
Abstract
Introduction
Cannabinoids are a group of terpenophenolic compounds derived from the Cannabis sativa L. plant. There is a growing body of evidence from cell culture and animal studies in support of cannabinoids possessing anticancer properties.
Method
A database search of peer reviewed articles published in English as full texts between January 1970 and April 2021 in Google Scholar, MEDLINE, PubMed and Web of Science was undertaken. References of relevant literature were searched to identify additional studies to construct a narrative literature review of oncological effects of cannabinoids in pre-clinical and clinical studies in various cancer types.
Results
Phyto-, endogenous and synthetic cannabinoids demonstrated antitumour effects both in vitro and in vivo. However, these effects are dependent on cancer type, the concentration and preparation of the cannabinoid and the abundance of receptor targets. The mechanism of action of synthetic cannabinoids, (−)-trans-Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol (CBD) has mainly been described via the traditional cannabinoid receptors; CB1 and CB2, but reports have also indicated evidence of activity through GPR55, TRPM8 and other ion channels including TRPA1, TRPV1 and TRPV2.
Conclusion
Cannabinoids have shown to be efficacious both as a single agent and in combination with antineoplastic drugs. These effects have occurred through various receptors and ligands and modulation of signalling pathways involved in hallmarks of cancer pathology. There is a need for further studies to characterise its mode of action at the molecular level and to delineate efficacious dosage and route of administration in addition to synergistic regimes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since time immemorial, the Cannabis plant has been used as a source of fibre, herbal remedy, medicinal and religious purposes (Kalant 2001; Goncalves et al. 2020). In the mid-nineteenth century, O’Shaughnessy and Moreau reported positive effects of cannabis on muscle spasms, vomiting, convulsions, rheumatism, tetanus, and rabies (O’Shaughnessy 1843; Zuardi 2006). However, during the twentieth century, its utilisation in Western medicine started to decline as a result of political prejudices and economic interests rather than scientific or medical reasons (Zuardi 2006). Over recent years, cannabis and its derivatives have been used for treating chemotherapy induced nausea and vomiting, epilepsy and multiple sclerosis amongst other indications (Parker et al. 2011; Kleckner 2019). Increasing data from and in vivo studies have started to show evidence of cannabis in modulating signalling pathways involved in cancer cell proliferation, autophagy, apoptosis and inhibition of angiogenesis and metastasis (Velasco et al. 2016). Emerging reports have also indicated synergistic effects of cannabinoids in combination with antineoplastic drugs (Moreno et al. 2019; Dariš et al. 2019; Fogli et al. 2006; Velasco et al. 2012).
The cannabis plant has been termed as a “storehouse” of several pharmacologically relevant compounds (Andre et al. 2016). The unique qualities of each cannabis variety or chemovar are the result of varying concentrations of numerous classes of bioactive molecules, most notably, cannabinoids as shown in Fig. 1, terpenoids and flavonoids (Chakravarti et al. 2014). Cannabinoids interact directly with cannabinoid receptors, which include G-protein coupled receptors (cannabinoid receptor 1, CB1 and cannabinoid receptor 2, CB2), ligand-gated ion channels (i.e. vanilloid cell surface channels) and nuclear receptors (i.e. peroxisome proliferator-activated receptor gamma, PPARγ) (Moreno et al. 2019; Śledziński et al. 2018) comprising the endogenous endocannabinoid system (ECS) (Zou and Kumar 2018). Three major classifications of cannabinoids include phytocannabinoids (plant-based), such as Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol (CBD), endocannabinoids (or endogenous cannabinoids) which include anandamide (AEA) and 2-arachidonolyglycerol (2-AG) and synthetic cannabinoids that mimic the cannabinoid groups (1) and (2) (Pertwee 2006; Lu and Mackie 2016). Endocannabinoids play a crucial role in mediating physiological functions including metabolic, cardiovascular regulation, reproduction, inflammatory response, immune system and analgesia (Guindon and Hohmann 2012; Kaur et al. 2016). AEA and 2-AG are degraded by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) enzymes (Pisanti et al. 2013). Modulation of their activity may have potential therapeutic implications and inhibitors are under active investigation as pharmaceuticals. Synthetic cannabinoids have been studied extensively and some have been shown to be highly bioactive than their natural counterparts, some common ones include WIN55, 212–2 (potent CB1 receptor agonist), JWH-018, JWH-073, JWH-133 (CB receptor agonists) and SR141716 or Rimonabant (CB1 receptor antagonist) (Morales et al. 2017), overview shown in Fig. 2.

The chemical structures of Cannabigerol (CBG), Cannabidiol (CBD), Tetrahydrocannabinol (Δ9-THC), Cannabichromene (CBC) and Cannabinol (CBN)-type neutral, varinic and acidic phytocannabinoids. More than 120 phytocannabinoids have been isolated from Cannabis sativa L. which can be distinguished into eleven chemical subtypes (Gonçalves et al. 2020; ElSohly 2017). Their common chemical features include a dibenzopyran ring and a hydrophobic alkyl chain (Morales et al. 2017). Aside from Δ9-THC and CBD, there has been a current focus on the therapeutic properties of some minor, varinic and acidic cannabinoids (Andre et al. 2016; Franco et al. 2020). Created with BioRender.com

Overview of the components of the endocannabinoid system (ECS) which include endogenous endocannabinoids; Anandamide (AEA) and 2-Arachidonoylglycerol (2-AG), its major receptors classified into cannabinoid receptors 1 and 2, and non-cannabinoid receptors; GPR55, GPR35, GPR119, GPR18, GPR12, ion channels including transient receptor potential cation channel subfamily members; TRPM8, TRPV1, TRPV2, peroxisome-proliferator-activated receptors (PPAR). A third component of the system are its enzymes/transporters responsible for the synthesis and degradation of endocannabinoids including serum albumin, ceramide, cholesterol, diacylglycerol lipase (DAGL), phospholipase C (PLC), monoacylglycerol lipase (MAGL), fatty acid amide hydrolase (FAAH). Created with BioRender.com
Several studies have reported the varying affinities of phytocannabinoids for the classical CB1 and CB2 receptors with agonistic and antagonistic behaviours (Morales et al. 2017; Zhao and Abood 2013). However, it is now emerging that cannabinoids can interact with multiple orphan G-protein coupled receptors (GPCRs) including GPR12, GPR18, GPR35, GPR55, GPR119, opioid and serotonin receptors (Morales et al. 2017; Zhao and Abood 2013; Console-Bram et al. 2014; Brown et al. 2017; Soderstorm et al. 2017; Ferro et al. 2018; Guerrero-Alba 2019). The interaction of GPCRs is crucial for maintaining the ECS as it allows the production of endocannabinoids from cells through activation of Gq/11 or Gs proteins causing the activation of the cannabinoid receptor (Gyombolai et al. 2012). Furthermore, the downstream receptor-mediated effects of endocannabinoids also contribute to the plasticity of the ECS (Lu and Mackie 2016).
Since the first report of cannabinoids anticancer effects (Munson et al. 1975), there have been many studies investigating phytocannabinoids, endogenous and synthetic ones in multiple cancer models. Various signalling pathways and changes to internal conditions which favour antitumour activity by cannabinoids have been observed. CBD amongst other cannabinoids has shown to increase the de novo synthesis of ceramide through upregulation of a plethora of enzymes each catalysing specific biochemical steps. Ceramide synthases are one of the major group of enzymes involved and reports have revealed an upregulation of its six isoforms; CerS 1–6 (Ceramide Synthases 1–6) in cancer via cannabinoids (Gomez et al. 2002; Gustafsson et al. 2009; Schiffman et al. 2009). However, it is not clear whether specific isoform(s) upregulation correlates to the cancer type and whether this is also specific to the type of cannabinoid. An interesting finding from a report has shown siRNA-induced knockdown of ceramide synthase 1 (CerS1 isoform) prevented gemcitabine-induced caspase 9 activation (Senkal et al. 2007; Levy and Futerman 2010). This could be explored further when considering cannabinoids action synergistically with chemotherapy drugs as ceramide may have the ability to sensitize the cancer cells to chemotherapy agents. Another major area of cannabinoids action has been through modulating the cell cycle. In a recent report in gastric cancer cells, CBD-induced cell cycle arrest at the G0–G1 phase and retardation in this phase corresponded to a reduction in CDK2/cyclin E protein levels (Zhang et al. 2019). Apoptotic changes are prevalent in cannabinoids mechanism of action which include morphological changes to the cells and cytoplasmic vacuolization, an increase in cleaved caspase-3 and -9 levels and activation of the mitochondrial apoptotic pathway (Zhang et al. 2019; Schoeman et al. 2020). Endoplasmic reticulum (ER) stress which occurs following ceramide synthesis causes downstream apoptotic changes and increases in proapoptotic proteins, such as BAD and Bax, also resulting in an increase in reactive oxygen species (ROS) signalling (Zhang et al. 2019). Δ9-THC in glioma cells has shown to induce upregulation of the p8 protein (involved in ER stress and metastasis) via de novo synthesis of ceramide (Carracedo et al. 2006). From the literature available, it is evident that there is an interplay between cannabinoids downstream effects.
Overall cannabinoids induce apoptosis to inhibit proliferation, downregulate the vascular endothelial growth factor (VEGF) pathway affecting angiogenesis and dampen metastasis by inhibiting cell adhesion and migration through modifying matrix metalloproteinase 2, 9 (MMP2, 9), tissue inhibitor of matrix metalloproteinases 1 (TIMP1), inhibitor of DNA binding 1 (ID1) and inducing ER stress (Velasco et al. 2016). Cancer cells do not exist in isolation and the tumour microenvironment (TME) has also been an imperative target for cancer therapy as it can influence the propensity for tumour growth, metastasis and resistance to therapy. The TME is composed of a host of factors including cancer-associated fibroblasts (CAFs), immune and inflammatory cells, lymph and blood vasculature, neuroendocrine cells, and extracellular matrix (ECM) (Wang et al. 2017). Cancer stem cells (CSCs), a subpopulation of stem cells expressing CD44, CD24 and CD133, are tumorigenic with demonstrated resistance to certain chemotherapeutics and also play a role in metastasis (Yu et al. 2012). Reports have shown the involvement of cannabinoids in inhibiting CAFs and CSCs in prostate and breast cancer models (Sharma et al. 2014; Mohammadpour et al. 2017; Pietrovito et al. 2020). The aforementioned effects, however, occur at varying degrees which depend on the cancer cell line, the expression levels of cannabinoid receptors, the type of cannabinoid compound and dosage.
The aim of this review is to analyse pre-clinical work and outline previous and forthcoming clinical research studies exploring cannabinoids in cancer treatment. Below, we outline the research encompassing endogenous and non-endogenous cannabinoids in which we review the proposed mechanisms of action culminated from studies into various cancers and discuss the need for more clinical studies to explore the possible therapeutic efficacy of cannabinoids as a possible treatment for cancer.
Method
Research question
This narrative review was conducted of available literature reporting the treatment effects of all cannabinoids as either a single agent or co-administered with other antitumour therapies in all cancer types. The aim of this review is to analyse and evaluate pre-clinical and clinical research determining the use of cannabinoids as a potential anti-cancer therapy.
Search strategy and inclusion criteria
A broad electronic search was conducted on Google Scholar, MEDLINE, PubMed and Web of Science articles published in English between 1st January 1970 and 30th April 2021. Investigations of cannabinoids use in oncology clinical trials were searched using the database, clinicaltrials.gov.uk with the key words; “Cannabinoids and Cancer”, “Cannabis and Cancer”, “Tetrahydrocannabinol and Cancer”, “CBD and Cancer” and “THC and Cancer”. The literature search was performed by two independent researchers (N.M. and S.E.) and if any discrepancies were identified then these were resolved by a senior author (M.S.). The reference lists of all publications were screened for further relevant references. The free text search included articles citing both original research and literature reviews. Inclusion criteria encompassed all reports identifying cannabinoids use in pre-clinical cancer models which includes in vitro, in vivo and in ovo experimental models, as well as clinical research. In addition, reports of potential mechanisms of action and signalling pathways involved were also included. Where literature reviews were identified, the relevant cited studies were also identified and included for de novo analysis.
Data extraction and presentation
Two independent researchers (N.M. and S.E.) performed the data extraction. Primary research papers reporting half maximal inhibitory concentration (IC50) and concentrations where the described effects were observed in pre-clinical cancer models were included in separate tables for in vitro and in vivo investigations. Concentration values are presented as micro-molar concentrations (μM) with their standard deviation (S.D.), standard error (S.E), or range except when unreported in the original study.
Results
Mechanism of action and signalling pathways
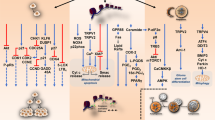
The ECS is a complex system composed of different ligands, receptors and ion channels resulting in many signalling pathways subject to modulation from external cannabinoids as shown in Fig. 3. It is therefore no surprise that there remains ambiguity in its precise role within cancer pathophysiology (Wu 2019). Many pre-clinical studies and histological analysis of patient tumours, suggest that an upregulation in the CB1 and CB2 receptors, endogenous ligands and over-activation of the ECS correlates with more aggressive tumours (Dariš et al. 2019) although other reports have concluded the contrary (Jung et al. 2013; Tutino et al. 2019). Cancer is a heterogenous disease and current evidence should be interpreted on the basis that different tumour types have been shown to exhibit various levels of CB receptors as well as ECS components. The role of the endogenous endocannabinoids and CB receptors within each cancer system is specific to the underlying cancer, therefore conflicting data can be presented across different cancers. It has also been reported that some cannabinoids have shown oncological effects independent of known CB receptors (Moreno et al. 2019; Fogli et al. 2006) implying that there may be undiscovered cannabinoid receptors implicated in cancer pathophysiology.

Overview of the downstream activation and crosstalk of signalling pathways of cannabinoid and non-cannabinoid receptors. Activation of the cannabinoid receptors CB1 and CB2 (red arrows) via cannabinoids stimulate ERK1/2 signalling which activates p27 and p21 causing a decrease in cyclins D and E, cdc2 and cdk2 through an increase in pRb, leading to cell cycle arrest. Inhibition of the P13K pathway leads to a decrease in Akt which inhibits cell proliferation. Biosynthesis of ceramide takes place at the endoplasmic reticulum through a series of biochemical steps involving many enzymes which help to convert dihydroceramides (DhCers) into ceramide. An increase in ceramide level in turn increases the stress protein p8/Nupr1 and TRIB3 which activates upregulation of ATF4 and CHOP proteins. A decrease in Akt leads to a downregulation in mTORC1 signalling causing autophagy. Activation of TRPM8 (purple arrows) leads to an increase in ROS production which also induces ER stress. Stimulation of non-cannabinoid receptor GPR55 (blue arrows) through LPI via the subunit Gαq subunit stimulates the production of PLC to release Ca2+ and DAG which leads to the activation of MAPK/ERK signalling. This causes gene transcription by activation of transcription factors CREB and NF-κß. Gα12/13 subunit activates the RhoA/ROCK pathway which regulates PLC, actin cytoskeleton and p38/ATF2 activity. ATF2/p38 inhibits antiapoptotic proteins and enhances the interaction between Beclin-1 and Vps34 which is also inhibited by BCL-2 further enhancing ROS production by activation of the intrinsic apoptotic pathway (Velasco et al. 2012, 2016).Created with BioRender.com. TRPV1,2 transient receptor potential cation channel subfamily V member 1,2, TRPM8 transient receptor potential cation channel subfamily members (melastatin) 8, GPR55 orphan G-protein coupled receptor 55, ROS reactive oxygen species, ER endoplasmic reticulum, p8 protein p8 (Nuclear Protein 1, NUPR1), CHOP CCAAT/-enhancer-binding protein homologous protein, ATF4 activating transcription factor 4, TRIB3 tribbles pseudokinase 3, Akt protein kinase B, mTORC1 mammalian target of rapamycin C1, p21 cyclin-dependent kinase inhibitor 1, p27 cyclin-dependent kinase inhibitor 1B, CDK cyclin-dependent kinase, pRb retinoblastoma protein; Nuclear factor-kappaß (NF-κß), LPI Lysophosphatidylinositol, DAG diacylglycerol, BAD BCL2-associated agonist of cell death, ROCK rho-associated protein kinase, PLC phospholipase C
The characterisation of cannabinoids mechanism of action has been discerned from in vitro and in vivo studies. Reports of their oncological effects have been observed through modulating the hallmarks of cancer (Hanahan and Weinberg 2000, 2011) whilst ∆9-THC trends in inducing apoptosis and cytotoxicity through CB receptor-dependent pathways; CBD exhibits its activity via orphan GPCRs and non-GPRCs-mediated signalling (Velasco et al. 2012, 2016; Afrin et al. 2020).
Studies have reported positive upregulation of ceramide sphingolipid metabolism, leading to the subsequent arrest of the cell cycle and apoptosis via downstream activation of signals through extracellular regulated kinase (ERK) upon cannabinoid action (Calvaruso et al. 2012). Additional studies have also concluded ∆9-THC’s role in regulating sphingolipid metabolism via serine palmitoyl transferase (SPT) (Śledziński et al. 2018) and recent reports have concluded other enzymes of the metabolism of sphingolipids to be regulated by cannabinoids (Shaw et al. 2018). Dihydroceramides which are metabolic intermediates of the de novo synthesis pathway have been involved in the mechanisms of promoting autophagy-mediated cancer cell death (Hernández-Tiedra et al. 2016). ∆9-THC increases the dihydroceramide:ceramide ratio in the endoplasmic reticulum of glioma cells causing pre-apoptotic changes (Hernández-Tiedra et al. 2016).
Activation of the CB receptors causes the induction of the ER stress-related response and promotes the upregulation of the transcription factor p8 (Nupr1), this further simulates the following transcription factors, activating transcription factor 4 (ATF-4), C/EBP-homologous protein (CHOP) and pseudokinase tribbles-homologue 3 (TRIB3) (Velasco et al. 2016). The inhibitory interaction of TRIB3 and a pro-survival kinase Akt is favoured which leads to the inhibition of the mammalian target of rapamycin target 1 (mTORC1) favouring cell autophagy. Autophagy is upstream of apoptosis in cannabinoid-induced cell death as shown in studies where blocking autophagy prevented cannabinoid-induced apoptosis (Salazar et al. 2009; Vara et al. 2011). An increase in ceramide level has also been associated with ER stress in cannabinoid-induced apoptosis in tumour cells (Salazar et al. 2009). In addition, other environmental stimuli may also promote ER stress which can lead to the activation of the apoptotic pathway. These include a decrease in intracellular Ca2+, viral infections, chemotherapy agents and oxidative stress (Schröder and Kaufman 2005; Śledziński et al. 2018).
The mitogen-activated protein kinase (MAPK) pathway has also been reported in numerous studies to be involved in cannabinoid response. Serine/threonine protein kinases are mainly involved in this pathway and act to convert extracellular stress into different cellular responses including, cell cycle arrest, apoptotic cell death and cytokine production via phosphorylation. The involvement of the MAPK pathway in cancer is complex as its response to different stimuli can produce conflicting outcomes. Brief activation of the ERK cascade leads to cell survival and proliferation, whilst chronic activation is pro-apoptotic (Howlett 2005; Javid et al. 2016).
CBD has been demonstrated to affect a diverse set of cellular targets. First, it inhibits FAAH and FABP (Fatty Acid-Binding Protein). FAAH is responsible for the breakdown of anandamide, whilst FABP aids the transport of anandamide to from extracellular spaces to intracellular targets, such FAAH or nuclear PPAR. Both effects result in indirect activation of CB1 and CB2 receptors through increased extracellular concentration of anandamide (Lee et al. 2007; Pistis and O’Sullivan 2017). Second, CBD activates the 5-HT1A serotonin receptor, PPARγ and the transient receptor potential cation subfamily channels; TRPV1, TRPV2 and TRPA1. CBD is also an antagonist of GPR55, transient receptor potential cation channel subfamily M member 8 (TRPM8) and T-type Ca2+ channels. Finally, CBD has also been reported to inhibit adenosine reuptake via multiple proposed mechanisms (Lee et al. 2007; Ibeas Bih et al. 2015; McPartland 2018). Antagonization of GPR55 via CBD has been reported to reduce proliferation of pancreatic tumour cells and its activation has been reported to lead to metastasis in triple-negative breast cancer when stimulated by LPI (Zhao and Abood 2013; Ferro et al. 2018; Andradas et al. 2016; Falasca and Ferro et al. 2018; Pellati et al. 2018). Below we summarise pre-clinical studies which include both in vitro and in vivo experimental results in various cancer models with summaries included in Tables 1 and 2.
Pancreatic adenocarcinoma
In vitro
A study analysing the in vitro effects of synthetic receptor agonists of CB1 and CB2, WIN55, 212–2, ACEA and JWH-015 found they each induced a high level of apoptosis of MIA PaCa-2 cells (Console-Bram et al. 2014). The same study showed that a CB1 antagonist, N-(piperidin-1-1yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251), induced apoptosis and transcriptional changes of the genes involved in the janus kinase/signal transducers, activators of transcription signalling network (JAK/STAT) and MAPK signalling pathways in the MIA PaCa-2 pancreatic cancer cell line through activation independent of the CB1 receptor-independent pathways (Fogli et al. 2006). AM251, which expresses molecular similarities with cyclo-oxygenase-2 (COX-2) inhibitor celecoxib, demonstrated a synergistic interaction with 5-fluorouracil (5-FU) increasing their anti-cancer activity when administered in appropriate ratios as demonstrated by a combination index of 0.52 (Fogli et al. 2006).
Dando et al. report arachidonoyl cyclopropylamide (ACPA) and GW, CB1 and CB2 selective agonists, respectively, inhibited proliferation and invasion of PANC-1 cells (Dando et al. 2013). Activation of the receptors via cannabinoid receptor agonists showed an elevation in 5′ adenosine monophosphate-activated protein kinase (APMK) activation via a ROS-dependent increase of AMP/ATP ratio promoting cell autophagy and subsequent inhibition of cell growth (Dando et al. 2013; Brandi et al. 2013). ∆9-THC has been shown to induce a reduction in cell viability via apoptosis in a dose-dependent manner, specifically via the de novo synthesized ceramide up-regulation of the p8 and ATF-4, TRIB3 ER stress genes in MIA PaCa-2 and PANC-1 cells (Carracedo et al. 2006). The p8 protein has been shown to increase with ceramide treatment and potentiates anticancer effects (Javid et al. 2016). In support of this, MIA PaCa-2 cells treated with ∆9-THC caused an increase in p8 mRNA levels in vitro. Knockdown of the p8 gene prevented apoptosis by ∆9-THC in these cells (Carracedo et al. 2006). In addition to p8 and TRIB3 stress-related genes, further ER stress-inducing genes have been identified and associated with apoptosis, such as CHOP and ATF-4, where mRNA levels were elevated following ∆9-THC treatment (Ohoko et al. 2005).
Cannabinoids in combination with chemotherapy agents have shown promising results in pancreatic cancer cell line studies. One study reported the increase in gemcitabine activity by synergism with CB1 and CB2 receptor ligands by a NF-κß-dependent mechanism (Donadelli et al. 2011). This synergistic inhibition of tumour growth was most marked in gemcitabine-resistant cell lines (Donadelli et al. 2011). Gemcitabine increased cannabinoid-induced autophagy through a ROS-mediated mechanism and cannabinoids enhanced the apoptotic effect of gemcitabine (Donadelli et al. 2011). Ferro and co-workers reported the anticancer effects of blocking the putative GPR55 receptor in pancreatic cancer cells via CBD. A cross between GPR55 homozygous knockout and mice which do not harbour the TP53 mutation did not reveal any statistical difference in survival. Investigators analysed the possible role that p53 may play in regulating GPR55. In pancreatic ductal adenocarcinoma cell lines, they report a negative regulation of GPR55 with TP53 status, where overexpression of wild-type p53 in the AsPC-1 cell line (harbouring a TP53 mutation) caused a reduction in GPR55 expression. Further analysis revealed the negative regulation was through modulation of the micro-RNA miR34b-3p. Pharmacological inhibition of GPR55 via CBD in various pancreatic cell lines, inhibited anchorage-dependent growth. Treatment with CID16020046 (CID), an antagonist of GPR55, revealed similar results in AsPC-1 and HPFA-II and cell cycle arrest at the G1–S phase in PANC-1 and HPFA-II in a dose-dependent manner. Cyclin D1, activation of tumour-suppressor protein (RB) was also reduced in CBD treatment and an inhibition of MEK/ERK and ERK-dependent pathways was also observed. The study demonstrates a novel pathway by which gemcitabine may be potentiating anticancer effects through inhibiting GPR55 via CBD antagonization (Ferro et al. 2018).
In vivo
Administration of ∆9-THC at 15 mg/kg/day into a xenograft model of MIA PaCa-2 pancreatic tumour growth showed a reduction in the tumour burden (Carracedo et al. 2006). A synthetic cannabinoid, WIN55, 212–2 was found to increase the expression of downstream targets of the ER stress-related pathway involved in apoptosis in pancreatic cancer in comparison to healthy controls, demonstrating apoptotic selectivity effect of cannabinoids to cancer cells (Carracedo et al. 2006).
The role of other cannabinoid receptors including GPR55 has been speculated to be involved in regulating many cancer types including pancreatic cancer. A study by Ferro et al. revealed genetic ablation of GPR55 in a KPC mouse model of pancreatic ductal adenocarcinoma (PDAC) significantly prolonged survival and KPC mice treated with CBD and gemcitabine as a combination treatment survived three times longer than control or gemcitabine single treatment (Ferro et al. 2018). Immunohistochemistry analysis of the tumours revealed CBD inhibition of GPR55 affected signalling pathways involved in gemcitabine resistance. CBD was able to counteract the effect of gemcitabine on ERK phosphorylation and downregulated the enzyme’s ribonucleotide reductases 1 and 2 (RRM1/2), a marker for gemcitabine resistance (Ferro et al. 2018). In line with this, gemcitabine-treated tumours from KPC mice expressed high levels of RRM1 and reduced levels were observed in KPCG mice upon treatment with CBD (Ferro et al. 2018). The counteractions of CBD on gemcitabine only occurred when both drugs were administered together, suggesting synergistic effects of CBD on gemcitabine’s mode of action in vivo (Ferro et al. 2018). Donadelli et al. also reported an enhanced effect with combination therapy. CB1 antagonist, Rimonabant, combined with gemcitabine reduced tumour growth when compared to single therapy in vivo (Donadelli et al. 2011). An increase in ROS and autophagy pathways were observed which may explain the synergistic effects they observed (Donadelli et al. 2011).
The translation of preclinical data to the clinic remains to be somewhat unclear as many factors in cannabinoids pharmacokinetics, bioactivity and efficacy remain undetermined (Ladin et al. 2016; Millar et al. 2018). In addition, their low aqueous solubility and poor stability (sensitivity to light, temperature and oxidation) make developing effective formulations a problem (Fraguas-Sánchez et al. 2020). The route of cannabinoid administration remains uncertain as the oral bioavailability is very low and is subject to a significant first-pass effect in the body (Millar et al. 2018). Therefore, alternative routes of administration are required, although it has been reported that intratumour (IT) administration of low doses of cannabinoids has improved efficacy of the drug as well as survival (Ngwa et al. 2017, 2018; Yasmin-Karim et al. 2018). Successful administration has been reported when cannabinoids were combined with radiotherapy in treating pancreatic cancer (Yasmin-Karim et al. 2018).
A recent study has reported the use of CBD and ∆9-THC inhibited proliferation of pancreatic cancer and stellate cells. PDL-1 (a key target for immune checkpoint blockade) expression was reduced in mice tumours via the PAK-1-dependent pathway (p-21 activated kinase 1) activated by Kirsten rat sarcoma (KRAS). Their findings suggest a novelty for the cannabinoids in which KRAS, an undruggable target expressed in many lethal cancers can be supressed through targeting PAK1 and the suppression of PDL-1 could be enhanced for immune checkpoint blockade therapy in pancreatic cancers (Yang et al. 2020).
Brain cancer
In vitro
Investigation into human glioma cell lines U87 and U373 administered with CBD led to a decrease in mitochondrial oxidative metabolism, cell viability and antiproliferative effects correlated to induction of apoptosis (Massi et al. 2004). Solinas et al. investigated CBD in U87-MG and T98G glioma cell lines and reported inhibition of cell proliferation and invasiveness, a downregulation of ERK and Akt signalling and a decrease in the hypoxia-inducible factor HIF-1α expression (Solinas et al. 2013). In the following neuroblastoma cell lines, SK-N-SH, IMR-32, NUB-6 and LAN-1, CBD and ∆9-THC treatment induced antitumorigenic activity by decreasing cell viability and invasiveness, arrest of the cell cycle at the G1/G0 phase and an increase in activated caspase-3, albeit CBD was more potent in these effects when compared to ∆9-THC (Fisher et al. 2016). Salazar et al. investigated ∆9-THC in the astrocytoma cell line U87MG and in vivo where they report autophagy induction via the upregulation of p8 leading to apoptosis and inhibition of Akt and mTORC1 (Salazar et al. 2009).
A recent study has reported in the following human glioma cell lines A172, U251, U87 MG, U118 MG and LN18, CBD induced autophagic rather than apoptotic cell death. Specifically, CBD caused mitochondrial dysfunction and lethal mitophagy arrest mechanistically via TRPV4 with an influx of calcium (Huang et al. 2021). Further analysis revealed ER stress and in particular the ATF4-DDIT3-TRIB3-AKT-MTOR axis downstream of TRPV4 was involved in CBD’s mitophagy effect. Combination of CBD and temozolomide (TMZ) in neurosphere cultures and mouse models conveyed synergistic effects in reducing tumour burden and improving survival rates (Torres et al. 2011). Their findings suggest a novel TRPV4-CBD-mitophagy pathway in glioma and combination of CBD and TMZ as a potential to explore in future clinical studies. Additionally, Vrechi and colleagues show CBD stimulates autophagy signal transduction via crosstalk of ERK1/2 and AKT kinases and that CBD-induced autophagy was reduced in presence of CB receptors and TRPV1 receptor antagonists, AM251, AM630 and capsazepine in neuroblastoma and murine astrocyte cell lines (Vrechi et al. 2021).
Kolbe et al. recently investigated the effects of cannabinoids in glioblastoma multiforme (GBM) cells derived from primary human tumour samples and to identify possible receptors involved. Their findings revealed ∆9-THC reduced the number of Ki67 immuno-reactive nuclei, through GPR55. Their findings suggest that the sensitivity of cannabinoids and receptor-dependent signalling pathways should be considered to reflect the heterogeneity amongst GBM forms which is critical for when evaluating this translationally to clinic (Kolbe et al. 2021). Mutation-driven cancers are important to take into account when tailoring specific treatments. In a recent paper, Ellert-Miklaszewska et al. investigated the use of synthetic cannabinoids in GBM which have frequent TP53 or PTEN genetic defects rendering it from chemotherapy treatments. Their experimental work showed synthetic cannabinoids not only reduce tumour cells but that p53 could also act as an activator or inhibitor of autophagy and apoptosis and this depends on subcellular localisation and the mutant variant of p53 (Ellert-Miklaszewska et al. 2021).
In vivo
In a glioma mouse model treated with CBD daily at 0.5 mg/mouse, Massi and colleagues reported a significant reduction in xenografted human U87 tumour growth in vivo (Massi et al. 2004). A further study investigating CBD’s action in tumours from derived glioma stem cells (GSCs) which known to be resistant to therapies, reported in vivoan increase in the production of ROS leading to the inhibition of cell survival and an increase in the survival rate of mice bearing the GSC xenograft (Singer et al. 2015). They also observed activation of the p-p38 pathway and a downregulation of stem cell regulators including Sox2, Id-1 (a transcription factor involved in cell growth, senescence and differentiation) and p-STAT3 which inhibited the self-renewal of the cells (Singer et al. 2015). Although CBD inhibited glioma progression, a fraction of therapeutic resistance to CBD in a subset of glioma cells was due to the upregulation of antioxidant response genes (Singer et al. 2015). SK-N-SH neuroblastoma cell line induced in nude mice treated with CBD and ∆9-THC led to a reduction in tumour burden and an observed increase in activated caspase-3 (Fisher et al. 2016). Various forms of cannabinoids have been trialled and tested to measure the most efficacious form for oncological effects and these include a pure (P) form versus a botanical drug substance (BDS) which is an active form of the drug that has been cultivated usually available as a powder, tablet or elixir. In a study by Scott et al. using P and BDS forms for both CBD and ∆9-THC, they report efficacious activity for CBD-P in comparison to CBD-BDS and vice versa for ∆9-THC (Scott et al. 2014). As discussed earlier in their in vitrofindings, they report similar outcomes in their orthotopic murine model of glioma and in particular they observed a significant decrease in tumour volumes when both cannabinoids were administered with irradiation, p < 0.001 (Scott et al. 2014). These findings support the anticancer effects of cannabinoid treatment in glioma as a single therapy and also as an addition in combination treatment.
Cannabinoids share the common anticancer effect of apoptosis in their mode of action; however, it has also become apparent that autophagy is also involved. The process of apoptosis and autophagy interplay, where the survival function of autophagy negatively regulates apoptosis and inhibition of apoptosis blocks autophagy (Marino et al. 2014). Salazar and co-workers investigated ∆9-THC in a murine model of astrocytoma and found that autophagy is upstream of apoptosis in cannabinoid-induced cell death as shown by blocking autophagy, prevented cannabinoid-induced apoptosis (Salazar et al. 2009). ∆9-THC induced the effects of stimulation of ceramide synthesis de novo, ER stress, upregulation of p8 and TRIB3, phosphorylation of eIF2α on Ser51 via the activation of the CB1 receptor (Salazar et al. 2009). A human glioblastoma-induced murine model investigating GICs (glioma initiating cells; a subpopulation of cells responsible for the aggressiveness of GBM) was treated with ∆9-THC, CBD and TMZ in varying combinations. They reported an effective tumour reduction when CBD and ∆9-THC with TMZ were co-administered and that treatment with a high ratio of CBD was most efficacious (López-Valero et al. 2018).
Breast cancer
In vitro
McKallip et al. investigated the effects of ∆9-THC in human breast cancer cell lines MDA-MB-231, MCF-7 and mouse mammary carcinoma 4T-1. They reported a low expression of cannabinoid receptors; CB1 and CB2 in these cell lines. ∆9-THC did not affect cell viability in MCF-7 and 4T-1 cell lines but increased the size of a 4T1 primary tumour and enhanced metastasis in vivo. ∆9-THC exposure caused an increase in IL-4 and IL-10 cytokines and suppression of cell-mediated Th1 response by enhancement of Th2 cytokines due to upregulation in Th2-related genes. These findings suggest exposure to ∆9-THC may increase susceptibility to breast cancer which does not express cannabinoid receptors and is resistant to ∆9-THC-induced apoptosis (McKallip et al. 2005). In another study by Caffarel and colleagues ∆9-THC was investigated in the following human breast cancer cell lines; MCF-7, EVSA-T, MDA-MB-231, MDA-MB468, T-47D and SKBr3. They reported a reduction in human breast cancer cell proliferation by arrest of the cell cycle at the G2–M phase via down-regulation of the cyclin-dependent kinase (CDK1 or Cdc2) protein and an induction of apoptosis via the CB2 cannabinoid receptor which was highly expressed in the EVSA-T cell line. CB2 expression was also found to be correlated with tumours that had a low response to conventional therapies and that were also positive for certain prognostic markers including oestrogen, progesterone receptors and the presence of ERBB2/HER-2 oncogene. The psychotropic effects of cannabinoids are mediated via the CB1 rather than CB2, suggesting a cannabinoid therapy that would target the CB2 receptor would be beneficial (Caffarel et al. 2006). In a follow-up study investigating the ∆9-THC antiproliferative mechanism, exposure to ∆9-THC upregulated JunD expression, a proto-oncogene which belongs to the AP-1 transcription factor family, in the tumour cells. In addition, they also identified the involvement of the cyclin-dependent kinase inhibitor p27 and testin (a tumour-suppressor gene) as candidate targets of JunD. Stress protein p8, however was involved in ∆9-THC antiproliferative action in a JunD-independent manner, suggesting a multimodal mechanism of action (Caffarel et al. 2008).
In an interesting report by Blasco-Benito et al., they found ∆9-THC was able to disrupt the HER2–CB2R complex by selective binding to CB2R. Additionally, they concluded the antitumour efficacy of a botanical drug preparation to be more potent than pure ∆9-THC for both cell lines and animal studies (Blasco-Benito et al. 2019). Ligresti et al. investigated the anticancer properties of plant-based cannabinoids including CBD, CBG, CBC, CBDA and ∆9-THCA in addition to assessing the use of enriched CBD or ∆9-THC cannabis extracts over pure cannabinoids (Ligresti et al. 2006). Within the breast cancer cell lines, MDA-MB-231 and MCF-7, treated with the above cannabinoids, CBD was the most potent in its antiproliferative activity (Ligresti et al. 2006). They also report CBD mediated its apoptotic effects via the following routes: the direct or indirect activation of the receptors CB2 and TRPV1, receptor-independent elevation of intracellular Ca2+ and ROS generation (Ligresti et al. 2006).
Synthetic agonists or antagonists of cannabinoid receptors have been used to study the role of the ECS in cancer signalling and growth. Sarnataro and co-workers investigated the effects of Rimonabant, a CB1 antagonist, in the invasive human breast cancer line MDA-MB-231 and in the less-invasive lines, T47D and MCF-7 (Sarnataro et al. 2006). Treatment with Rimonabant caused antiproliferative effects characteristic of G1–S-phase cell cycle arrest accompanied by a downregulation in cyclins D and E with associated upregulation of cyclin-dependent kinase inhibitor p27KIP1. No observed apoptosis or necrosis occurred in vitro (Sarnataro et al. 2006). Additionally, within the invasive cells, these effects were found to be associated with lipid raft/caveolae as previously shown by the group (Sarnataro et al. 2005). Rimonabant caused complete displacement of the CB1 receptor from lipid rafts and the depletion of cholesterol by methyl-β-cyclodextrin (MCD) prevented these effects (Sarnataro et al. 2006). In cells overexpressing the CB1 receptor, Rimonabant inhibited MAPK signalling and decreased ERK1/2 activity (Sarnataro et al. 2006). Pre-treatment with MCD before Rimonabant administration caused a depletion in cholesterol and this reverted the inhibitory effects on ERK1/2 via Rimonabant, suggesting an interplay between the CB1 receptor and lipid raft motility in breast tumour growth (Sarnataro et al. 2006). JWH-015, an agonist of the CB2 receptor, in human MCF-7 mammary carcinoma cells reduced viability by inducing apoptosis independent of Gαi signalling or by pharmacological blockade of CB1, GPR55, TRPV1 or TRPA1 receptors and instead these effects were calcium-dependent and caused changes in MAPK/ERK signalling (Hanlon et al. 2016).
CBD has also been shown to downregulate Id-1 in the aggressive human breast cancer line MDA-MB-231 through modulation of ERK and ROS pathways leading to a decrease in Id-1 expression and also upregulated Id-2 (a transcriptional regulator) (McAllister et al. 2011). Shrivastava et al. observed a complex interplay between apoptosis and autophagy in CBD-treated invasive breast cancer cells, MDA-MB-231 (Shrivastava et al. 2011). In particular, CBD induced ER stress which led to the inhibition of AKT and mTOR signalling in vitroindicated by low levels of phosphorylated cyclin D1, mTOR and 4EBP1 (Shrivastava et al. 2011). Further analysis revealed CBD inhibited the association between beclin1 (central role in autophagy) and BCL-2 known to inhibit autophagy through cleavage of Beclin-1 and enhanced the interaction between Beclin-1 and Vps34 favouring autophagy (Shrivastava et al. 2011). Electron microscopy revealed morphological changes to MDA-MB-231 CBD-treated cells which included nuclear condensation, margination, increased vacuolization, decrease in intracellular organelles and enlarged mitochondria evident of apoptotic activity (Shrivastava et al. 2011). They hypothesized that the event changes in inducing autophagy may also cause apoptosis as the cleavage product from Beclin-1 translocates to the mitochondria and induces cytochrome C (Shrivastava et al. 2011). These observations and hypothesis suggest CBD may be able to control the complex interplay between autophagy and apoptosis in these breast cancer cells (Shrivastava et al. 2011). CBD also increased ROS levels and blockage of ROS inhibited apoptotic and autophagy pathways (Shrivastava et al. 2011). These effects were independent of cannabinoid and vanilloid receptor activation (Shrivastava et al. 2011).
Many drugs have failed in clinics for many of the aggressive cancers due to the recalcitrant TME. The TME plays a major role in contributing to the growth and invasion of cancer and in particular tumour-associated macrophages (TAMs) which are a class of immune cells contributing to the immunosuppressive TME through interchange of its two forms: M1 (anti-tumorigenic) and M2 (pro-tumorigenic) (Lin et al. 2019). Elbaz and colleagues investigated CBD in triple-negative breast cancer (TNBC) cell lines SUM159, MDA-MB-231-SCP2, MVT-1, 4T1.2 and in murine leukaemia RAW264.7. They observed CBD inhibited EGF-induced proliferation and chemotaxis in the cell lines, activated EGFR, ERK, Akt, and NF-κß pathways in addition to inhibition of matrix metallopeptidase 2 and 9 (MMP2 and MMP9) secretion (Elbaz et al. 2015). A cancer education experiment (conditioned media from CBD-treated cancer cells) showed a significant reduction in the number of migrated RAW 264.7 cells towards this medium which also contained lower levels of granulocyte–macrophage colony-stimulating factor (GM-CSF) and chemokine ligand 3 (CCL3) cytokines, crucial for macrophage recruitment and activation (Elbaz et al. 2015). They observed a reduction in tumour growth and metastasis and inhibition of the recruitment of total and M2 macrophages to the stroma of the primary tumour and secondary lung metastasis (Elbaz et al. 2015).
Cannabinoid’s effect on ER has been evident in many cancer studies, however, the exact mechanism by which this occurs remain elusive. In a recent study by de la Harpe et al., they found CBD selectively targeted MCF7 cells via oxidative stress-induced ER stress and UPR (unfolded protein response) activation, and these effects were caused by calcium influx via the TRPV1 receptor as opposed to MDA-MB-231 cells. This suggests the difference in CBD treatment was dependent on localization of TRPV1 (de la Harpe et al., 2021).
In vivo
One of the factors to consider in cannabinoid treatment is the abundance of cannabinoid receptors in the tissue of interest. In a study investigating the effects of ∆9-THC in a murine model of mammary carcinoma, it was found that the murine mammary carcinoma cell line 4T1 first did not express detectable levels of the cannabinoid receptors CB1 and CB2 and second, these cells were resistant to the cytotoxicity of ∆9-THC. They also show treatment with ∆9-THC led to an increase in tumour growth and metastasis due to an increase in production of IL-4 and IL-10 which suppressed the cell-mediated Th1 response by enhancing Th2-associated cytokines (McKallip et al. 2005). This finding was supported by the injection of anti-IL-4 and anti-IL-10 monoclonal antibodies which partially reversed the immune suppression of ∆9-THC in 4T1 cells (McKallip et al. 2005).
A study investigating the effects of the endogenous cannabinoid, Met-F-AEA (a metabolically stable anandamide analogue) in a highly invasive murine breast cancer model reported a significantly reduced amount and size of metastatic nodes and this effect was antagonized by the selective CB1 antagonist Rimonabant (Grimaldi et al. 2006). Molecular interrogation in treated cells with the endogenous cannabinoid caused a decrease in tyrosine phosphorylation of focal adhesion kinase (FAK) and steroid receptor coactivator (Src) and these effects were mitigated by Rimonabant (Grimaldi et al. 2006). They concluded CB1 receptor agonists by modulating FAK phosphorylation inhibited tumour cell invasion and metastasis and therefore CB1 receptor activation may represent a novel therapeutic target for the treatment of breast carcinoma and metastasis (Grimaldi et al. 2006). Rimonabant has also been reported to significantly reduce tumour volume in vivo in the invasive human MDA-MD-231 murine model and this effect occurred via the CB1R lipid raft/caveolae-mediated mechanism (Sarnataro et al. 2006).
In a human MDA-MB-231 breast carcinoma xenografted tumour model, both CBD and CBD enriched extract treatment induced apoptosis, inhibited the growth of tumours and metastasis in vivo (Ligresti et al. 2006). CBD has also been shown to modulate transcriptional activity downstream in breast cancer. A study by McAllister and colleagues investigated CBD treatment of a murine model of metastatic breast cancer and found CBD inhibited Id-1 gene expression in the primary tumour and lung metastasis in vivo through modulation of the ERK and ROS pathways (McAllister et al. 2011). Caffarel et al. have shown using a genetically engineered animal model of ErbB2-driven metastatic breast cancer (MMTV-neu mice), ∆9-THC and JWH-133 (selective CB2 agonist) reduce metastatic progression via AKT pathway inhibition (Caffarel et al. 2010).
Cannabinoids mechanistic actions have been reported to be CB-independent with studies reporting other channels through which they may activate their oncological effects, such as GPR55 or vanilloid channels. Hanlon and co-workers report using JWH-015, a CB2 agonist, significantly reduced tumour burden and metastasis of murine mammary carcinoma 4T1 cells in immunocompetent mice and these effects were dependent on calcium and induced changes to MAPK/ERK signalling which were independent of G-protein-coupled signalling, CB or vanilloid receptors (McAllister et al. 2011).
Other gastrointestinal (GI) cancers
In vitro
In a study investigating human colorectal cancer cells using the lines DLD-1, CaCo-2 and SW620, treatment with Rimonabant significantly reduced cell proliferation and induced death. In DLD-1 cells, treatment resulted in G2–M-phase cell cycle arrest without inducing apoptosis or necrosis (Aviello et al. 2012). Further investigation revealed an increase in mitotic catastrophe characterized by changes in the following, cyclin B1, PARP-1 (involved in DNA repair) Aurora B (involved in the attachment of the mitotic spindle in prophase), phosphorylated p38/MAPK and Chk1 (checkpoint kinase 1) in a time-dependent manner (Aviello et al. 2012). Rimonabant, can therefore mediate cancer tumour growth via mitotic catastrophe inducing cell-cycle arrest during spindle assembly and DNA-damage checkpoints (Aviello et al. 2012).
In hepatocellular carcinoma cell lines, HepG2 and Huh-7, treatment with ∆9-THC and JWH-015 (synthetic CB2 receptor agonist) reduced cell viability through activation of the CB2 receptor. Autophagy was subsequently induced by the upregulation of TRIB3, stimulation of adenosine monophosphate-activated kinase (AMPK) and Akt/mTORC1 inhibition (Vara et al. 2011).
In human colorectal cell lines, Caco-2 and HCT116, CBD treatment protected DNA from oxidative damage, reduced cell proliferation and increased endocannabinoid levels via CB1, TRPV1 and PPARγ (Romano et al. 2014). In addition, CBD treatment of colorectal carcinoma cell line DLD-1, reduced cell proliferation (Macpherson et al. 2014). An interesting study investigated the antiproliferative effects of CBD in Caco-2 cell line in various oxygen environments and found the antitumour effects of CBD to be greater in PhysO2 than AtmosO2. They conclude that CBD induced a mitochondrial production of ROS in PhysO2 cells, suggesting that the cellular redox environment can influence how CBD induced antiproliferative effects in PhysO2 to AtmosO2 cells (Nallathambi et al. 2018). This study demonstrates the important role microenvironments play in cell cultures when studying the pharmacokinetics and mechanism of drugs. Macpherson and colleagues report the increase in sensitivity to CBD-induced antiproliferative effects through changes to cell energetics, from a drop in oxygen and a loss in mitochondrial membrane integrity in cells under the atmospheric condition to the increase in ROS in mitochondria under low oxygen conditions (Nallathambi et al. 2018).
Purified cannabinoids have been mainly reported in inducing apoptosis, inhibiting proliferation and metastasis in many cancer types, however, other forms such as unheated extracts of the plants have been less studied. Nallathambi and colleagues identified unheated extract fractions (F7: THCA, F3: CBGA) from C. sativa which displayed cytotoxic effects in colorectal cancer cell lines, HCT116 and CCD-18Co and adenomatous polyps but reduced activity on normal colon cell lines (Nallathambi et al. 2018). Combination treatment analysed by the Bliss independence model, exhibited more potent cytotoxic effects which included cell cycle arrest, cell death and a reduction in genes involved in the Wnt signalling pathway (Proto et al. 2017).
In vivo
Rimonabant treatment in a mouse model of azoxymethane-induced colon carcinogenesis caused a significant reduction in aberrant crypt foci formation, which is a neoplastic precursor to colorectal cancer and additionally observed inhibitory effects with changes to mitotic and DNA-damage checkpoints in their cell lines as mentioned previously (Aviello et al. 2012). Another study investigated the synthetic cannabinoids effects on the Wnt/β-catenin pathway, a signalling pathway involved in the formation of colorectal cancer (Borelli et al. 2014). The administration of rimonabant in HCT116 xenografts caused a significant reduction in tumour growth and destabilized the nuclear localization of β-catenin in vivo by inhibiting the canonical Wnt pathway (Borelli et al. 2014). This study suggests a novel use for cannabinoids in treating colorectal cancer harbouring mutations in β-catenin.
In a murine model of hepatocellular carcinoma, treatment with JWH-015 and ∆9-THC, both cannabinoids reduced subcutaneous xenograft growth; however, this effect was not observed when autophagy was pharmacologically inhibited (Vara et al. 2011) indicating the importance of cell death in both cannabinoids reducing tumour burden in vivo. Furthermore, administration of the cannabinoids also led to a reduction in ascites (abnormal build-up of fluid in the abdomen) formation (Vara et al. 2011). In support of the mechanisms observed in the HCC cell lines, Salazar et al. investigated ∆9-THC in the astrocytoma cell line U87MG and in vivo where they report autophagy induction via the upregulation of p8 leading to apoptosis and inhibition of Akt and mTORC1 (Salazar et al. 2009).
The effect of CBD in gastrointestinal cancers has also been studied. In a study by Aviello et al., CBD treatment in an azoxymethane (AOM)-induced murine model of colon cancer, reduced aberrant crypt foci, polyps, tumour growth and led to a decrease in expression of inducible nitric oxide synthase (iNOS) and phosphorylated Akt with an upregulation in caspase-3 (Aviello et al. 2012). CBG’s anticancer effect has been observed in colon cancer models. Borelli et al. evaluated the antineoplastic effects in xenograft models of colon cancer and observed a reduction tumour growth, however due to the limitation in the model, they further tested CBG in an AOM colon murine model which mimics the tumour in situ and found CBG completely abolished the formation of aberrant crypt foci and reduced the number of tumours (Borelli et al. 2014). In addition, Romano et al. tested the effects of the BDS form of CBD, which contains a high content of CBD on colorectal cancer growth in both xenograft and AOM models. They also observed a reduction in tumour growth, preneoplastic lesions and polyps (Macpherson et al. 2014).
Prostate cancer
In vitro
∆9-THC induced apoptosis in a PC-3 prostate cancer cell line in a dose-dependent manner (Sreevalsan et al. 2011). CBD’s pro-apoptotic nature has been shown to be phosphate-dependent in prostate and colon cancer cells (De Petrocellis et al. 2013). In LNCaP (prostate) and SW480 (colon) cancer cell lines, the growth and mRNA expression of several phosphatases inhibited cannabinoid-induced PARP cleavage (De Petrocellis et al. 2013). De Petrocellis et al. investigated CBD’s effect in prostate carcinoma cell lines; LNCaP, 22RV1 (positive for androgen receptor), DU-145 and PC-3 (negative for androgen receptor). CBD treatment significantly decreased cell viability and potentiated the effects of bicalutamide and docetaxel (standard drugs for treatment of prostate carcinoma) against LNCaP and DU-145 xenograft tumours and when given alone reduced LNCaP xenograft size. CBD administered between 1 and 10 µM induced apoptosis and markers of intrinsic apoptotic pathways (PUMA, CHOP expression and intracellular Ca2+). In LNCaP cells, the pro-apoptotic effect of CBD was only partly due to TRPM8 antagonism and was accompanied by down-regulation of AR, p53 activation and elevation of ROS. LNCaP cells differentiated to androgen-insensitive neuroendocrine-like cells were more sensitive to CBD-induced apoptosis (De Petrocellis et al. 2013).
Gynaecological cancers
In vitro
The effects of ∆9-THC were also investigated in aggressive endometrial cancer. Zhang et al. report in HEC-1B and An3ca aggressive endometrial cancer cell lines a high level of cannabinoid receptor expression and treatment with ∆9-THC inhibited cell viability and motility by inhibiting epithelial-mesenchymal transition (EMT) in addition to down-regulation of the MMP9 gene in inhibiting metastasis. These findings suggest regulation and targeting of the MMP9-related pathways via ∆9-THC treatment may inhibit metastasis in this aggressive cancer type (Zhang et al. 2018). A recent study investigated the oncological effects of CBD as a monotherapy and in combination with chemotherapy drugs in ovarian cancer, administered as Poly lactic-co-glycolytic acid (PGLA)-microparticles (Fraguas-Sánchez et al. 2020). Their results show the combination of paclitaxel (PTX) with CBD to be effective in vitro and in ovo (Fraguas-Sánchez et al. 2020). CBD administered as microparticles was more efficacious than in single solution and in ovo, PTX resulted in a 1.5-fold tumour growth inhibition whereas in combination with CBD this increased to a twofold decrease, suggesting a promising therapy to explore in treating ovarian cancer as it provides the advantageous effect of using a lower dose of the antineoplastic drug whilst maintaining the same efficacy (Fraguas-Sánchez et al. 2020).
Clinical studies
The anticancer effects of cannabinoids have so far been limited to preclinical studies and translation to the clinic has remained stagnant. The first report of the use of cannabinoids on cancer patients was a pilot study that investigated ∆9-THC on nine terminal patients with recurrent glioblastoma where standard therapy remained unhopeful as a curative (Guzmán et al. 2006). These patients underwent intracranial administration of ∆9-THC, as this route was deemed the safest and patients did not exhibit any of the associated psychoactive effects (Guzmán et al. 2006). In-depth analysis of two patients’ tumours revealed molecular effects associated with cannabinoids antitumour action, which included decreased cell proliferation, stimulation of apoptosis and autophagy (Guzmán et al. 2006). Although positive effects were observed, the small case number hinders any statistically significant conclusions to be drawn from this study.
A recently published completed clinical study investigated the safety and preliminary efficacy of nabiximols oromucosal cannabinoid spray and dose intense (DIT) TMZ in patients with first recurrence glioblastoma (Twelves et al. 2021). The study included an open label arm where patients received nabiximols (n = 6) and a randomised, double-blind, and placebo-controlled arm (n = 12 and n = 9). Up to 12 sprays/days with DIT for 12 months were administered and the safety, efficacy and pharmacokinetics of TMZ were observed. Study reports a 33% of nabiximols and placebo-treated patients were progression free for 6 months and survival at 1 year for nabiximols was 83% and 44% for placebo patients and no effects of nabiximols on TMZ were reported. Although nabiximols spray was tolerable in GBM patients, a major limitation to the study was the small size of enrolled patients, specifically 21 across 9 sites and there was no predetermined power calculation to the study to define the minimum number of patients for statistical power (Twelves et al. 2021). Nevertheless, the observations warrant the need for further clinical trials to help establish safe and efficacious routes of administration, patient sub-stratification and to explore its possible synergistic effects with other antitumour agents as shown in pre-clinical data. Table 3 summarises clinical trials investigating cannabinoids including synthetic versions, CBD and ∆9-THC in cancer treatment.
Conclusion
Plant-based, endogenous and synthetic cannabinoid compounds have shown merits in not only alleviating the unwanted side effects of antineoplastic drug regiments, but have also shown promising evidence in decreasing tumour burden, and one in vivo study so far concludes increasing survival rates in mice. The antitumour effects of cannabinoids trend in modulating processes which include apoptosis and autophagy through first stimulating de novo synthesis of ceramide which induces activation of ER stress-related signalling proteins further leading to the inhibition of the AKT/mTORC1 axis promoting cell cycle arrest and additional mechanisms, such as cell death and aging. Other pathways involved mechanistically are activation of MAPK/ERK signalling through calcium induction. Strategies that would optimize the anticancer effects of cannabinoids through interference of these signalling cross-talks may prove useful for therapeutic intervention. Nevertheless, we found that these effects were reached differently downstream depending on the type of cancer, the dosage of the compound and which receptor/ligands were activated. We also found the co-administration of cannabinoids with chemotherapy drugs enhanced the potency of these effects. These synergistic effects should be targeted for translation to clinical application, especially in cancers which are refractory to chemotherapy. Various extracted forms of cannabinoids from C. sativa have shown varying cytotoxic effects which should be explored in more detail in future studies as majority of the evidence originates from studies investigating mainly ∆9-THC and CBD’s actions. Whilst the emerging evidence of phytocannabinoid anticancer effects are promising, there remains a paucity of clinical evaluation which must be overcome.
References
Afrin F, Chi M, Eamens AL, Duchatel RJ, Douglas AM, Schneider J, Gedye C, Woldu AS, Dun MD (2020) Can hemp help? Low-THC cannabis and non-THC cannabinoids for the treatment of cancer. Cancers (basel) 12(4):1033. https://doi.org/10.3390/cancers12041033
Akimov MG, Gamisonia AM, Dudina PV, Gretskaya NM, Gaydaryova AA, Kuznetsov AS, Zinchenko GN, Bezuglov VV (2021) GPR55 receptor activation by the N-acyl dopamine family lipids induces apoptosis in cancer cells via the nitric oxide synthase (nNOS) over-stimulation. Int J Mol Sci 22(2):622. https://doi.org/10.3390/ijms22020622
Andradas C, Blasco-Benito S, Castillo-Lluva S, Dillenburg-Pilla P, Diez-Alarcia R, Juanes-García A, García-Taboada E, Hernando-Llorente R, Soriano J, Hamann S, Wenners A, Alkatout I, Klapper W, Rocken C, Bauer M, Arnold N, Quintanilla M, Megías D, Vicente-Manzanares M, Urigüen L, Gutkind JS, Guzmán M, Pérez-Gómez E, Sánchez C (2016) Activation of the orphan receptor GPR55 by lysophosphatidylinositol promotes metastasis in triple-negative breast cancer. Oncotarget 7(30):47565–47575. https://doi.org/10.18632/oncotarget.10206
Andre CM, Hausman J-F, Guerriero G (2016) Cannabis sativa: the plant of the thousand and one molecules. Front Plant Sci 7:19. https://doi.org/10.3389/fpls.2016.00019
Armstrong JL, Hill DS, McKee CS, Hernandez-Tiedra S, Lorente M, Lopez-Valero I, Eleni Anagnostou M, Babatunde F, Corazzari M, Redfern CPF, Velasco G, Lovat PE (2015) Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J Invest Dermatol 135(6):1629–1637. https://doi.org/10.1038/jid.2015.45
Aviello G, Romano B, Borrelli F, Capasso R, Gallo L, Piscitelli F, Di Marzo V, Izzo AA (2012) Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J Mol Med (berl) 90(8):925–934. https://doi.org/10.1007/s00109-011-0856-x
Barbado MV, Medrano M, Caballero-Velázquez T, Álvarez-Laderas I, Sánchez-Abarca LI, García-Guerrero E, Martín-Sánchez J, Rosado IV, Piruat JI, Gonzalez-Naranjo P, Campillo NE, Páez JA, Pérez-Simón JA (2017) Cannabinoid derivatives exert a potent anti-myeloma activity both in vitro and in vivo. Int J Cancer 140(3):674–685. https://doi.org/10.1002/ijc.30483
Blasco-Benito S, Moreno E, Seijo-Vila M, Tundidor I, Andradas C, Caffarel MM, Caro-Villalobos M, Urigüen L, Diez-Alarcia R, Moreno-Bueno G, Hernández L, Manso L, Homar-Ruano P, McCormick PJ, Bibic L, Bernadó-Morales C, Arribas J, Canals M, Casadó V, Canela EI, Guzmán M, Pérez-Gómez E, Sánchez C (2019) Therapeutic targeting of HER2-CB2R heteromers in HER2-positive breast cancer. Proc Natl Acad Sci USA 116(9):3863–3872. https://doi.org/10.1073/pnas.1815034116
Blázquez C, Carracedo A, Barrado L, Real PJ, Fernández-Luna JL, Velasco G, Malumbres M, Guzmán M (2006) Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J 20(14):2633–2635. https://doi.org/10.1096/fj.06-6638fje
Borrelli F, Pagano E, Romano B, Panzera S, Maiello F, Coppola D, De Petrocellis L, Buono L, Orlando P, Izzo AA (2014) Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis 35(12):2787–2797. https://doi.org/10.1093/carcin/bgu205
Brandi J, Dando I, Palmieri M, Donadelli M, Cecconi D (2013) Comparative proteomic and phosphoproteomic profiling of pancreatic adenocarcinoma cells treated with CB1 or CB2 agonists. Electrophoresis 34(9–10):1359–1368. https://doi.org/10.1002/elps.201200402
Brown KJ, Laun AS, Song Z-H (2017) Cannabidiol, a novel inverse agonist for GPR12. Biochem Biophys Res Commun 493(1):451–454. https://doi.org/10.1016/j.bbrc.2017.09.001
Caffarel MM, Sarrió D, Palacios J, Guzmán M, Sánchez C (2006) Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res 66(13):6615–6621. https://doi.org/10.1158/0008-5472.CAN-05-4566
Caffarel M, Andradas C, Mira E, Pérez-Gómez E, Cerutti C, Moreno-Bueno G, Flores JM, García-Real I, Palacios J, Mañes S, Guzmán M, Sánchez C (2010) Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol Cancer 9:196. https://doi.org/10.1186/1476-4598-9-196
Caffarel M, Moreno-Bueno G, Cerutti C et al (2008) JunD is involved in the antiproliferative effect of Δ9-tetrahydrocannabinol on human breast cancer cells. Oncogene 27:5033–5044. https://doi.org/10.1038/onc.2008.145
Calvaruso G, Pellerito O, Notaro A, Giuliano M (2012) Cannabinoid-associated cell death mechanisms in tumor models (review). Int J Oncol 41(2):407–413. https://doi.org/10.3892/ijo.2012.1476
Carracedo A, Lorente M, Egia A, Blázquez C, García S, Giroux V, Malicet C, Villuendas R, Gironella M, González-Feria L, Piris MA, Iovanna JL, Guzmán M, Velasco G (2006) The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 9(4):301–312. https://doi.org/10.1016/j.ccr.2006.03.005
Chakravarti B, Ravi J, Ganju RK (2014) Cannabinoids as therapeutic agents in cancer: current status and future implications. Oncotarget 5(15):5852–5872. https://doi.org/10.18632/oncotarget.v5i15
Console-Bram L, Brailoiu E, Brailoiu GC, Sharir H, Abood ME (2014) Activation of GPR18 by cannabinoid compounds: a tale of biased agonism. Br J Pharmacol 171(16):3908–3917. https://doi.org/10.1111/bph.12746
Dando I, Donadelli M, Costanzo C, Dalla Pozza E, D’Alessandro A, Zolla L, Palmieri M (2013) Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis 4(6):e664. https://doi.org/10.1038/cddis.2013.151
Dariš B, Tancer Verboten M, Knez Ž, Ferk P (2019) Cannabinoids in cancer treatment: therapeutic potential and legislation. Bosn J Basic Med Sci 19(1):14–23. https://doi.org/10.17305/bjbms.2018.3532
de la Harpe A, Beukes N, Frost CL (2021) CBD activation of TRPV1 induces oxidative signaling and subsequent ER stress in breast cancer cell lines. Biotechnol Appl Biochem. https://doi.org/10.1002/bab.2119
De Petrocellis L, Ligresti A, Schiano Moriello A, Iappelli M, Verde R, Stott CG, Cristino L, Orlando P, Di Marzo V (2013) Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: pro-apoptotic effects and underlying mechanisms. Br J Pharmacol 168(1):79–102. https://doi.org/10.1111/j.1476-5381.2012.02027
Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, Bifulco M, Caraglia M, Palmieri M (2011) Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis 2(4):e152. https://doi.org/10.1038/cddis.2011.36
Elbaz M et al (2015) Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol Oncol 9(4):906–919. https://doi.org/10.1016/j.molonc.2014.12.010
Ellert-Miklaszewska A, Ciechomska IA, Kaminska B (2021) Synthetic cannabinoids induce autophagy and mitochondrial apoptotic pathways in human glioblastoma cells independently of deficiency in TP53 or PTEN tumor suppressors. Cancers 13(3):419. https://doi.org/10.3390/cancers13030419
Ellert-Miklaszewska A, Ciechomska IA, Kaminska B (2021) Synthetic cannabinoids induce autophagy and mitochondrial apoptotic pathways in human glioblastoma cells independently of deficiency in TP53 or PTEN tumor suppressors. Cancers (basel) 13(3):419. https://doi.org/10.3390/cancers13030419
ElSohly MA et al (2017) Phytochemistry of Cannabis sativa L. Prog Chem Org Nat Prod. https://doi.org/10.1007/978-3-319-45541-9_1
Falasca M, Ferro R (2016) Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv Biol Regul. https://doi.org/10.1016/j.jbior.2015.10.003
Ferro R et al (2018) GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 37(49):6368–6382. https://doi.org/10.1038/s41388-018-0390-1
Fiore D, Ramesh P, Proto MC, Piscopo C, Franceschelli S, Anzelmo S, Medema JP, Bifulco M, Gazzerro P (2018) Rimonabant kills colon cancer stem cells without inducing toxicity in normal colon organoids. Front Pharmacol 8:949. https://doi.org/10.3389/fphar.2017.00949
Fisher T, Golan H, Schiby G, PriChen S, Smoum R, Moshe I, Peshes-Yaloz N, Castiel A, Waldman D, Gallily R, Mechoulam R, Toren A (2016) In vitro and in vivo efficacy of non-psychoactive cannabidiol in neuroblastoma. Curr Oncol 23(2):S15-22. https://doi.org/10.3747/co.23.2893
Fogli S, Nieri P, Chicca A, Adinolfi B, Mariotti V, Iacopetti P, Breschi MC, Pellegrini S (2006) Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett 580(7):1733–1739. https://doi.org/10.1016/j.febslet.2006.02.024
Fraguas-Sánchez AI, Fernández-Carballido A, Simancas-Herbada R, Martin-Sabroso C, Torres-Suárez AI (2020) CBD loaded microparticles as a potential formulation to improve paclitaxel and doxorubicin-based chemotherapy in breast cancer. Int J Pharm 25(574):118916. https://doi.org/10.1016/j.ijpharm.2019.118916
Franco R, Rivas-Santisteban R, Reyes-Resina I, Casanovas M, Pérez-Olives C, Ferreiro-Vera C, Navarro G, Sánchez de Medina V, Nadal X (2020) Pharmacological potential of varinic-, minor-, and acidic phytocannabinoids. Pharmacol Res 158:104801. https://doi.org/10.1016/j.phrs.2020.104801
Gazzerro P, Malfitano AM, Proto MC, Santoro A, Pisanti S, Caruso MG, Notarnicola M, Messa C, Laezza C, Misso G, Caraglia M, Bifulco M (2010) Synergistic inhibition of human colon cancer cell growth by the cannabinoid CB1 receptor antagonist rimonabant and oxaliplatin. Oncol Rep 23(1):171–175
Gómez del Pulgar T, Velasco G, Sánchez C, Haro A, Guzmán M (2002) De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem J 363(Pt 1):183–8. https://doi.org/10.1042/0264-6021:3630183
Gonçalves ECD, Baldasso GM, Bicca MA, Paes RS, Capasso R, Dutra RC (2020) Terpenoids, cannabimimetic ligands, beyond the Cannabis plant. Molecules 25(7):1567. https://doi.org/10.3390/molecules25071567
Grimaldi C, Pisanti S, Laezza C, Malfitano AM, Santoro A, Vitale M, Caruso MG, Notarnicola M, Iacuzzo I, Portella G, Di Marzo V, Bifulco M (2006) Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res 312(4):363–373. https://doi.org/10.1016/j.yexcr.2005.10.024
Guerrero-Alba R et al (2019) Some prospective alternatives for treating pain: the endocannabinoid system and its putative receptors GPR18 and GPR55. Front Pharmacol 9:1496. https://doi.org/10.3389/fphar.2018.01496
Guindon J, Hohmann A (2012) The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. https://doi.org/10.2174/187152709789824660
Gustafsson K, Sander B, Bielawski J, Hannun YA, Flygare J (2009) Potentiation of cannabinoid-induced cytotoxicity in mantle cell lymphoma through modulation of ceramide metabolism. Mol Cancer Res MCR 7(7):1086–1098. https://doi.org/10.1158/1541-7786.MCR-08-0361
Guzmán M, Duarte MJ, Blázquez C, Ravina J, Rosa MC, Galve-Roperh I, Sánchez C, Velasco G, González-Feria L (2006) A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer 95(2):197–203. https://doi.org/10.1038/sj.bjc.6603236
Gyombolai P et al (2012) Regulation of endocannabinoid release by G proteins: a paracrine mechanism of G protein-coupled receptor action. Mol Cell Endocrinol. https://doi.org/10.1016/j.mce.2011.10.011
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell. https://doi.org/10.1016/S0092-8674(00)81683-9
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell. https://doi.org/10.1016/j.cell.2011.02.013
Hanlon KE, Lozano-Ondoua AN, Umaretiya PJ, Symons-Liguori AM, Chandramouli A, Moy JK, Kwass WK, Mantyh PW, Nelson MA, Vanderah TW (2016) Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent. Breast Cancer (dove Med Press) 15(8):59–71. https://doi.org/10.2147/BCTT.S100393
Hart S, Fischer OM, Axel U (2004) Cannabinoids induce cancer cell proliferation via tumor necrosis factor α-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res 64(6):1943–1950. https://doi.org/10.1158/0008-5472.CAN-03-3720
Hernández-Tiedra S, Fabriàs G, Dávila D, Salanueva ÍJ, Casas J, Montes LR, Antón Z, García-Taboada E, Salazar-Roa M, Lorente M, Nylandsted J, Armstrong J, López-Valero I, McKee CS, Serrano-Puebla A, García-López R, González-Martínez J, Abad JL, Hanada K, Boya P, Goñi F, Guzmán M, Lovat P, Jäättelä M, Alonso A, Velasco G (2016) Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 12(11):2213–2229. https://doi.org/10.1080/15548627.2016.1213927
Hirao-Suzuki M, Takeda S, Koga T, Takiguchi M, Toda A (2020) Cannabidiolic acid dampens the expression of cyclooxygenase-2 in MDA-MB-231 breast cancer cells: possible implication of the peroxisome proliferator-activated receptor β/δ abrogation. J Toxicol Sci 45(4):227–236. https://doi.org/10.2131/jts.45.227
Howlett AC (2005) Cannabinoid receptor signaling. Handb Exp Pharmacol 168:53–79. https://doi.org/10.1007/3-540-26573-2_2
Huang T, Xu T, Wang Y, Zhou Y, Yu D, Wang Z, He L, Chen Z, Zhang Y, Davidson D, Dai Y, Hang C, Liu X, Yan C (2021) Cannabidiol inhibits human glioma by induction of lethal mitophagy through activating TRPV4. Autophagy 25:1–15. https://doi.org/10.1080/15548627.2021.1885203
Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ (2015) Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics 12(4):699–730. https://doi.org/10.1007/s13311-015-0377-3
Javid FA, Phillips RM, Afshinjavid S, Verde R, Ligresti A (2016) Cannabinoid pharmacology in cancer research: a new hope for cancer patients? Eur J Pharmacol 775:1–14. https://doi.org/10.1016/j.ejphar.2016.02.010
Jung CK, Kang WK, Park JM, Ahn HJ, Kim SW, Taek OS, Choi KY (2013) Expression of the cannabinoid type I receptor and prognosis following surgery in colorectal cancer. Oncol Lett 5(3):870–876. https://doi.org/10.3892/ol.2012.1081
Kalant H (2001) Medicinal use of cannabis: history and current status. Pain Res Manage 6(2):80–91. https://doi.org/10.1155/2001/469629
Kalenderoglou N, Macpherson T, Wright KL (2017) Cannabidiol reduces leukemic cell size—but is it important? Front Pharmacol 8:144. https://doi.org/10.3389/fphar.2017.00144
Kaur R, Ambwani RS, Singh S (2016) Endocannabinoid system: a multi-facet therapeutic target. Curr Clin Pharmacol. https://doi.org/10.2174/1574884711666160418105339
Khan MI, Sobocińska AA, Brodaczewska KK, Zielniok K, Gajewska M, Kieda C, Czarnecka AM, Szczylik C (2018) Involvement of the CB2 cannabinoid receptor in cell growth inhibition and G0/G1 cell cycle arrest via the cannabinoid agonist WIN 55,212–2 in renal cell carcinoma. BMC Cancer 18(1):583. https://doi.org/10.1186/s12885-018-4496-1
Kleckner AS et al (2019) Opportunities for cannabis in supportive care in cancer. Ther Adv Med Oncol. https://doi.org/10.1177/1758835919866362
Kolbe MR, Hohmann T, Hohmann U, Ghadban C, Mackie K, Zöller C, Prell J, Illert J, Strauss C, Dehghani F (2021) THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers (basel) 13(5):1064. https://doi.org/10.3390/cancers13051064
Ladin DA, Soliman E, Griffin L, Van Dross R (2016) Preclinical and clinical assessment of cannabinoids as anti-cancer agents. Front Pharmacol 7(7):361. https://doi.org/10.3389/fphar.2016.00361
Lee JLC, Bertoglio LJ, Guimarães FS, Stevenson CW (2017) Cannabidiol regulation of emotion and emotional memory processing: relevance for treating anxiety-related and substance abuse disorders. Br J Pharmacol 174(19):3242–3256. https://doi.org/10.1111/bph.13724
Levy M, Futerman AH (2010) Mammalian ceramide synthases. IUBMB Life 62(5):347–356. https://doi.org/10.1002/iub.319
Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, Laezza C, Portella G, Bifulco M, Di Marzo V (2006) Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther 318(3):1375–1387. https://doi.org/10.1124/jpet.106.105247
Lin Y, Xu J, Lan H (2019) Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 12:76. https://doi.org/10.1186/s13045-019-0760-3
López-Valero I, Saiz-Ladera C, Torres S, Hernández-Tiedra S, García-Taboada E, Rodríguez-Fornés F, Barba M, Dávila D, Salvador-Tormo N, Guzmán M, Sepúlveda JM, Sánchez-Gómez P, Lorente M, Velasco G (2018) Targeting glioma initiating cells with A combined therapy of cannabinoids and temozolomide. Biochem Pharmacol 157:266–274. https://doi.org/10.1016/j.bcp.2018.09.007
Lorente M, Torres S, Salazar M, Carracedo A, Hernández-Tiedra S, Rodríguez-Fornés F, García-Taboada E, Meléndez B, Mollejo M, Campos-Martín Y, Lakatosh SA, Barcia J, Guzmán M, Velasco G (2011) Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ 18(6):959–973. https://doi.org/10.1038/cdd.2010.170
Lu H-C, Mackie K (2016) An introduction to the endogenous cannabinoid system. Biol Psychiat 79(7):516–525. https://doi.org/10.1016/j.biopsych.2015.07.028
Macpherson T, Armstrong JA, Criddle DN, Wright KL (2014) Physiological intestinal oxygen modulates the Caco-2 cell model and increases sensitivity to the phytocannabinoid cannabidiol. Vitro Cell Dev Biol Anim 50(5):417–426. https://doi.org/10.1007/s11626-013-9719-9
Marcu JP, Christian RT, Lau D, Zielinski AJ, Horowitz MP, Lee J, Pakdel A, Allison J, Limbad C, Moore DH, Yount GL, Desprez PY, McAllister SD (2010) Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol Cancer Ther 9(1):180–189. https://doi.org/10.1158/1535-7163.MCT-09-0407
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15(2):81–94. https://doi.org/10.1038/nrm3735
Marshall AD, Lagutina I, Grosveld GC (2011) PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res 71(24):7471–7481. https://doi.org/10.1158/0008-5472.CAN-11-0924
Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D (2004) Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J Pharmacol Exp Ther 308(3):838–845. https://doi.org/10.1124/jpet.103.061002
Matas D, Juknat A, Pietr M, Klin Y, Vogel Z (2007) Anandamide protects from low serum-induced apoptosis via its degradation to ethanolamine. J Biol Chem 282(11):7885–7892. https://doi.org/10.1074/jbc.M608646200
McAllister SD, Murase R, Christian RT, Lau D, Zielinski AJ, Allison J, Almanza C, Pakdel A, Lee J, Limbad C, Liu Y, Debs RJ, Moore DH, Desprez PY (2011) Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat 129(1):37–47. https://doi.org/10.1007/s10549-010-1177-4 (Erratum in: Breast Cancer Res Treat. 2012 May;133(1):401–4)
McKallip RJ, Nagarkatti M, Nagarkatti PS (2005) Δ-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol 174(6):3281–3289. https://doi.org/10.4049/jimmunol.174.6.3281
McKallip RJ, Jia W, Schlomer J, Warren JW, Nagarkatti PS, Nagarkatti M (2006) Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol Pharmacol 70(3):897–908. https://doi.org/10.1124/mol.106.023937
McPartland JM (2018) ‘Cannabis systematics at the levels of family, genus, and species. Cannabis Cannabinoid Res. https://doi.org/10.1089/can.2018.0039
Millar SA, Stone NL, Yates AS, O’Sullivan SE (2018) A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharmacol 26(9):1365. https://doi.org/10.3389/fphar.2018.01365
Mimeault M, Pommery N, Wattez N, Bailly C, Hénichart JP (2003) Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 56:1–12. https://doi.org/10.1002/pros.10190
Mohammadpour F et al (2017) Anti-invasion effects of cannabinoids agonist and antagonist on human breast cancer stem cells. Iran J Pharm Res. https://doi.org/10.22037/ijpr.2017.2143
Morales P, Reggio PH, Jagerovic N (2017) An overview on medicinal chemistry of synthetic and natural derivatives of cannabidiol. Front Pharmacol 8:422. https://doi.org/10.3389/fphar.2017.00422
Morell C, Bort A, Vara D, Ramos-Torres A, Rodríguez-Henche N, Díaz-Laviada I (2016) The cannabinoid WIN 55,212–2 prevents neuroendocrine differentiation of LNCaP prostate cancer cells. Prostate Cancer Prostatic Dis 19(3):248–257. https://doi.org/10.1038/pcan.2016.19
Moreno E, Cavic M, Krivokuca A, Casadó V, Canela E (2019) The endocannabinoid system as a target in cancer diseases: are we there yet? Front Pharmacol 10:339. https://doi.org/10.3389/fphar.2019.00339
Müller L, Radtke A, Decker J, Koch M, Belge G (2017) The synthetic cannabinoid WIN 55,212–2 elicits death in human cancer cell lines. Anticancer Res 37(11):6341–6345. https://doi.org/10.21873/anticanres.12086
Munson AE, Harris LS, Friedman MA, Dewey WL, Carchman RA (1975) Antineoplastic activity of cannabinoids. J Natl Cancer Inst 55(3):597–602. https://doi.org/10.1093/jnci/55.3.597
Nallathambi R, Mazuz M, Namdar D, Shik M, Namintzer D, Vinayaka AC, Ion A, Faigenboim A, Nasser A, Laish I, Konikoff FM, Koltai H (2018) Identification of synergistic interaction between cannabis-derived compounds for cytotoxic activity in colorectal cancer cell lines and colon polyps that induces apoptosis-related cell death and distinct gene expression. Cannabis Cannabinoid Res 3(1):120–135. https://doi.org/10.1089/can.2018.0010
Ngwa W, Kumar R, Moreau M, Dabney R, Herman A (2017) Nanoparticle drones to target lung cancer with radiosensitizers and cannabinoids. Front Oncol 7:208. https://doi.org/10.3389/fonc.2017.00208
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC (2018) Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 18(5):313–322. https://doi.org/10.1038/nrc.2018.6
O’Shaughnessy WB (1843) On the preparations of the Indian Hemp, or gunjah*. Prov Med Surg J. https://doi.org/10.1136/bmj.1.2629.1171
Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H (2005) TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J 24(6):1243–1255. https://doi.org/10.1038/sj.emboj.7600596
Orellana-Serradell O, Poblete CE, Sanchez C, Castellón EA, Gallegos I, Huidobro C, Llanos MN, Contreras HR (2015) Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol Rep 33(4):1599–1608. https://doi.org/10.3892/or.2015.3746
Ortega A, García-Hernández VM, Ruiz-García E, Meneses-García A, Herrera-Gómez A, Aguilar-Ponce JL, Montes-Servín E, Prospero-García O, Del Angel SA (2016) Comparing the effects of endogenous and synthetic cannabinoid receptor agonists on survival of gastric cancer cells. Life Sci 15(165):56–62. https://doi.org/10.1016/j.lfs.2016.09.010
Parker LA, Rock EM, Limebeer CL (2011) Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol. https://doi.org/10.1111/j.1476-5381.2010.01176.x
Pellati F, Borgonetti V, Brighenti V, Biagi M, Benvenuti S, Corsi L (2018) Cannabis sativa L. and nonpsychoactive cannabinoids: their chemistry and role against oxidative stress, inflammation, and cancer. Biomed Res Int 2018:1691428. https://doi.org/10.1155/2018/1691428
Pertwee RG (2006) Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. https://doi.org/10.1038/sj.bjp.0706406
Pietrovito L et al (2020) Treatment with cannabinoids as a promising approach for impairing fibroblast activation and prostate cancer progression. Int J Mol Sci. https://doi.org/10.3390/ijms21030787
Pisanti S et al (2013) The endocannabinoid signaling system in cancer. Trends Pharmacol Sci. https://doi.org/10.1016/j.tips.2013.03.003
Pistis M, O’Sullivan SE (2017) The role of nuclear hormone receptors in cannabinoid function. Adv Pharmacol. https://doi.org/10.1016/bs.apha.2017.03.008
Powles T, te Poele R, Shamash J, Chaplin T, Propper D, Joel S, Oliver T, Liu WM (2005) Cannabis-induced cytotoxicity in leukemic cell lines: the role of the cannabinoid receptors and the MAPK pathway. Blood 105(3):1214–1221. https://doi.org/10.1182/blood-2004-03-1182
Preet A, Ganju R, Groopman J (2008) Δ9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 27:339–346. https://doi.org/10.1038/sj.onc.1210641
Proto MC, Fiore D, Piscopo C et al (2017) Inhibition of Wnt/β-Catenin pathway and Histone acetyltransferase activity by Rimonabant: a therapeutic target for colon cancer. Sci Rep 7:11678. https://doi.org/10.1038/s41598-017-11688-x
Qamri Z, Preet A, Nasser MW, Bass CE, Leone G, Barsky SH, Ganju RK (2009) Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol Cancer Ther 8(11):3117–3129. https://doi.org/10.1158/1535-7163.MCT-09-0448
Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M, Hinz B (2013) COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol Cancer Ther 12(1):69–82. https://doi.org/10.1158/1535-7163.MCT-12-0335
Ravi J, Elbaz M, Wani NA, Nasser MW, Ganju RK (2016) Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog 55(12):2063–2076. https://doi.org/10.1002/mc.22451
Romano B, Borrelli F, Pagano E, Cascio MG, Pertwee RG, Izzo AA (2014) Inhibition of colon carcinogenesis by a standardized Cannabis sativa extract with high content of cannabidiol. Phytomedicine 21(5):631–639. https://doi.org/10.1016/j.phymed.2013.11.006
Ruiz L, Miguel A, Diaz-Laviada I (1999) v9-Tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. https://doi.org/10.1016/S0014-5793(99)01073-X
Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M et al (2009) Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Investig 119(5):1359–1372. https://doi.org/10.1172/jci37948
Sanchez MG, Sanchez AM, Ruiz-Llorente L, Diaz-Laviada I (2003) Enhancement of androgen receptor expression induced by (R)-methanandamide in prostate LNCaP cells. FEBS Lett 555:561–566. https://doi.org/10.1016/s0014-5793(03)01349-8
Santoro A, Pisanti S, Grimaldi C, Izzo AA, Borrelli F, Proto MC, Malfitano AM, Gazzerro P, Laezza C, Bifulco M (2009) Rimonabant inhibits human colon cancer cell growth and reduces the formation of precancerous lesions in the mouse colon. Int J Cancer 125(5):996–1003. https://doi.org/10.1002/ijc.24483
Sarfaraz S, Afaq F, Adhami VM, Malik A, Mukhtar H (2006) Cannabinoid receptor agonist-induced apoptosis of human prostate cancer cells LNCaP proceeds through sustained activation of ERK1/2 leading to G1 cell cycle arrest. J Biol Chem 281:39480–39491
Sarnataro D, Grimaldi C, Pisanti S, Gazzerro P, Laezza C, Zurzolo C, Bifulco M (2005) Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett 579(28):6343–6349. https://doi.org/10.1016/j.febslet.2005.10.016
Sarnataro D, Pisanti S, Santoro A, Gazzerro P, Malfitano AM, Laezza C, Bifulco M (2006) The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol Pharmacol 70(4):1298–1306. https://doi.org/10.1124/mol.106.025601
Schiffmann S, Sandner J, Birod K, Wobst I, Angioni C, Ruckhäberle E, Kaufmann M, Ackermann H, Lötsch J, Schmidt H, Geisslinger G, Grösch S (2009) Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 30(5):745–752. https://doi.org/10.1093/carcin/bgp061
Schoeman R, Beukes N, Frost C (2020) Cannabinoid combination induces cytoplasmic vacuolation in MCF-7 breast cancer cells. Molecules (basel, Switzerland) 25(20):4682. https://doi.org/10.3390/molecules25204682
Schröder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74(1):739–789. https://doi.org/10.1146/annurev.biochem.73.011303.074134
Scott KA, Dalgleish AG, Liu WM (2014) The combination of cannabidiol and D 9-tetrahydrocannabinol enhances the anticancer effects of radiation in an orthotopic murine glioma model. Mol Cancer Ther. https://doi.org/10.1158/1535-7163
Senkal CE, Ponnusamy S, Rossi MJ, Bialewski J, Sinha D, Jiang JC, Jazwinski SM, Hannun YA, Ogretmen B (2007) Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol Cancer Ther 6(2):712–722. https://doi.org/10.1158/1535-7163.MCT-06-0558
Sharma M et al (2014) In vitro anticancer activity of plant-derived cannabidiol on prostate cancer cell lines. Pharmacol Pharm. https://doi.org/10.4236/pp.2014.58091
Shaw J, Costa-Pinheiro P, Patterson L, Drews K, Spiegel S, Kester M (2018) Novel sphingolipid-based cancer therapeutics in the personalized medicine era. Adv Cancer Res 140:327–366. https://doi.org/10.1016/bs.acr.2018.04.016
Shrivastava A, Kuzontkoski PM, Groopman JE, Prasad A (2011) Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther 10(7):1161–1172. https://doi.org/10.1158/1535-7163
Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S, Soroceanu L (2015) Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis 6(1):e1601. https://doi.org/10.1038/cddis.2014.566
Śledziński P, Zeyland J, Słomski R, Nowak A (2018) The current state and future perspectives of cannabinoids in cancer biology. Cancer Med 7(3):765–775. https://doi.org/10.1002/cam4.1312
Soderstrom K, Soliman E, Dross RV (2017) Cannabinoids modulate neuronal activity and cancer by CB1 and CB2 receptor-independent mechanisms. Front Pharmacol 8:720. https://doi.org/10.3389/fphar.2017.00720
Soliman E, Henderson KL, Danell AS, Van Dross R (2016) Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: Role of cyclooxygenase-2 and novel J-series prostamides. Mol Carcinog 55(2):117–130. https://doi.org/10.1002/mc.22257
Solinas M, Massi P, Cinquina V, Valenti M, Bolognini D, Gariboldi M, Monti E, Rubino T, Parolaro D (2013) Cannabidiol, a non-psychoactive cannabinoid compound, inhibits proliferation and invasion in U87-MG and T98G glioma cells through a multitarget effect. PLoS One. 8(10):e76918. https://doi.org/10.1371/journal.pone.0076918
Sreevalsan S, Joseph S, Jutooru I, Chadalapaka G, Safe SH (2011) Induction of apoptosis by cannabinoids in prostate and colon cancer cells is phosphatase dependent. Anticancer Res 31(11):3799–3807
Torres S, Lorente M, Rodríguez-Fornés F, Hernández-Tiedra S, Salazar M, García-Taboada E, Barcia J, Guzmán M, Velasco G (2011) A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther 10(1):90–103. https://doi.org/10.1158/1535-7163.MCT-10-0688
Tutino V, Caruso MG, De Nunzio V, Lorusso D, Veronese N, Gigante I, Notarnicola M, Giannelli G (2019) Down-regulation of cannabinoid type 1 (CB1) receptor and its downstream signaling pathways in metastatic colorectal cancer. Cancers 11(5):708. https://doi.org/10.3390/cancers11050708
Twelves C, Sabel M, Checketts D et al (2021) A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br J Cancer 124:1379–1387. https://doi.org/10.1038/s41416-021-01259-3
Vara D, Salazar M, Olea-Herrero N et al (2011) Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ 18:1099–1111. https://doi.org/10.1038/cdd.2011.32
Velasco G, Sánchez C, Guzmán M (2012) Towards the use of cannabinoids as antitumour agents. Nat Rev Cancer 12(6):436–444. https://doi.org/10.1038/nrc3247
Velasco G, Sánchez C, Guzmán M (2016) Anticancer mechanisms of cannabinoids. Curr Oncol 23:23–32. https://doi.org/10.3747/co.23.3080
Vrechi TAM, Leão AHFF, Morais IBM et al (2021) Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci Rep 11:5434. https://doi.org/10.1038/s41598-021-84879-2
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8(5):761–773. https://doi.org/10.7150/jca.17648
Wu J (2019) Cannabis, cannabinoid receptors, and endocannabinoid system: yesterday, today, and tomorrow. Acta Pharmacol Sin 40(3):297–299. https://doi.org/10.1038/s41401-019-0210-3
Xian X, Huang L, Zhang B, Wu C, Cui J, Wang Z (2016) WIN 55,212–2 inhibits the epithelial mesenchymal transition of gastric cancer cells via COX-2 signals. Cell Physiol Biochem 39(6):2149–2157. https://doi.org/10.1159/000447910
Xu D, Wang J, Zhou Z, He Z, Zhao Q (2015) Cannabinoid WIN55, 212–2 induces cell cycle arrest and inhibits the proliferation and migration of human BEL7402 hepatocellular carcinoma cells. Mol Med Rep 12(6):7963–7970. https://doi.org/10.3892/mmr.2015.4477
Yang Y, Huynh N, Dumesny C, Wang K, He H, Nikfarjam M (2020) Cannabinoids inhibited pancreatic cancer via P-21 activated kinase 1 mediated pathway. Int J Mol Sci 21(21):8035. https://doi.org/10.3390/ijms21218035
Yasmin-Karim S, Moreau M, Mueller R, Sinha N, Dabney R, Herman A, Ngwa W (2018) Enhancing the therapeutic efficacy of cancer treatment with cannabinoids. Front Oncol 24(8):114. https://doi.org/10.3389/fonc.2018.00114
Yu Z, Pestell TG, Lisanti MP, Pestell RG (2012) Cancer stem cells. Int J Biochem Cell Biol 44(12):2144–2151. https://doi.org/10.1016/j.biocel.2012.08.022
Zhang Y, Zheng W, Shen K, Shen W (2018) ∆9-tetrahydrocannabinol inhibits epithelial-mesenchymal transition and metastasis by targeting matrix metalloproteinase-9 in endometrial cancer. Oncol Lett 15(6):8527–8535. https://doi.org/10.3892/ol.2018.8407
Zhang X, Qin Y, Pan Z, Li M, Liu X, Chen X, Qu G, Zhou L, Xu M, Zheng Q, Li D (2019) Cannabidiol induces cell cycle arrest and cell apoptosis in human gastric cancer SGC-7901 cells. Biomolecules 9(8):302. https://doi.org/10.3390/biom9080302
Zhao P, Abood ME (2013) GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci. https://doi.org/10.1016/j.lfs.2012.06.039
Zhu LX, Sharma S, Stolina M, Gardner B, Roth MD, Tashkin DP, Dubinett SM (2000) Delta-9-tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J Immunol 165(1):373–380. https://doi.org/10.4049/jimmunol.165.1.373
Zou S, Kumar U (2018) Cannabinoid receptors and the endocannabinoid system: signaling and function in the central nervous system. Int J Mol Sci 19(3):833. https://doi.org/10.3390/ijms19030833
Zuardi AW (2006) History of cannabis as a medicine: a review. Rev Bras Psiquiatr. https://doi.org/10.1590/S1516-44462006000200015
Funding
There were no sources of funding associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Code of availability
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mangal, N., Erridge, S., Habib, N. et al. Cannabinoids in the landscape of cancer. J Cancer Res Clin Oncol 147, 2507–2534 (2021). https://doi.org/10.1007/s00432-021-03710-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03710-7