
Abstract
Background
Targeted treatment of chronic myelogenous leukemia using imatinib has dramatically improved patient outcome. However, residual disease can be detected in the majority of patients treated with imatinib. Compensatory activation of MAP kinases (MAPK1/2) in response to BCR-ABL-inhibitors has been reported as a potential cytokine-dependent resistance mechanism leading to the rescue of leukemic progenitor cells.
Methods
Differential MAPK-modulating activity of clinically approved tyrosine kinase inhibitors was assessed in vitro using BCR-ABL-transformed cells. CD34+-enriched progenitors of newly diagnosed chronic myelogenous leukemia patients were exposed to tyrosine kinase inhibitors. MAPK-signaling was studied by Western blot technique. Proliferation assays were used to analyze response to antileukemic treatment.
Results
The ABL-inhibitors imatinib and nilotinib activate MAPKs in CD34+ chronic myelogenous leukemia progenitor cells, whereas treatment with the SRC/ABL-inhibitor dasatinib does not affect MAPK-activation at clinically relevant concentrations. Similar results are seen in BCR-ABL-transformed cells in the presence of interleukin-3 (IL-3). Experiments using BCR-ABL-mutant T315I, a resistance mutation not amenable to tyrosine kinase inhibitor binding, demonstrate that ABL-inhibitor-induced MAPK-activation does not depend on BCR-ABL-inhibition and cannot be prevented by selective SRC-inhibition. However, BCR-ABL-T315I enhances MAPK-activation, suggesting a T315I-dependent positive feedback of MAPK-activation. An autocrine IL-3-loop as trigger for aberrant T315I-dependent MAPK-activation was excluded.
Conclusions
Aberrant MAPK-activation triggered by ABL-inhibitors and positively regulated by BCR-ABL kinase mutation T315I might be an experimental explanation for the clinical observation that patients carrying high-resistance mutations show a highly aggressive course of their disease when tyrosine kinase inhibitor treatment is not discontinued in time.




Similar content being viewed by others

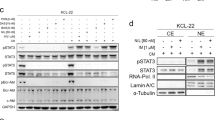
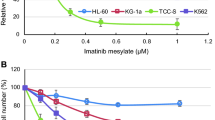
References
Apperley JF (2007) Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol 8:1018–1029
Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ (2008) Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol 15:1109–1118
Bhatia R, McGlave PB, Dewald GW, Blazar BR, Verfaillie CM (1995) Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood 85:3636–3645
Burchert A, Wang Y, Cai D et al (2005) Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 19:1774–1782
Chang F, Steelman LS, Lee JT et al (2003) Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 17:1263–1293
Chu S, Holtz M, Gupta M, Bhatia R (2004) BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood 103:3167–3174
Copland M, Hamilton A, Elrick LJ et al (2006) Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 107:4532–4539
Cortez D, Reuther G, Pendergast AM (1997) The Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1-to-S phase transition in hematopoietic cells. Oncogene 15:2333–2342
Di Cristofano A, Niki M, Zhao M, Karnell FG, Clarkson B, Pear WS, Van Aelst L, Pandolfi PP (2001) p62(dok), a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210(bcr-abl). J Exp Med 3:275–284
Dorsey JF, Cunnick JM, Lanehart R et al (2002) Interleukin-3 protects Bcr-Abl-transformed hematopoietic progenitor cells from apoptosis induced by Bcr-Abl tyrosine kinase inhibitors. Leukemia 16:1589–1595
Druker BJ, Tamura S, Buchdunger E et al (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Medic 2:561–566
Druker BJ, Guilhot F, O’Brien SG et al (2006) Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355:2408–2417
Fabian MA, Biggs WH III, Treiber DK et al (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23:329–336
Fedorov O, Marsden B, Pogacic V et al (2007) A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci USA 104:20523–20528
Griswold IJ, MacPartlin M, Bumm T et al (2006) Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol 26:6082–6093
Hanfstein B, Mueller MC, Kreil S et al (2011) Dynamics of mutant BCR-ABL-positive clones after cessation of tyrosine kinase inhibitor therapy. Haematologica 96(3):360–366
Hantschel O, Wiesner S, Guttler T et al (2005) Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell 19:461–473
Hentschel J, Rubio I, Eberhart M et al (2011) BCR-ABL- and Ras-independent activation of Raf as a novel mechanism of Imatinib resistance in CML. Int J Oncol 39:585–591
Hochhaus A, La Rosée P (2004) Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia 18:1321–1331
Jabbour E, Kantarjian H, Jones D et al (2008) Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood 112:53–55
Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL (2007) Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 109:4016–4019
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, Moiraghi B, Shen Z, Mayer J, Pasquini R, Nakamae H, Huguet F, Boqué C, Chuah C, Bleickardt E, Bradley-Garelik MB, Zhu C, Szatrowski T, Shapiro D, Baccarani M (2010) Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 24:2260–2270
Konig H, Copland M, Chu S et al (2008a) Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res 68:9624–9633
Konig H, Holtz M, Modi H et al (2008b) Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 22:748–755
La Rosee P, Corbin AS, Stoffregen EP, Deininger MW, Druker BJ (2002a) Activity of the Bcr-Abl kinase inhibitor PD180970 against clinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesylate (Gleevec, STI571). Cancer Res 62:7149–7153
La Rosee P, O’Dwyer ME, Druker BJ (2002b) Insights from pre-clinical studies for new combination treatment regimens with the Bcr-Abl kinase inhibitor imatinib mesylate (Gleevec/Glivec) in chronic myelogenous leukemia: a translational perspective. Leukemia 16:1213–1219
La Rosee P, Johnson K, Corbin AS et al (2004) In vitro efficacy of combined treatment depends on the underlying mechanism of resistance in imatinib-resistant Bcr-Abl-positive cell lines. Blood 103:208–215
La Rosee P, Holm-Eriksen S, Konig H et al (2008) Phospho-CRKL monitoring for the assessment of BCR-ABL activity in imatinib-resistant chronic myeloid leukemia or Ph + acute lymphoblastic leukemia patients treated with nilotinib. Haematologica 93:765–769
Lahaye T, Riehm B, Berger U et al (2005) Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer 103:1659–1669
Liu J, Joha S, Idziorek T et al (2008) BCR-ABL mutants spread resistance to non-mutated cells through a paracrine mechanism. Leukemia 22:791–799
Matulonis U, Salgia R, Okuda K, Druker B, Griffin JD (1993) Interleukin-3 and p210 BCR/ABL activate both unique and overlapping pathways of signal transduction in a factor-dependent myeloid cell line. Exp Hematol 21:1460–1466
McCubrey JA, Steelman LS, Abrams SL et al (2006) Roles of the RAF/MEK/ERK and PI3 K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 46:249–279
McCubrey JA, Steelman LS, Chappell WH et al (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773:1263–1284
Miething C, Feihl S, Mugler C et al (2006) The Bcr-Abl mutations T315I and Y253H do not confer a growth advantage in the absence of imatinib. Leukemia 20:650–657
Muller MC, Lahaye T, Hochhaus A (2002) Resistance to tumor specific therapy with imatinib by clonal selection of mutated cells. Dtsch Med Wochenschr 127:2205–2207
Nicolini FE, Hayette S, Corm S et al (2007) Clinical outcome of 27 imatinib mesylate-resistant chronic myelogenous leukemia patients harboring a T315I BCR-ABL mutation. Haematologica 92:1238–1241
Nishioka C, Ikezoe T, Yang J, Yokoyama A (2010) Long-term exposure of leukemia cells to multi-targeted tyrosine kinase inhibitor induces activations of AKT, ERK and STAT5 signaling via epigenetic silencing of the PTEN gene. Leukemia 9:1631–1640
Quintas-Cardama A, Cortes J (2009) Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113:1619–1630
Ricci C, Scappini B, Divoky V et al (2002) Mutation in the ATP-binding Pocket of the ABL Kinase Domain in an STI571-resistant BCR/ABL-positive Cell Line. Cancer Res 62:5995–5998
Rix U, Hantschel O, Durnberger G et al (2007) Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110:4055–4063
Saudemont A, Hamrouni A, Marchetti P et al (2007) Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res 67:4491–4498
Sharma SV, Gajowniczek P, Way IP et al (2006) A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell 10:425–435
Soverini S, Colarossi S, Gnani A et al (2006) Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res 12:7374–7379
Steelman LS, Pohnert SC, Shelton JG et al (2004) JAK/STAT, Raf/MEK/ERK, PI3 K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18:189–218
Thiesing JT, Ohno-Jones S, Kolibaba KS, Druker BJ (2000) Efficacy of STI571, an abl tyrosine kinase inhibitor, in conjunction with other antileukemic agents against bcr-abl-positive cells. Blood 96:3195–3199
Wang Y, Cai D, Brendel C et al (2007) Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL + progenitors via JAK-2/STAT-5 pathway activation. Blood 109:2147–2155
Weisberg E, Manley PW, Breitenstein W et al (2005) Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7:129–141
Williams RT, den Besten W, Sherr CJ (2007) Cytokine-dependent imatinib resistance in mouse BCR-ABL + , Arf-null lymphoblastic leukemia. Genes Dev 21:2283–2287
Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O (2004) The two major imatinib resistance mutations E255 K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun 319:1272–1275
Yu C, Krystal G, Varticovksi L et al (2002) Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res 62:188–199
Acknowledgments
We would like to thank Mrs Susanne Brendel and Mrs Silke Will for expert technical assistance. PL was funded by the Max Eder program, Deutsche Krebshilfe, Germany. AH received research support by the German José-Carreras Leukämiestiftung (H03/01), Novartis Pharmaceuticals, Basel, CH, and Bristol-Myers-Squibb, New York, NY, USA.
Conflict of interest
We declare that we have no conflict of interest
Author information
Authors and Affiliations
Corresponding author
Additional information
Nicolai Härtel and Thomas Klag contributed equally to this work.
Electronic supplementary material

{kind=link}
Rights and permissions
About this article
Cite this article
Härtel, N., Klag, T., Hanfstein, B. et al. Enhanced ABL-inhibitor-induced MAPK-activation in T315I-BCR-ABL-expressing cells: a potential mechanism of altered leukemogenicity. J Cancer Res Clin Oncol 138, 203–212 (2012). https://doi.org/10.1007/s00432-011-1086-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-011-1086-x