration curve, and decision curve analysis (DCA) curve were used to verify the stability and accuracy of the final predictive model, and a nomogram was then drawn.
Results
The MEblack, MEpurple, and MEblue modules were significantly associated with the metastasis of UM patients (P value < 0.001, = 0.001, = 0.022, respectively). Four genes (UBXN2B, OTUD3, KAT8, LAMTOR2) were obtained by Pearson correlation analysis, weighted gene correlation network analysis (WGCNA), univariate Cox, and LASSO-Cox. And a novel prognostic risk score was established. Immune-related prognostic signature can well classify UM patients into high-risk and low-risk groups. Kaplan–Meier curve showed that the OS of high-risk patients was worse than that of low-risk patients. In addition, the risk score played an important role in evaluating the signaling pathway and immune cell infiltration of UM patients in high-risk and low-risk groups. Both the training set and validation set of the model showed good predictive accuracy in the degree of differentiation and calibration (e.g., 1-year overall survival: AUC = 0.930 (0.857–1.003)). Finally, a nomogram was established to serve in clinical practice.
Significance
UM key gene signature and prognosis predictive model might provide insights for further investigation of the pathogenesis and development of UM at the molecular level, and provide theoretical basis for determining new prognostic markers of UM and immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the most common primary intraocular malignancy among adults (Jager et al. 2020), cancer cells of uveal melanoma (UM) originate from melanocytes within the uveal tract of the eye. In the United States, the mean age-adjusted incidence attains 5.1 per million (Singh et al. 2011). About 85% of the tumor cases derive from the choroid whereas the remaining cases derive from the iris (3–5%) and ciliary body (5–8%) (Damato 2012). The early symptoms of UM are characterized by blurred vision, central dark spots, eye pain, and scleral congestion. Some certain advances have been made in the treatment of UM (Kashyap et al. 2016). As suggested by the National Comprehensive Cancer Network (NCCN) guideline, clinically, UM is usually treated by the combination of enucleation, local resection, stereotactic radiotherapy, brachytherapy, and phototherapy (Rao et al. 2020). However, over the past decades, the mortality rate is still rather high (Spagnolo et al. 2016). Metastasis approximately occurs in 50% of patients diagnosed with primary UM (Kujala et al. 2003), with the liver being the most common area involved. Once metastasis occurs, patients with metastatic UM manifest a median overall survival (OS) of less than 12 months (Patel 2013). Further, the response rates of monotherapy were consistently in the single-digit percentage range in a panel of previous studies while the combined immune checkpoint blockade (ICB) revealed higher response rates and better survival outcomes, albeit at the cost of high immune-related toxicity (Heppt et al. 2017a, 2017b, 2019; Kottschade et al. 2016; Algazi et al. 2016; Zimmer et al. 2015). Tebentafusp is a novel treatment modality and the only approved systemic therapy for uveal melanoma (Balushi et al. 2023). Notably, Tebentafusp achieved the best results compared to combined ICB and other systemic treatments; admittedly, the efficacy of this drug needs to be confirmed in more phase 3 clinical trials (Petzold et al. 2023).
To date, there are few studies on the co-gene expression patterns of UM (Kashyap et al. 2016; Zhang et al. 2014), which may limit the exploration of hub genes associated with the prognosis of the disease.
At present, immunotherapies including ICB, cancer vaccines, and chimeric antigen receptor T cell immunotherapy (CAR-T) have become a novel clinical strategy for treating various malignant tumors (Igarashi and Sasada 2020). Nevertheless, the overall therapeutic effect of immunotherapy for UM patients remains not satisfactory (Fu et al. 2022). The adverse response of UM to immunotherapy highlights the lack of knowledge about how metastatic UM develops immune resistance or evades immune surveillance. Interestingly, previous studies have identified that the special anatomic site of UM may contribute to its distinctive immune resistance (Bronkhorst and Jager 2012; Niederkorn 2012). As an immune-privileged organ, the eye serves to restrict the amount of inflammation caused by immune to occur in this region. To be specific, the eye aqueous humor contains a large number of anti-inflammatory and immunosuppressive cytokines, including transforming growth factor-β (TGF-β) and macrophage migration inhibitory factor (MIF) (Niederkorn 2012). Thus, it is urgent to better figure out the physiologic immunosuppressive microenvironment to improve individualized treatment for patients with UM.
In the present study, we sought to start with characterizing the immunophenotype of UM, and then identify the gene modules associated with metastasis using weighted gene correlation network analysis (WGCNA) analysis. Key genes were further identified by univariate Cox, least absolute shrinkage and selection operator (LASSO)-Cox analysis to construct a risk score. Moreover, CIBERSORT analysis was used to evaluate and quantify the degree of immune cell infiltration among low- and high-risk UM patients, and then a predictive model was constructed by combining clinical variables.
Materials and methods
Dataset collection
mRNA expression profiles and the corresponding clinical characteristics of UM samples were extracted from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds) including GSE22138. Based on the GPL570 platform, this dataset included 63 UM samples. The array probe name was converted to a matching gene name according to the platform annotation information. In addition, the mRNA expression profiles along with the clinical traits of 80 UM samples were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). We used the GSE22138 dataset as the training set whereas we utilized the TCGA-UM datasets as the testing sets. The expression data of genes in the training set and testing sets were standardized through the “limma” package. After merging, R software “sva” (Xie et al. 2021a) package was used to eliminate batch effect.
The collection of immune-related mRNA
According to the IMPORT database (https://www.immport.org/resources) list of immune-related genes (IRGs), IRGs were screened. The expression of annotated genes in GSE22138 was crossed with the expression of IRGs (Pearson correlation coefficient was greater than 0.3, and P value was less than 0.001). Ultimately, 23,516 immune-related mRNAs were obtained.
Construction of the co‑expression modules
Using the WGCNA package of R software (Langfelder and Horvath 2008), Pearson correlation coefficients between genes were calculated, and appropriate soft threshold β was selected to construct scale-free networks. The optimal soft threshold for adjacency calculation was determined using graphical methods. The topological overlap matrix (TOM) was then constructed from the adjacency matrix. The genes with similar expression patterns were divided into a class and constituted a gene co-expression module. The minimum number of genes in the co-expression modules was set as 50 for the high reliability of the results. Based on TOM matrix, the average linkage hierarchical clustering method was used to cluster genes, and the minimum module was set according to the criteria of hybrid dynamic cutting tree. A weighted co-expression network was constructed to screen hub genes (Chen et al. 2019). The clinical traits of patients in this study included age, sex, presence or absence of metastasis, and time of death. The gene significance obtained in WGCNA meant the correlation between a gene and a clinical trait whereas high gene significance meant this gene was highly correlated with the clinical trait.
Construction and verification of the risk score signature
The univariate Cox regression analysis was performed to screen out the potential hub genes. The survival-related genes (P < 0.05) were then enrolled into the subsequent LASSO analysis. The minimum value of the partial likelihood deviance represented the optimal performance of the model. Consequently, the lambda value with the lowest corresponding deviance was chosen, and this algorithm will output a best model with minimum number of genes. The penalty parameter lambda was detected using tenfold cross-validation (Friedman et al. 2010). Furthermore, independence analysis of the signature was conducted through multivariate Cox regression analyses, and P < 0.05 was taken as statistically significant. Four genes with prognostic significance were screened, and the risk score was constructed using the four genes. The risk score was calculated using the following formula:
In this formula, Coefi is the coefficient and Expi is the expression value of immune-related mRNA. By the survminer R package (Ramos et al. 2017), we calculated an optimal cut-off value for best division of low- and high-risk groups who differed in their survival time both in the training set and validation set. Kaplan–Meier survival analysis was used to determine the difference in OS between the two groups.
Four gene expression level analysis
To further characterize the four genes used to construct the risk score, we performed gene expression profiling. Cancer Cell Line Encyclopedia (CCLE) (https://sites.broadinstitute.org/ccle) database was applied to obtain mRNA expression levels of four genes in cell lines (Nusinow et al. 2020). We further investigated gene expression in 33 types of cancer and normal tissues from TCGA datasets using the enhanced version of tumor immune estimation resource (TIMER2.0) (http://timer.cistrome.org/) (Li et al. 2020).
Gene set enrichment analysis (GSEA) enrichment analysis
The single-sample GSEA (ssGSEA) analysis was performed to identify differences in the set of genes expression between the high-risk and low-risk groups. Subsequently, we performed ssGSEA (Subramanian et al. 2005) to obtain further insights into the differences in enrichment pathways between high-risk and low-risk groups. In ssGSEA analysis, four R packages “limma”, “GSVA”, “GSEABase”, “ggpubr”, and “shape2” were used.
Analysis of immune cell infiltration
To predict the proportion of infiltrating immune cells in tumor tissue, the CIBERSORT bioinformatics computational tool was used. Deconvolution algorithm was used to calculate the infiltration of different types of immune cells between high-risk and low-risk groups. The “corrplot” package was used to deconvolute the correlation between 22 types of tumor-infiltrating immune cells in UM.
Construction and verification of prognostic-related predictive model
The predictive model was constructed based on age, gender, and risk score. In addition, we used time-dependent receiver operating characteristic (ROC), calibration curve, and decision curve analysis (DCA) curve analysis to assess the predictive value of this signature. To facilitate the prediction of 1-, 3-, and 5-year OS of UM patients, we developed nomogram using the “survival” and “regplot” R packages.
Statistical analysis
All statistical P values were two sided, and P < 0.05 was considered statistically significant. All data analysis was performed in R 4.0.1 software (64-bit; https://www.r-project.org/).
Results
Extraction and screening of immune-related mRNAs
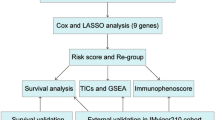
The process of data extraction and processing is shown in Fig. 1. First, we obtained expression profile data and corresponding clinical information of 63 tumor samples from GSE22138. Human GPL570 platform was used to annotate genes and obtain 23,520 mRNA expression data. Subsequently, 2013 IRGs were obtained from the IMPORT database. By Pearson correlation analysis (Pearson correlation coefficient greater than 0.3, P value less than 0.001), a total of 23,516 immune-associated mRNAs were identified for subsequent analysis.

Flow chat of the study. GEO gene expression omnibus; GTF gene transfer format; UM uveal melanoma; TCGA The Cancer Genome Atlas; LASSO least absolute shrinkage and selection operator; GSEA gene set enrichment analysis; ROC receiver operating characteristic; DCA decision curve analysis
Construction of the co‑expression modules
The first step of WGCNA analysis is to calculate the soft threshold β. It can be seen from Fig. 2 that when the soft threshold reaches 5, the curve tends to be gentle, and the constructed network is more in line with the scale-free network characteristic (Fig. 2A, B). The 18 co-expressed gene modules were obtained by WGCNA analysis. Based on the module genes, the module cluster tree was constructed. The cluster tree of genes showed the module division process (Fig. 2C). Pearson correlation coefficient and P value between each module and clinical traits were calculated (Fig. 2D). In this paper, the coefficient threshold of the module-clinical trait relationship was set at 0.3 ((> 0.3 means positively related to metastasis and < − 0.3 means negatively associated with metastasis)). As shown in the figure, the Black, Purple, and Blue modules were significantly positively correlated with metastasis (r = 0.44, P < 0.001; r = 0.445, P = 0.001; r = 0.31, P = 0.022), and the genes contained in these three modules were included in the next analysis (Fig. 2E–G).

Construction of the co-expression modules via WGCNA in the training set. WGCNA weighted gene co-expression network analysis; (cor), correlation. A Soft threshold β screening process 1; B Soft threshold β screening process 2; C Clustering tree of genes (a total of 18 coexpressed gene modules are obtained, which are represented by different colors at the bottom); D Heat map of correlation between modules and clinical traits (horizontal axis represents clinical traits, different colors on the left vertical axis represent different modules, each grid marks the correlation coefficient, and the corresponding P-value is in parentheses. The darker the color, the greater the degree of correlation); E Scatter plots of gene and model correlation and gene and trait correlation (blue module); F Scatter plots of gene and model correlation and gene and trait correlation (black modules); G Scatter plots of gene-model correlation and gene-trait correlation (purple module)
Prognostic gene screening and construction of risk score
First, univariate Cox analysis was performed to obtain 82 prognostic mRNAs (P < 0.05). Then the LASSO regression analysis was iterated 1000 times, the data features were reduced in dimension, and 13 optimal candidate genes were obtained (Fig. 3A, B), (for example, EEF1A2, GJA1, KAT8, LAMTOR2, LIG3, ZNF707, NSMF, OTUD3, PDK1, SLC39A1, SLC5A6, STAG3L3, UBXN2B, respectively.). The forest map showed the relationship between the 13 selected candidate genes and prognosis (Fig. 3C). Then multivariate Cox analysis was performed, and finally four genes (UBXN2B, OTUD3, KAT8, LAMTOR2) were included to establish the risk score.

The screening out of gene-based risk score signature for patients with UM in the training set. UM uveal melanoma
After construction of the four-gene-based OS risk score signature, UM patients in the training set (GSE22138) were divided into low- and high-risk groups using the risk scores calculated by the optimal cut-off value. As shown in the figure, with the increase of risk score, the survival time presented a trend of shortening. Moreover, the proportion of death in the high-risk group was higher than that in the low-risk group (Fig. 4A, B). This may indicate that our gene risk score could nicely predict the progression and aggressiveness of UM. The Kaplan–Meier (KM) curve demonstrated that in both the training set and the validation set, high-risk patients had significantly poorer OS than low-risk ones (log-rank P < 0.05) (Fig. 5A, B). Subsequently, we performed the forest plot to evaluate the prognostic value of the risk score calculated by the formula mentioned above. And we found that the risk score was a prognostic factor for UM in both the training and testing sets (training set: hazard ratio (HR) = 1.11 (1.05, 1.06), P < 0.001; testing set: HR = 1.12 (1.05, 1.19), P < 0.001) (Supplementary Fig. 1).

The survival status of the patients in high-risk and low-risk groups

Survival analysis
Analysis of gene expression profile
First, we examined the mRNA expression level of these four genes (UBXN2B, OTUD3, KAT8, LAMTOR2) in CCLE UVM cell lines. For example, we found that the mRNA expression level of UBXN2B was highest in WM3772F, followed by MEL270 and 921. In terms of OTUD3, the mRNA expression level was highest in 921, followed by MEL202 and MEL270. Details can be seen in Supplementary Fig. 2. Taking TCGA data, compared with adjacent normal tissues, the UBXN2B expression level was significantly upregulated in breast invasive carcinoma (BRCA), cholangiocarcinoma (CHOL), esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), head and neck squamous cell carcinoma (HNSC)-human papilloma virus (HPV), liver hepatocellular carcinoma (LIHC), and stomach adenocarcinoma (STAD), but downregulated in kidney chromophobe (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), lung squamous cell carcinoma (LUSC), skin cutaneous melanoma (SKCM), and thyroid carcinoma (THCA). Details can be seen in Supplementary Fig. 3.
GSEA enrichment analysis
The results of GSEA enrichment analysis for high-risk and low-risk groups were as follows. The high-risk groups were mainly concentrated in antigen processing and antigen presentation, cytokine receptor interaction, natural killer cytotoxicity, oocyte meiosis, primary immunodeficiency, systemic lupus erythematosus, and toll-like receptor signaling pathways. In contrast, the low-risk group was mainly concentrated in basal cell carcinoma, cytochrome P450 metabolism, Hedgehog signaling pathway, histidine metabolism, olfactory conduction, phenylalanine metabolism, taurine metabolism, tight junction, tryptophan metabolism, and tyrosine metabolism (Fig. 6).

GSEA based on the immune-related mRNAs risk model. GSEA gene set enrichment analysis
Analysis of immune cell infiltration
By CIBERSORT analysis, the infiltration of 22 kinds of immune cells in low-risk group and high-risk group was obtained. As shown in the figure, the infiltrating levels of γδT cells were significantly higher in the high-risk group compared with the low-risk group (P < 0.05). Furthermore, using the TIMER website (https://cistrome.shinyapps.io/timer/), high γδT cells infiltration was found to be associated with worse OS of UM patients (P < 0.05). These results indicated that the bad prognosis might be partly due to the high infiltration of this type of immune cell (Fig. 7).

Boxplots of differences in immune cell infiltration between high-risk and low-risk groups
Construction of prognostic-related predictive model
To further verify the clinical significance of risk score, two clinical variables, gender and age, were added for multivariate Cox analysis. In the training set, the survival rates of 1-, 3-, and 5 year were predicted, and the time-dependent area under the curves (AUCs) of the model were above 0.930 (Fig. 8A, B). To be specific, the AUCs of the ROC curve were 0.930 at 1-year, 0.947 at 3-year, and 0.951 at 5-year in the training cohort. In the validation set, the model also showed a good degree of differentiation. The calibration curves of 1 year, 3 years, and 5 years in the training set were close to the diagonal line, which indicated that the model had a good calibration degree (Fig. 9A, B). However, the performance of calibration curve in the validation set was not as good as that of the verification set, which may be due to the fact that the sample size was not large enough and thus, the classifier was weak. In the training set, the 1-, 3- and 5-year DCA curves were all between None and ALL, showing a good net benefit, whereas in the verification set, the 1-year and 3-year DCA curves performed better (Fig. 9C, D). To facilitate clinical use, this paper further constructed a nomogram composed of age, gender, and risk score. After calculating the score of each variable and combining the total score, the survival rate of 1 year, 3 years, and 5 years can be calculated (Fig. 10).

ROC of risk model in the dataset. ROC receiver operating characteristic; AUC area under the curve

Calibration and DCA curve in the dataset. DCA decision curve analysis; OS overall survival

Nomogram was plotted for the prediction of overall survival time
Discussion
Despite its low incidence, UM is still the most common primary intraocular malignant tumor in adults, and is characterized by its high mortality (> 95%), high metastasis (> 50%), and poor prognosis, making the exploration for effective biomarkers for assessing prognosis crucial (Marseglia et al. 2021). In this study, WGCNA were used to screen out metastasis-related co-expression modules from the GEO database. The Black, Purple, and Blue modules were significantly associated with the progression and metastasis in UM. Then we used univariate Cox, LASSO-Cox regression to screen out four genes associated with prognosis. The GSEA analysis demonstrated that multiple immune-related pathways were upregulated in the high-risk group. Subsequently, we used these four prognosis-related genes to construct a risk score signature, which was found to be an independent factor for predicting OS. Moreover, combined with other age, gender, a clinical predictive model was established. Both the survival curves and ROC analysis results showed the robustness and reliability of the predictive model for prognosis prediction of UM patients. The AUCs of the ROC curve in the training cohort for 1-year, 3-year, and 5-year were 0.930, 0.947, and 0.951, respectively. This predictive model helps simplify clinical individualized monitoring and treatment strategies for UM patients.
Four genes included in the predictive model have not been adequately studied in UM. As indicated by previous study, downregulation of OTUD3 was associated with poor prognosis in patients with esophageal cancer. The specific mechanism may be that OTUD3 directly interacts with zinc finger protein ZFP36, and stabilizes ZFP36 by inhibiting FBXW7-mediated polyubiquitination of K48 junction (Ntunzwenimana et al. 2021). Xie et al. (2021b) found that OTUD3 was significantly overexpressed in hepatocellular carcinoma, and high expression of OTUD3 was correlated with tumor size, distant metastasis, and poor TNM stage. Wang et al. (2021) analyzed the relation between OTUD3 and lymphatic metastasis, and found that nicotine-mediated OTUD3 downregulation inhibited VEGF-C mRNA decay to promote lymphatic metastasis of human esophageal cancer. Qiu et al. (2023) found that acetylation degradation of KAT8 could affect colorectal cancer invasion and migration. Dong et al. (2019) found that MYST1/KAT8/MOF promoted the progression of glioblastoma by activating epidermal growth factor receptor (EGFR) signaling. Previous study also identified KAT8 as a key molecule important for cancer cell survival. To be specific, they found that KAT8 regulated G2/M cell cycle arrest through AKT/ERK-cyclin D1 signaling (Zhang et al. 2013). In addition, LAMTOR3 expression was significantly decreased in renal clear cell carcinoma compared with normal renal tissue. LAMTOR3 may be a potential marker for the diagnosis and treatment of renal clear cell carcinoma (Gong et al. 2022). Song et al. (2015) found that LAMTOR3 polymorphisms may be a potential biomarker of genetic susceptibility to gastric cancer. In a mouse model of pancreatic cancer, the siRNA-FA-PEG-COL nanoparticles against LAMTOR, which strongly inhibited retroperitoneal invasion and significantly inhibited peritoneal dissemination compared to the other nanoparticles, improved prognosis of the mice (Taniuchi et al. 2019).
Two clinical variables, age and gender, were also included. Kaliki et al. (2013) found that the older patients with UM had worse prognosis. In addition, they divided UM patients into young adults (< 20 years old), middle-aged (21–60 years old), and senior citizens (> 60), and found that younger people had a lower risk of metastasis compared to middle-aged and older adults. In a survival analysis of 119 patients, Rietschel et al. (2005) found that women had a significantly lower risk of disease-specific death than men. Similarly, a study of 723 patients with UM found that male patients had a worse prognosis compared to female patients (Zloto et al. 2013). Therefore, two variables including age and gender were included in the clinical predictive model for further analysis. Previous studies have tried to reveal the immune infiltration pattern of this special tumor (Luo and Ma 2020; Pan et al. 2020). In this study, we found that γδT infiltration levels were significantly higher in high-risk UM samples than in low-risk ones (P < 0.05). Consistent with previous reports (Wang et al. 2020; Lei and Zhang 2021), γδT were identified to be related to poor prognosis in UM patient. Previous studies also have focused on prognosis prediction based on the gene expression profiles of UM. For instance, Vaquero-Garcia et al. (2017) used variables including chromosome characteristics, age, sex, tumor site, and tumor size to predict patients’ prognosis, and the accuracy of model attained 85%. Using TCGA as the training cohort and GSE22138 as the validation cohort, (Luo and Ma 2020) identified 21 prognostic genes related to microenvironment. Xue et al. (2019) conducted univariate and multivariate Cox analysis on the TCGA cohort to establish an 18-gene prognostic model for patients with OS. NDUFB9, NDUFV2, Cyc1, and CTNNB1 were screened out by Choi et al. (2020) as prognostic predictors of UM patients. Luo et al. (2020) performed univariate Cox regression and LASSO-Cox to construct ten gene signatures, and used the GSE22138 cohort to verify the signatures. Compared to Luo et al., the model in our paper is more accurate in predicting 1-year and 3-year OS (AUCs of 0.892 and 0.737, respectively).
Our study had some strengths. First, we identified metastasis-related genes by WGCNA, and then constructed a gene signature based on these genes for better prognosis prediction. Notably, metastasis is significantly associated with the survival of patients. To the best of our knowledge, there is limited literature on the immune signature of metastatic UM tumors (Qin et al. 2017; Rothermel et al. 2016; Krishna et al. 2017; Javed et al. 2017). Second, the model was derived from the GSE22138 cohort and verified by the external data set TCGA cohort. Third, clinical variables including age and gender were added together with risk score to establish a model for predicting UM prognosis.
Admittedly, there are some limitations in our current study. First, these results were obtained from public databases, and therefore need to be validated by experiment. Second, the sample size is not large enough, and further data training of large multi-center samples is needed to improve the performance of the model.
Conclusion
In conclusion, through WGCNA, our study demonstrated that three co-expression modules were associated with metastasis among UM patients. Through LASSO-Cox analysis, four genes were finally identified for inclusion in the risk score. Furthermore, patients in high-risk group have significantly higher levels of an immune infiltrate (γδT cell) compared with patients in low-risk group. Notably, this model needs to be validated in a larger dataset, and may be conducive to therapeutic decision-making in the clinical setting.
Availability of data and materials
The datasets analyzed during the current study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) (GSE22138) and TCGA database (https://portal.gdc.cancer.gov/) (TCGA-UVM). All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- UM:
-
Uveal melanoma
- NCCN:
-
National comprehensive cancer network
- OS:
-
Overall survival
- ICB:
-
Immune checkpoint blockade
- TGF -β:
-
Transforming growth factor-β
- MIF:
-
Migration inhibitory factor
- WGCNA:
-
Weighted gene correlation network analysis
- LASSO:
-
Least absolute shrinkage and selection operator
- GEO:
-
Gene expression omnibus
- TCGA:
-
The Cancer Genome Atlas
- IRGs:
-
Immune-related genes
- TOM:
-
Topological overlap matrix
- GSEA:
-
Gene set enrichment analysis
- ssGSEA:
-
Single-sample GSEA
- ROC:
-
Receiver operating characteristic
- DCA:
-
Decision curve analysis
- KM:
-
Kaplan–Meier
- HR:
-
Hazard ratio
- AUCs:
-
Area under the curves
- EGFR:
-
Epidermal growth factor receptor
References
Algazi AP, Tsai KK, Shoushtari AN, Munhoz RR, Eroglu Z, Piulats JM, Ott PA, Johnson DB, Hwang J, Daud AI et al (2016) Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 122(21):3344–3353
Balushi KA, Hadhrami AA, Balushi HA, Das S, Lawati AA (2023) Tebentafusp as a promising drug for the treatment of uveal melanoma. Curr Drug Targets. https://doi.org/10.2174/0113894501280380231214105255
Bronkhorst IH, Jager MJ (2012) Uveal melanoma: the inflammatory microenvironment. J Innate Immun 4(5–6):454–462
Chen S, Yang D, Lei C, Li Y, Sun X, Chen M, Wu X, Zheng Y (2019) Identification of crucial genes in abdominal aortic aneurysm by WGCNA. PeerJ 7:e7873
Choi S, Ha M, Lee JS, Heo HJ, Kim GH, Oh SO, Lee JJ, Goh TS, Kim YH (2020) Novel prognostic factor for uveal melanoma: bioinformatics analysis of three independent cohorts. Anticancer Res 40(7):3839–3846
Damato B (2012) Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye (lond) 26(9):1157–1172
Dong Z, Zou J, Li J, Pang Y, Liu Y, Deng C, Chen F, Cui H (2019) MYST1/KAT8 contributes to tumor progression by activating EGFR signaling in glioblastoma cells. Cancer Med 8(18):7793–7808
Friedman J, Hastie T, Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33(1):1–22
Fu Y, Xiao W, Mao Y (2022) Recent advances and challenges in uveal melanoma immunotherapy. Cancers 14(13):3094
Gong Y, Lv Y, Xu F, Xiu Y, Lu Y, Liu Z, Deng L (2022) LAMTOR3 is a prognostic biomarker in kidney renal clear cell carcinoma. J Clin Lab Anal 36(9):e24648
Heppt MV, Steeb T, Schlager JG, Rosumeck S, Dressler C, Ruzicka T, Nast A, Berking C (2017a) Immune checkpoint blockade for unresectable or metastatic uveal melanoma: a systematic review. Cancer Treat Rev 60:44–52
Heppt MV, Heinzerling L, Kähler KC, Forschner A, Kirchberger MC, Loquai C, Meissner M, Meier F, Terheyden P, Schell B et al (2017b) Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur J Cancer (oxford, England: 1990) 82:56–65
Heppt MV, Amaral T, Kähler KC, Heinzerling L, Hassel JC, Meissner M, Kreuzberg N, Loquai C, Reinhardt L, Utikal J et al (2019) Combined immune checkpoint blockade for metastatic uveal melanoma: a retrospective, multi-center study. J Immunother Cancer 7(1):299
Igarashi Y, Sasada T (2020) Cancer vaccines: toward the next breakthrough in cancer immunotherapy. J Immunol Res 2020:5825401
Jager MJ, Shields CL, Cebulla CM, Abdel-Rahman MH, Grossniklaus HE, Stern MH, Carvajal RD, Belfort RN, Jia R, Shields JA et al (2020) Uveal melanoma. Nat Rev Dis Primers 6(1):24
Javed A, Arguello D, Johnston C, Gatalica Z, Terai M, Weight RM, Orloff M, Mastrangelo MJ, Sato T (2017) PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 9(16):1323–1330
Kaliki S, Shields CL, Mashayekhi A, Ganesh A, Furuta M, Shields JA (2013) Influence of age on prognosis of young patients with uveal melanoma: a matched retrospective cohort study. Eur J Ophthalmol 23(2):208–216
Kashyap S, Meel R, Singh L, Singh M (2016) Uveal melanoma. Semin Diagn Pathol 33(3):141–147
Kottschade LA, McWilliams RR, Markovic SN, Block MS, Villasboas Bisneto J, Pham AQ, Esplin BL, Dronca RS (2016) The use of pembrolizumab for the treatment of metastatic uveal melanoma. Melanoma Res 26(3):300–303
Krishna Y, McCarthy C, Kalirai H, Coupland SE (2017) Inflammatory cell infiltrates in advanced metastatic uveal melanoma. Hum Pathol 66:159–166
Kujala E, Mäkitie T, Kivelä T (2003) Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol vis Sci 44(11):4651–4659
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9:559
Lei S, Zhang Y (2021) Identification of survival-related genes and a novel gene-based prognostic signature involving the tumor microenvironment of uveal melanoma. Int Immunopharmacol 96:107816
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS (2020) TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 48(W1):W509-w514
Luo H, Ma C (2020) Identification of prognostic genes in uveal melanoma microenvironment. PLoS ONE 15(11):e0242263
Luo H, Ma C, Shao J, Cao J (2020) Prognostic implications of novel ten-gene signature in uveal melanoma. Front Oncol 10:567512
Marseglia M, Amaro A, Solari N, Gangemi R, Croce E, Tanda ET, Spagnolo F, Filaci G, Pfeffer U, Croce M (2021) How to make immunotherapy an effective therapeutic choice for uveal melanoma. Cancers 13(9):2043
Niederkorn JY (2012) Ocular immune privilege and ocular melanoma: parallel universes or immunological plagiarism? Front Immunol 3:148
Ntunzwenimana JC, Boucher G, Paquette J, Gosselin H, Alikashani A, Morin N, Beauchamp C, Thauvette L, Rivard M, Dupuis F et al (2021) Functional screen of inflammatory bowel disease genes reveals key epithelial functions. Genome Med 13(1):181
Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jané-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M et al (2020) Quantitative proteomics of the cancer cell line encyclopedia. Cell 180(2):387-402.e316
Pan H, Lu L, Cui J, Yang Y, Wang Z, Fan X (2020) Immunological analyses reveal an immune subtype of uveal melanoma with a poor prognosis. Aging 12(2):1446–1464
Patel SP (2013) Latest developments in the biology and management of uveal melanoma. Curr Oncol Rep 15(6):509–516
Petzold A, Steeb T, Wessely A, Koch EAT, Vera J, Berking C, Heppt MV (2023) Is tebentafusp superior to combined immune checkpoint blockade and other systemic treatments in metastatic uveal melanoma? A comparative efficacy analysis with population adjustment. Cancer Treat Rev 115:102543
Qin Y, de Macedo MP, Reuben A, Forget MA, Haymaker C, Bernatchez C, Spencer CN, Gopalakrishnan V, Reddy S, Cooper ZA et al (2017) Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: a pilot study. Oncoimmunology 6(6):e1321187
Qiu B, Li S, Li M, Wang S, Mu G, Chen K, Wang M, Zhu WG, Wang W, Wang J et al (2023) KAT8 acetylation-controlled lipolysis affects the invasive and migratory potential of colorectal cancer cells. Cell Death Dis 14(2):164
Ramos M, Schiffer L, Re A, Azhar R, Basunia A, Rodriguez C, Chan T, Chapman P, Davis SR, Gomez-Cabrero D et al (2017) Software for the integration of multiomics experiments in bioconductor. Can Res 77(21):e39–e42
Rao PK, Barker C, Coit DG, Joseph RW, Materin M, Rengan R, Sosman J, Thompson JA, Albertini MR, Boland G et al (2020) NCCN guidelines insights: uveal melanoma, version 1.2019. J Natl Compr Cancer Netw: JNCCN 18(2):120–131
Rietschel P, Panageas KS, Hanlon C, Patel A, Abramson DH, Chapman PB (2005) Variates of survival in metastatic uveal melanoma. J Clin Oncol : off J Am Soc Clin Oncol 23(31):8076–8080
Rothermel LD, Sabesan AC, Stephens DJ, Chandran SS, Paria BC, Srivastava AK, Somerville R, Wunderlich JR, Lee CC, Xi L et al (2016) Identification of an immunogenic subset of metastatic uveal melanoma. Clin Cancer Res: off J Am Assoc Cancer Res 22(9):2237–2249
Singh AD, Turell ME, Topham AK (2011) Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology 118(9):1881–1885
Song P, Wang W, Tao G, Chu H, Wang M, Wu D, Tong N, Gong W, Zhou J, Zhang Z et al (2015) A miR-29c binding site genetic variant in the 3’-untranslated region of LAMTOR3 gene is associated with gastric cancer risk. Biomed Pharmacother = Biomed Pharmacotherapie 69:70–75
Spagnolo F, Picasso V, Spano L, Tanda E, Venzano C, Queirolo P (2016) Update on metastatic uveal melanoma: progress and challenges. BioDrugs: Clin Immunother Biopharma Gene Ther 30(3):161–172
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545–15550
Taniuchi K, Yawata T, Tsuboi M, Ueba T, Saibara T (2019) Efficient delivery of small interfering RNAs targeting particular mRNAs into pancreatic cancer cells inhibits invasiveness and metastasis of pancreatic tumors. Oncotarget 10(30):2869–2886
Vaquero-Garcia J, Lalonde E, Ewens KG, Ebrahimzadeh J, Richard-Yutz J, Shields CL, Barrera A, Green CJ, Barash Y, Ganguly A (2017) PRiMeUM: a model for predicting risk of metastasis in uveal melanoma. Invest Ophthalmol vis Sci 58(10):4096–4105
Wang Y, Xu Y, Dai X, Lin X, Shan Y, Ye J (2020) The prognostic landscape of adaptive immune resistance signatures and infiltrating immune cells in the tumor microenvironment of uveal melanoma. Exp Eye Res 196:108069
Wang M, Li Y, Xiao Y, Yang M, Chen J, Jian Y, Chen X, Shi D, Chen X, Ouyang Y et al (2021) Nicotine-mediated OTUD3 downregulation inhibits VEGF-C mRNA decay to promote lymphatic metastasis of human esophageal cancer. Nat Commun 12(1):7006
Xie X, Wang EC, Xu D, Shu X, Zhao YF, Guo D, Fu W, Wang L (2021a) Bioinformatics analysis reveals the potential diagnostic biomarkers for abdominal aortic aneurysm. Front Cardiovasc Med 8:656263
Xie P, Chen Y, Zhang H, Zhou G, Chao Q, Wang J, Liu Y, Fang J, Xie J, Zhen J et al (2021b) The deubiquitinase OTUD3 stabilizes ACTN4 to drive growth and metastasis of hepatocellular carcinoma. Aging 13(15):19317–19338
Xue M, Shang J, Chen B, Yang Z, Song Q, Sun X, Chen J, Yang J (2019) Identification of prognostic signatures for predicting the overall survival of uveal melanoma patients. J Cancer 10(20):4921–4931
Zhang S, Liu X, Zhang Y, Cheng Y, Li Y (2013) RNAi screening identifies KAT8 as a key molecule important for cancer cell survival. Int J Clin Exp Pathol 6(5):870–877
Zhang Y, Yang Y, Chen L, Zhang J (2014) Expression analysis of genes and pathways associated with liver metastases of the uveal melanoma. BMC Med Genet 15:29
Zimmer L, Eigentler TK, Kiecker F, Simon J, Utikal J, Mohr P, Berking C, Kämpgen E, Dippel E, Stadler R et al (2015) Open-label, multicenter, single-arm phase II DeCOG-study of ipilimumab in pretreated patients with different subtypes of metastatic melanoma. J Transl Med 13:351
Zloto O, Pe’er J, Frenkel S (2013) Gender differences in clinical presentation and prognosis of uveal melanoma. Invest Ophthalmol vis Sci 54(1):652–656
Acknowledgements
We acknowledge TCGA and GEO database for providing their platforms and contributors for uploading their meaningful datasets.
Funding
This study was supported by the National Natural Science Foundation of China (81900870); Young Talent Fund of University Association for Science and Technology in Shaanxi, China (20220305); Project funded by China Postdoctoral Science Foundation (2021MD703956); Clinical Research Program of Air Force Medical University (2022LC2247); and Clinical Application Research Project of Xijing Hospital (JSYXM02).
Author information
Authors and Affiliations
Contributions
JW and ZFZ: conceptualization, data curation, investigation, methodology, software, visualization, writing—original draft; MML: conceptualization, data curation, formal analysis; JXS: methodology, project administration, supervision—review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Liu, M., Sun, J. et al. Immunogenic profiling of metastatic uveal melanoma discerns a potential signature related to prognosis. J Cancer Res Clin Oncol 150, 23 (2024). https://doi.org/10.1007/s00432-023-05542-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-023-05542-z