
Abstract
Bronchopulmonary dysplasia (BPD) is a common chronic respiratory disease in preterm infants caused by multifactorial etiology. Genetic factors are involved in the occurrence of BPD, but studies have found that candidate genes have poor reproducibility and are influenced by ethnic heterogeneity; therefore, more exploration is still needed. We performed whole-exon sequencing in 34 preterm infants with BPD and 32 non-BPD control neonates. The data were analyzed and interpreted by Fisher difference comparison, PLINK and eQTL association analysis, KEGG and GO enrichment analysis, STRING tool, Cytoscape software, ProtParam tool, HOPE online software, and GEOR2 analysis on NCBI GEO dataset. BPD has a highly heterogeneity in different populations, and we found 35 genes overlapped with previous whole-exon sequencing studies, such as APOB gene. Arterial and epithelial cell development and energy metabolism pathways affect BPD. In this study, 24 key genes were identified, and BIVM rs3825519 mutation leads to prolonged assisted ventilation in patients with BPD. A novel DDAH1 mutation site (NM_012137: exon1: c.89 T > G: p.L30R) was found in 9 BPD patients.
Conclusion: BIVM gene rs3825519 mutation may play a role in the pathogenesis of BPD by affecting cilia movement, and the DDAH1 and APOB genes mutations may have a pathogenic role in BPD.
What is Known: |
• Genetic factors are involved in the occurrence of bronchopulmonary dysplasia. |
• The candidate genes have poor reproducibility and are influenced by ethnic heterogeneity, therefore, more exploration is still needed. |
What is New: |
• We identified the role of susceptible SNPs in BPD in Shenzhen, China, and identified 24 key genes that influence the pathogenesis of BPD, and also found 35 genes overlapped with previous whole exon sequencing studies, such as APOB gene. |
• We found that BIVM and DDAH1 genes may play a pathogenic role in the pathogenesis of BPD. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bronchopulmonary dysplasia (BPD) is a common chronic respiratory disease in premature infants caused by complicated pathogenesis. In recent years, the survival rate of preterm newborns is increasing; however, the incidence of BPD is also increasing. In the USA, 63% of extremely preterm infants (EPI) are complicated with BPD [1], which has become the leading cause of death in preterm infants after 60 days [2]. In China, the incidence of BPD in EPI increased from 19.3 to 51.7% from 2010 to 2017 [3, 4]. Furthermore, BPD will cause a series of complications, such as airway hyperresponsive disease and recurrent infections in lower respiratory tract, which brings a heavy burden on society and families. Finding ways of early diagnosis for BPD is particularly important and urgent.
In twin studies, genetic factors were found to account for 53–82% of susceptibility to BPD [5, 6]. Numerous studies have attempted to identify the key genes involved in BPD; however, due to many reasons, the reproducibility of the candidate genes is poor. Whole-exon sequencing (WES), as a means of gene sequencing, can be used to explore common and rare coding gene mutations that may directly affect protein structure and function. There were five studies using WES to identify BPD mutations [7,8,9,10,11] included infants in Italy, the USA, France, and Shanghai in China. Considering the differences in race and the research in Shanghai was a single-center study, it is necessary to validate the candidate genes in these five studies and to explore whether there are new key genes of BPD in Chinese preterm newborns.
Materials and methods
Participants
We recruited 34 BPD patients and 32 non-BPD infants from 7 hospitals in Shenzhen from January 2020 to May 2022. The diagnostic criteria for BPD are based on the new consensus at 2018 NICHD Symposium [12]: preterm infants (gestational age (GA) ≤ 32 weeks) at 36 weeks of postmenstrual age were still dependent on FiO2 and respiratory support ≥ 3 days, and had interstitial lung disease (image confirmation). The exclusion criteria were as follows: (1) congenital inherited diseases and malformations, (2) lost to follow-up. Non-BPD premature infants who hospitalized in the same period and did not have BPD were selected. The clinical characteristics such as GA, feeding mode, day age, and birth mode were matched with the BPD group. The oral mucosal epithelium of participants and clinical data were collected. The study was approved by the Ethics Committee of Shenzhen University General Hospital (Approval No. 2020–001-02), and the Chinese Clinical Trial Registration (Registration No. ChiCTR2000033610) was completed. All participants’ guardians signed informed consent.
Exome sequencing
DNA were extracted by magnetic universal genomic DNA kit (TIANGEN DP705, Beijing, China), detected by Agilent 5400 fragments analyzer system (NYSE: A, California, USA), randomly interrupted by Covaris crusher (M220, Massachusetts, USA), purified by AMpure XP Reagent (Beckman A63881, California, USA). Library concentration was determined by fluorometer (Qubit 2.0, Thermo Fisher, USA). Exons were captured using the SureSelect Human All Exon V6 (Agilent Technologies, 5190–8865, California, USA) and streptavidin magnetic beads. The library was sequenced on NovaSeq 6000 platform (Illumina, California, USA). Sequencing reads quality was evaluated by FastQC package and removed the connector by cutadapt (v1.18), and discarded reads with N > 10% or the base number (the quality value Q ≤ 10) proportion > 50%; then, reads were uploaded to the CHI cloud analysis platform based on FANSe3 and were mapped to the human reference genome (GRCh37) to search mutations. Annovar software, hg19 refGene, the population frequencies of Exome Aggregation Consortium (ExAC, V.0.3.1), 1000g2015AUG and dbnsfp42a were used to annotate the mutation sites.
Data analysis
Principal component analysis was used to detect outliers and layered of group [13]. Five software (SIFT, Polyphen2_HDIV, Polyphen2_HVAR, Mutation Taster, and PROVEAN) were used to predict the harmfulness of mutations. Deleterious mutations were considered only if at least three software predictions were harmful. PLINK2 (v2.00a3lm) software was used to analyze the association between mutation sites and disease. Three Gene Ontology (GO) analysis, Kyoto Encyclopedia of Genes, and Genomes (KEGG) pathway enrichment analysis were used for the screened variant genes. Venn diagram was used to show coincidence genes with previous literature [14]. Gene expression and quantitative trait loci (eQTL) analysis used linear regression models to assess associations between the single-nucleotide polymorphisms (SNPs) and gene expression in 500-kbs upstream and downstream of those SNPs, and the data used in eQTL analysis were downloaded from the Genotype-Tissue Expression (GTEx) data portal. The SNP-set (sequence) kernel association test (SKAT-O) for R-package SKAT was used to obtain the association between mutations in a gene and disease.
Protein interaction network construction and protein structure prediction
We used STRING (Search Tool for the Retrieval of Interacting Genes Database, Version 11.5, https://string-db.org) tool to predict the relationship between different genes. Cytoscape software (Version 3.10.0) was used to visualize and analyze the interaction network. Degree ≥ 20 was set as the core gene. We used ProtParam tool (https://web.expasy.org/protparam/) to calculate the physical and chemical parameters of protein, which based on protein sequences given in NCBI. We used HOPE online software (https://www3.cmbi.umcn.nl/hope) to analyze the structure of the mutations.
Expression of the mutated gene in the patient
We downloaded previously published sequencing data from the NCBI GEO dataset (http://www.ncbi.nlm.nih.gov/gds). The GSE32472 dataset was from the peripheral blood of premature infants with GA ≤ 32 weeks and birth weight (BW) < 1500 g. There were 68 BPD patients and 43 non-BPD infants. The GSE188944 dataset is sequencing data from umbilical cord blood of BPD patients (n = 6) and preterm infants without BPD (n = 17) in Argentina (GA ≤ 35 weeks and BW < 1500 g). After GEOR2 analysis, P < 0.05 and |logFC|> 1 were screened to obtain differentially expressed genes, which were used to find the overlapping genes with the candidate genes in this study.
Statistical analyses
Approximately normally distributed data were described using mean ± standard deviation (SD) and evaluated using T-test analysis. Non-normally distributed data were described using medians and interquartile range (IQR) and evaluated using Fisher’s exact test. P values were adjusted using the Benjamini–Hochberg false discovery rate (FDR-BH), and P < 0.05 was considered statistically significant.
Results
Clinical characteristics of the patient
There were 34 BPD infants and 32 in non-BPD infants in the study. BPD patients’ GA and BW were statistical smaller and lighter than non-BPD infants, but there were no differences in gender, singleton, and multiple birth composition (Table 1). And, the results of principal component analysis showed there was no inherent diversity between these two groups (Fig. 1).

PCA plot of association test statistics in the WES-based discovery data
Preliminary findings from WES data
After stratification and annotation of gene mutation, there were 861,620 gene mutations (Fig. 2) and 27,745 were deleterious mutations. We compared the mutated genes between the two groups by Fisher’s exact test and screened out 457 differential SNPs, of which 336 (73.5%) were substitution mutations, 86(18.8%) were deletion mutations, and 37 (8.1%) were insertion mutations. Most of those SNPs were heterozygous mutations.

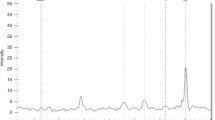
Manhattan plot of results from exome-wide association analysis of 34 BPD and 32 non-BPD infants
Few gene mutations consistent with previous research
In order to explore whether there are SNPs in line with previous reports, we searched databases (Phenopedia, DisGeNet, MalaCards, and GWAS catalog), and found 50 genes that have been studied for the relationship between SNP and BPD. There are 128 mutation sites in these 50 genes in our study (Supplementary Table 1). We through the interaction network to find the core genes were IL6, EGFR, MMP9, CD44, SERPINE1, and TLR4, which are inflammatory mediators, epidermal growth factor receptors, matrix metalloproteinase families, cell adhesion molecules, serine protease inhibitors, and toll-like receptors respectively (Fig. 3). In our study, IL6 (rs2069832, rs1474347), EGFR (rs11506105, rs17336919) and MMP9 (rs3787268), CD44 (rs34986068, rs7639388, rs3215691), and SERPINE1 (rs2227692, rs2854236) SNPs were more common to non-BPD group, suggesting these SNPs are not risk factors of BPD (Supplementary Table 2).

The mutant genes in our study were overlapped with those in previous studies. The genes with red color are core genes
In this study, the SNPs found only in the BPD group were SOD2 (superoxide dismutase 2) rs5746091 (10 cases, 29%), rs5746090 (9 cases, 26%), rs28662077 (6 cases, 19%), and DDAH1 (NM_012137:exon1:c.89 T > G:p.L30R, 9 cases, 26%), of which SOD2 SNPs were in intron regions, and BPD patients with SOD2 rs5746091 mutation had a larger GA (P = 0.015, 28.9 ± 1.8 weeks vs 27.0 ± 1.9 weeks) and heavier BW (P = 0.002, 1108 ± 235.0 g vs 882.1 ± 136.5 g) than non-mutated BPD patients, BPD patients with SOD2 rs5746090 mutation had a heavier BW than non-mutated BPD patients (P = 0.01, 1093.3 ± 243.3 g vs 896.4 ± 151.0 g), suggesting those two SOD2 mutations may be related to child’s GA and BW. DDAH1 (dimethylarginine dimethylaminohydrolase 1, NM_012137: exon1: c.89 T > G: p.L30R) SNP is a non-synonymous mutation and located in exon region, whose residue larger and more hydrophilic than the wild-type and residual charge changes from neutral to positive (Table 2, Fig. 4). This mutation in DDAH1 was predicted to be a harmful mutation that easily leads to loss of interactions with the ligand, suggesting that mutations of this gene may play an important role in BPD.

Predicted 3D structure of DDAH1 wild type and mutant. Close-up of the mutation. Wild-type, and mutant side chain are shown in green and red respectively, the rest of the protein is shown in gray
Compared with previous WES studies, we found no common overlapping genes in all studies (Fig. 5), but 35 genes in our study have been reported in previous WES studies. There were three overlapping genes (GIGYF2, KRT10, APOB) with the study in Shanghai [10]. Twenty-two genes (MAP3K6, NCOR2, FAM155A, PAPLN, MYO9A, ABCC6, ABCC11, PIEZO1, DNAH2, MYO15A, RNF213, ACP5, APOB, CSPG5, FGFRL1, DNAH5, SOBP, ROS1, TBP, GLI3, TMEM229A, GALNTL5) overlapped with Carrera’s work [8], two genes (MAB21L3, LIFR) overlapped with Hadchouel’s [9] studies, and 9 genes (FIGN, ITGA9, ANKRD6, CNOT4, NAV2, CIT, SLC8A3, TRIP11, SETBP1) overlapped with study by Li [7]. Among them, only apolipoprotein B (APOB) gene was found in three studies, and these genes are mainly involved in cellular components (FAM155A, PAPLN, TMEM229A, GALNTL5, SLC8A3), immune-related (GLI3, SOBP), microtubule and ciliary organization (MYO9A, MYO15A, DNAH2, DNAH5, FIGN), angiogenesis (MAP3K6, RNF213), fibroblasts (FGFRL1), and WNT signaling pathway (ANKRD6), which indicate these pathways are important to BPD.

Gene mutations strongly associated with BPD
Association analysis of mutation sites and groups using PLINK2 revealed 19,256 SNPs associated with BPD, most of which were in the intron region. Only 5 SNPs had P value < 5 × 10−5 (Table 3), and no reports found they are related to BPD. Among them, DEK/RNF144B gene rs6928572 has a population carrier frequency of 91% in the 1000 genome database and a low pathogenicity. The harmfulness of SNP on CREB3L1 gene is not high in various databases. None of the top 10 SNPs was in linkage disequilibrium, suggesting that there is no common effect on these genes (Fig. 6).

Linkage disequilibrium analysis among the top 10 SNPs associated with BPD
Functional enrichment of the differential genes in two groups
We found 3074 genes’ mutations were associated with BPD through SKAT-O test, and used GO- and KEGG-enriched pathways to find those genes in biological processes included arterial development, regulation of cellular metabolic processes, pancreas development, epithelial cell development, etc.; in cell composition, enriched pathways included photoreceptor disk membrane, dynactin complex, lamellar body, collagen type IV trimer, etc.; in molecular functions, enriched pathways included peptide, proton symporter activity, GTPase binding, RNA N6-methyladenosine dioxygenase activity, etc., suggesting that the mutated genes mainly cover blood vessels, epithelial cells, fibers, and energy metabolism. KEGG pathways are mainly involved in human diseases, metabolism, biological systems, etc. The top three are mainly methyl butanoate metabolism, phototransduction, alanine, aspartate, and glutamate metabolism (Fig. 7).

GO and KEGG enrichment analysis of differential genes associated with BPD. a GO enrichment analysis biological process, b GO enrichment analysis, cellular component, c GO enrichment analysis molecular function, d KEGG enrichment analysis
Twenty-four candidate genes may influence the pathogenesis of BPD
To further understand the effect of gene mutations on surrounding genes, we performed eQLT analysis for gene mutations with P < 0.01 and found that 24 deleterious mutations affected the expression of surrounding genes, namely TMEM17, CTD-2521M24.6, DYRK4, KLHDC4, AC007362.3, CNIH2, CTSF, SFI1, NGDN, IFNAR1, PLD4, F8A1, PRELID1, TMX4, RP11-510M2.2, STRCP1, ICAM3, SNRNP70, WWC3, DNM1P51, BIVM, LIFR, CASP1P2, and SMG8 (Table 4), these genes were the candidate genes in the pathogenesis of BPD. To confirm whether these genes are differentially expressed in BPD patients, through the NCBI GEO dataset, we found decreased expression of BIVM (immunoglobulin-like variable motif) in the cord blood of BPD patients, and BIMV rs3825519 mutation was also found in our study; therefore, we speculated BIMV rs3825519 mutation may lead to decreased expression of BIMV gene.
BIVM rs3825519 mutation was identified in BPD patients with prolonged assisted ventilation
BIVM rs3825519 mutation was found in fifteen BPD patients (44.1%, 15/34) and four non-BPD infants (12.5%,4/32), which had statistical difference (P = 0.006). We compared the GA, BW, and assisted ventilation duration between the BIVM rs3825519 mutation group (n = 15) and the wild-type group (n = 19) in BPD patients and found that BIVM mutation group had a longer assisted ventilation duration than the wild-type group (P = 0.02, 88.4 ± 49.8 days vs 54.5 ± 26.6 days). In the non-BPD group, the duration of assisted ventilation in the mutant was longer than in wild-type preterm infants (38.5 (29) days vs. 29 (18.5) days), but did not reach statistical difference, may be the sample size need to be expanded. BIVM rs3825519 mutation could lead to aggravation and prolong assisted ventilation of BPD patients, which further confirmed that BIVM gene could play a role in BPD (Table 5).
Approximately normally distributed data were described using mean ± standard deviation (SD), non-normally distributed data were described using medians and interquartile range (IQR) and non-BPD group with BIVM mutation was non-normally distributed data.
Discussion
Even though BPD is a common lung disease in premature infants, much remains unknown about the pathogenesis of BPD. Many reports have explored the role of SNPs in BPD, but the reproducibility of the results is poor, suggesting that the genetic variation in BPD may point to uncommon variants and complex genetic factors. In this study, we collected BPD and non-BPD infants for WES analysis. There was no difference in population composition between those two groups. Low GA and BW preterm infants are more likely to develop BPD, which is consistent with currently recognized risk factors. Gender and multiple births are not risk factors for BPD.
We found 457 deleterious SNPs, among them, substitution mutations were the most, followed by deletion mutations, insertion mutations were the fewest, and most of the variants were heterozygous, which was consistent with previous study [9]. One study found BPD patients had more haploinsufficient genes than all protein-coding genes in the human genome [7] and in our study also supported the hypothesis that BPD is dose sensitive to gene, which means more heterozygous mutations may increase the phenotypic advantage of BPD.
Fifty genes in this study had previously been studied in relation to BPD, among which were 6 core genes. However, the mutations carried by the core genes (IL6, EGFR, MMP9, CD44, SERPINE1) were more common in non-BPD, suggesting that these SNPs were not risk factors of BPD. Among them, IL6 rs2069832 is not associated with the incidence of BPD in Northern Ireland and Canadian populations [15], while this locus may have protective significance in this study, reducing the incidence of BPD disease. We found that SOD2 (rs5746091, rs5746090, rs28662077) was only carried in BPD patients and associated with higher GA and BW. Previous study [16] found other two SOD2 SNPs (rs4880 and rs5746136) were associated with lower GA and BW, but not related to the pathogenesis of BPD, contrary to this study. More research is needed to determine whether different mutations in SOD2 gene will make different functions of protein, thus has different influence on BPD. We also identified a novel DDAH1 mutation (NM_012137: exon1: c.89 T > G: p.L30R) in exon region, which is a non-synonymous and deleterious mutation affecting the properties and structure of proteins. Downregulation or reduced activity of DDAH1 leads to apoptotic activation and reduced angiogenesis [17], and rs480414 SNP in DDAH1 was previously found to be protective against the development of pulmonary hypertension in BPD patients [18]. Whether this new mutation affects the activity of DDAH1 and thus plays a pathogenic role deserves further study.
We found 35 genes in our study have been reported in previous WES studies, suggesting they are strongly associated with BPD. Among them, APOB gene was candidate gene in three studies, indicating a possible role in pathogenesis of BPD. Previous study found APOB gene variation associated with the survival rate of non-small cell lung cancer [19], but no research reported the correlation between APOB gene and BPD, which is worth digging into. There were only three overlapping genes between this study and the study of Shanghai [10], considering the differences in the study design, analysis strategy, and definition of BPD may lead to the different results. In this study, the diagnostic definition [12] in 2018 was used, and the definition of BPD in Shanghai used the 2001 [20] criteria. Those genes in our study that overlap with other studies were mainly involved in cellular components, immune-related, microtubule and ciliary organization, angiogenesis, and fibroblasts. On the other hand, we found that the genes associated with BPD disease are mainly concentrated on blood vessels, epithelial cells, fibers, and energy metabolism. This indicates that vascular development, epithelial cell development, collagen fiber, and energy metabolism are closely related to this disease.
We identified 24 candidate genes of BPD, supporting the hypothesis that BPD is a polygenic co-pathogenicity. These genes mainly are related to ciliary movement, and no association with BPD was reported. Through the GEO dataset, we found only BIVM gene expression was decreased in BPD patients, suggesting that BIVM gene is closely associated with BPD. BIMV gene was first identified in the variable region of immunoglobulin gene using electronic search technology, located in the 32–33 region of the long arm of 13 human chromosome, and predicted to encode a 503 amino acid protein, which is ubiquitously expressed in normal tissues and may have immunoglobulin-like functions [21]. Study found BIVM is expressed at the base of cilia and is a key gene in ciliopathies [22]. This study found that BPD patients with BIVM gene mutations needed longer assisted ventilation, which is a high-risk factor of BPD [23]. Therefore, we speculate that the BIVM rs3825519 mutation may affect the function of cilia and be involved in pathogenesis of BPD.
Our report has shortcomings, such as needing a separate sample to validate the results; thus, we plan to further verify the mutation and expression of the candidate genes in another cohort. Despite these limitations, in this study, the use of WES to explore genetic variation of BPD can increase understanding of genetic factors of BPD in China, to guide clinical prediction and intervention, and achieve individualized treatment of BPD.
Conclusion
For the first time, we identified the role of susceptible SNPs in BPD in Shenzhen, China, and identified 24 candidate genes that could influence the pathogenesis of BPD. And, we also found 35 genes in our study have been reported in previous WES studies, suggesting they have possible roles in pathogenesis of BPD, such as APOB gene. We found BIVM rs3825519 mutation may play a role in the pathogenesis of BPD by affecting ciliary motility. A novel DDAH1 mutation site (NM_012137: exon1: C.89 T> G: P.L30r) may be involved in the pathogenesis of BPD.
Data availability
All data relevant to the study are included in the article and its online supplementary material.
Abbreviations
- APOB:
-
Apolipoprotein B
- BIVM:
-
For basic, immunoglobulin-like variable motif-containing
- BW:
-
Birth weight
- DDAH1:
-
Dimethylarginine dimethylaminohydrolase 1
- GA:
-
Gestational age
- SNP:
-
Single-nucleotide polymorphism
- SOD2:
-
Superoxide dismutase 2
- WES:
-
Whole-exon sequencing
References
Bell EF, Hintz SR, Hansen NI, Bann CM, Wyckoff MH, DeMauro SB, Walsh MC, Vohr BR, Stoll BJ, Carlo WA et al (2022) Mortality, in-hospital morbidity, care practices, and 2-year outcomes for extremely preterm infants in the US, 2013–2018. JAMA 327:248–263. https://doi.org/10.1001/jama.2021.23580
Patel RM, Kandefer S, Walsh MC, Bell EF, Carlo WA, Laptook AR, Sanchez PJ, Shankaran S, Van Meurs KP, Ball MB et al (2015) Causes and timing of death in extremely premature infants from 2000 through 2011. N Engl J Med 372:331–340. https://doi.org/10.1056/NEJMoa1403489
Jiangsu Multicenter Study Collaborative Group for Breastmilk Feeding in Neonatal Intensive Care Units (2019). Clinical characteristics and risk factors of very low birth weight and extremely low birth weight infants with bronchopulmonary dysplasia: multicenter retrospective analysis [J]. Chin J Pediatr 57(01): 33–9. https://doi.org/10.3760/cma.j.issn.0578-1310.2019.01.009
Bronchopulmonary dysplasia of preterm infants Collaborative Group (2011). Incidence and risk factors of bronchopulmonary dysplasia in premature infants in 10 hospitals in China [J]. Chin J Pediatr (09): 655–62.
Bhandari V, Bizzarro MJ, Shetty A, Zhong X, Page GP, Zhang H, Ment LR, Gruen JR (2006) Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics 117:1901–1906. https://doi.org/10.1542/peds.2005-1414
Lavoie PM, Pham C, Jang KL (2008) Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics 122:479–485. https://doi.org/10.1542/peds.2007-2313
Li J, Yu KA-O, Oehlert J, Jeliffe-Pawlowski LL, Gould JB, Stevenson DK, Snyder M, Shaw GM, O’Brodovich HM (2015) Exome sequencing of neonatal blood spots and the identification of genes implicated in bronchopulmonary dysplasia. Am J Respir Crit Care Med 192(5):589–596. https://doi.org/10.1164/rccm.201501-0168OC
Carrera P, Di Resta C, Volonteri C, Castiglioni E, Bonfiglio S, Lazarevic D, Cittaro D, Stupka E, Ferrari M, Somaschini M et al (2015) Exome sequencing and pathway analysis for identification of genetic variability relevant for bronchopulmonary dysplasia (BPD) in preterm newborns: A pilot study. Clin Chim Acta 451:39–45. https://doi.org/10.1016/j.cca.2015.01.001
Hadchouel A, Decobert F, Besmond C, Delacourt C (2020) Exome sequencing of extreme phenotypes in bronchopulmonary dysplasia. Eur J Pediatr 179:579–586. https://doi.org/10.1007/s00431-019-03535-0
Dai D, Chen H, Dong X, Chen J, Mei M, Lu Y, Yang L, Wu B, Cao Y, Wang J et al (2021) Bronchopulmonary dysplasia predicted by developing a machine learning model of genetic and clinical information. Front Genet 12:689071. https://doi.org/10.3389/fgene.2021.689071
Hamvas A, Feng R, Bi Y, Wang F, Bhattacharya S, Mereness J, Kaushal M, Cotten CM, Ballard PL, Mariani TJ et al (2018) Exome sequencing identifies gene variants and networks associated with extreme respiratory outcomes following preterm birth. BMC Genet 19:94. https://doi.org/10.1186/s12863-018-0679-7
Higgins RD, Jobe AH, Koso-Thomas M, Bancalari E, Viscardi RM, Hartert TV, Ryan RM, Kallapur SG, Steinhorn RH, Konduri GG et al (2018) Bronchopulmonary dysplasia: executive summary of a workshop. J Pediatr 197:300–308. https://doi.org/10.1016/j.jpeds.2018.01.043
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909. https://doi.org/10.1038/ng1847
Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C (2014) Jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15:293. https://doi.org/10.1186/1471-2105-15-293
Huusko JM, Karjalainen MK, Mahlman M, Haataja R, Kari MA, Andersson S, Toldi G, Tammela O, Rämet M, Lavoie PM et al (2014) A study of genes encoding cytokines (IL6, IL10, TNF), cytokine receptors (IL6R, IL6ST), and glucocorticoid receptor (NR3C1) and susceptibility to bronchopulmonary dysplasia. BMC Med Genet 15:120. https://doi.org/10.1186/s12881-014-0120-7
Giusti B, Vestrini A, Poggi C, Magi A, Pasquini E, Abbate R, Dani C (2012) Genetic polymorphisms of antioxidant enzymes as risk factors for oxidative stress-associated complications in preterm infants. Free radic res 46:1130–1139. https://doi.org/10.3109/10715762.2012.692787
Trittmann JK, Almazroue H, Jin Y, Nelin LD (2019) DDAH1 regulates apoptosis and angiogenesis in human fetal pulmonary microvascular endothelial cells. Physiol rep 7:e14150. https://doi.org/10.14814/phy2.14150
Trittmann JK, Gastier-Foster JM, Zmuda EJ, Frick J, Rogers LK, Vieland VJ, Chicoine LG, Nelin LD (2016) A single nucleotide polymorphism in the dimethylarginine dimethylaminohydrolase gene is associated with lower risk of pulmonary hypertension in bronchopulmonary dysplasia. Acta paediatrica (Oslo, Norway : 1992) 105:e170–175. https://doi.org/10.1111/apa.13296
Deng W, Liu H, Luo S, Clarke J, Glass C, Su L, Lin L, Christiani DC, Wei Q (2020) APOB genotypes and CDH13 haplotypes in the cholesterol-related pathway genes predict non-small cell lung cancer survival. Cancer Epidemiology Biomarkers Prev 29(6):1204–1213. https://doi.org/10.1158/1055-9965.EPI-19-1262
Jobe AH, Bancalari E (2001) Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163:1723–1729. https://doi.org/10.1164/ajrccm.163.7.2011060
Yoder JA, Hawke NA, Eason DD, Mueller MG, Davids BJ, Gillin FD, Litman GW (2002) BIVM, a novel gene widely distributed among deuterostomes, shares a core sequence with an unusual gene in Giardia lamblia. Genomics 79:750–755. https://doi.org/10.1006/geno.2002.6768
Nevers Y, Prasad MK, Poidevin L, Chennen K, Allot A, Kress A, Ripp R, Thompson JD, Dollfus H, Poch O et al (2017) Insights into ciliary genes and evolution from multi-level phylogenetic profiling. Mol Biol Evol 34:2016–2034. https://doi.org/10.1093/molbev/msx146
Rutkowska M, Hożejowski R, Helwich E, Borszewska-Kornacka MK, Gadzinowski J (2019) Severe bronchopulmonary dysplasia - incidence and predictive factors in a prospective, multicenter study in very preterm infants with respiratory distress syndrome. J Matern Fetal Neonatal Med 32(12):1958–1964. https://doi.org/10.1080/14767058.2017.1422711
Acknowledgements
We express deep gratitude to the patients and their families for their willingness to take part in the study.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81971431).
Author information
Authors and Affiliations
Contributions
Xi Luo performed the experiments and performed analyses and wrote the manuscript, Min Zhao, Cheng Chen, and Fengji Lin reviewed all the cases and analyses initial data. Xiaodong Li, Jinxing Feng, Lei Dou and Haiyun Huang communicated with participants and collected clinical data. Shanqiu Xiao, Dong Liu and Junli He designed the participants’ collection instruments. Jialin Yu designed the study, and critically reviewed the manuscript. All authors approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Ethics Committee of Shenzhen University General Hospital (Approval No. 2020–001-02). Appropriate written informed consent was provided by the participants’ parents.
Consent for publication
All authors gave their consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Daniele De Luca
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, X., Zhao, M., Chen, C. et al. Identification of genetic susceptibility in preterm newborns with bronchopulmonary dysplasia by whole-exome sequencing: BIVM gene may play a role. Eur J Pediatr 182, 1707–1718 (2023). https://doi.org/10.1007/s00431-022-04779-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-022-04779-z