
Abstract
Preterm infants suffer from a higher incidence of acute diseases such as necrotising enterocolitis and sepsis. This risk can be mitigated through probiotic prophylaxis during admission. This reduction in risk is likely the result of acute modulation of the gut microbiome induced by probiotic species, which has been observed to occur up until discharge. We aimed to determine if this modulation, and the associated probiotic species, persisted beyond discharge. We conducted both a cross-sectional analysis (n = 18), at ~ 18 months of age, and a longitudinal analysis (n = 6), from admission to 18 months of the gut microbiome of preterm infants using both shotgun metagenomics and 16S rRNA profiling respectively. The 16S amplicon sequencing revealed that the microbial composition of the probiotic-supplemented infants changed dramatically over time, stabilising at discharge. However, species from the probiotic Infloran®, as well as positive modulatory effects previously associated with supplementation, do not appear to persist beyond discharge and once prophylaxis has stopped.
Conclusions: Although differences exist between supplemented and non-supplemented groups, the implications of these differences remain unclear. Additionally, despite a lack of long-term colonisation, the presence of probiotics during early neonatal life may still have modulatory effects on the microbiome assembly and immune system training.
What is Known: • Evidence suggests modulation of the microbiome occurs during probiotic prophylaxis, which may support key taxa that exert positive immunological benefits. • Some evidence suggests that this modulation can persist post-prophylaxis. | |
What is New: • We present support for long-term modulation in association with probiotic prophylaxis in a cohort of infants from North Queensland Australia. • We also observed limited persistence of the probiotic species post-discharge. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Preterm birth, defined by the World Health Organisation as < 37-week gestation [1], disrupts gut microbiome development [2]. The resulting preterm microbiome is characterised by low diversity and commensal microbe abundance, in combination with a greater number of pathogens [3, 4]. This characteristic preterm microbiome has been linked to increased disease burden in these infants [5]. This includes acute diseases like necrotising enterocolitis (NEC) and late-onset sepsis (LOS), and chronic diseases like asthma and both types 1 and 2 diabetes, all of which have been linked to the microbiome [6]. However, probiotic prophylaxis can mitigate the risk of these acute diseases [7]. As a result, probiotic prophylaxis has now become the standard of care for the most premature (< 32-week gestation) and small for gestational age infants (< 1500 g) in neonatal intensive care units (NICUs) across Australia.
Probiotic prophylaxis has been demonstrated to mitigate and treat several infectious and non-infectious diseases through modulation of the gut microbiome [8]. Probiotic supplementation has been effective in mitigating a number of infections including, Helicobacter pylori infection [9], rotavirus infection [10], obesity [11] and allergies [12]. In pre-term infants, probiotics have been shown to reduce the incidence of both NEC and LOS, with the benefits likely stemming from changes in the microbiome afforded by the presence of probiotic strains [8]. These strains, specifically from Bifidobacterium and Lactobacillus, have been shown to contribute to a Bifidobacterium-dominated microbiome, which, in turn, can positively modulate immune system activity and development [6, 13]. Although acute benefits of prophylactic supplementation have been noted in the most premature infants, data on both the long-term impact of probiotic supplementation and on non-supplemented infants remains sparse.
Certain probiotic species have been shown to persist beyond discharge [14], possibly continuing to exert positive effects on the development of the infant gut microbiome. However, this observation of probiotic persistence is not consistent [14, 15], and as infants have been shown to cluster in their microbial populations by NICU [16], caution should be taken when extrapolating from these single unit studies to another unit. We have demonstrated in a previous study that probiotic-prophylaxis had a significant positive modulatory effect on very-preterm infants over the course of their hospital admission [17]. We observed greater diversity in the gut microbiome of probiotic-supplemented preterm infants, relative to those not supplemented, at discharge from the hospital, suggesting that preterm infants who fall outside the criteria for probiotic prophylaxis (defined as < 32-week gestation and/or < 1500 g) may be missing out on the positive modulatory effects for healthy gut microbiome development. Our aim for this study was to investigate if these differences persist following discharge up to 1.5–2 years of age and conduct both a cross-sectional and longitudinal analysis of the gut microbiome of these preterm infants, using both shotgun metagenomics and 16S rRNA profiling. We were particularly interested to determine if the probiotic species, specifically L. acidophilus and B. bifidum, were persisting in the gut long-term. In addition, to determine if probiotics had a lasting modulatory effect on microbiome development, we compared these probiotic-supplemented infants to a group of infants who were born into the same nursery but did not receive probiotic supplementation. Lastly, we combined a subset of these newly acquired samples with data collected previously, to conduct a longitudinal examination of probiotic-supplemented infants.
Methods
Study design
This observational study involves both a longitudinal and cross-sectional component. As the main objective of this project was to examine if probiotic prophylaxis during admission has a lasting effect, we performed a cross-sectional analysis of 18 infants using shotgun metagenomics and compared those who had received probiotics against those who had not. As previously mentioned, a subset of this cohort (n = 6) had samples collected as part of an earlier study, and so we also performed a longitudinal analysis of these probiotic-supplemented infants using 16S rRNA amplicon sequencing, as this was the technique used previously.
Study population
A combination of 16S rRNA gene amplicon and shotgun metagenomic sequencing was used to characterise the faecal microbiome of preterm infants from North Queensland (NQLD), Australia. This region of Australia is disproportionately burdened by preterm birth and low birth weight [18], and its large indigenous population is more likely to experience prematurity relative to other Australians (13%), representing one in ten preterm births in Queensland [18]. The burden of preterm birth in NQLD, which has increased 5% over the last decade [18], places significant stress on the families and healthcare system in this region of Australia.
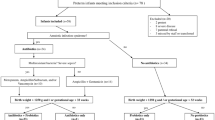
Infants recruited were previously admitted to the Townsville University Hospital’s (TUH) Neonatal Intensive Care Unit (NICU) and Special Care Nurseries (SCN). The TUH NICU is the only level six tertiary referral unit in NQLD, which is a specialised unit for dealing with complex pregnancies. The criteria for probiotic prophylaxis at the TUH NICU dictate that all high-risk preterm infants (defined as < 32-week gestation and/or < 1500 g) receive Infloran® [19], containing Lactobacillus acidophilus (1 × 109 CFU) and Bifidobacterium bifidum (1 × 109 CFU) on a daily basis, from the first day of feeding to > 34–36-week gestation. Inclusion criteria for the cohort included born < 32-week gestation and previously admitted to the NICU at the TUH for the probiotic group and > 32 weeks and admitted to the SCN at the TUH. The exclusion criteria were no parental consent, born > 32 weeks and contraindication to enteral feeds for the probiotic group, and no parental consent for the non-supplemented group. Ethics was obtained from the Townsville Hospital and Health Service Human Research Ethics, (HREC/QTHS/65181 and HREC/17/QTHS/7). Informed consent was obtained from parents/legal guardians of all subjects through the signing of a Parental Information Sheet and Consent Form (PICF), which can be found in the Supplementary material.
Recruitment and sample collection
Recruitment of infants previously admitted to the hospital and who were now between 18 months and 2 years of age was conducted by a neonatal nurse, who works in the nurseries, between January and August of 2021. Parents/guardians of previously admitted preterm infants were contacted via the phone, and upon verbal approval, mailed out a PICF and a collection kit. The collection kit included:
-
OMNIgene® GUT all in one system
-
Paid return postal package
-
Detailed instructions on sample collection and postage
-
Absorbent material and leak proof biohazard bag for postage requirements
-
Questionnaire
The samples of recruited infants were stored in the OMNIgene® GUT collection tube, and once mailed back to the research team, the tubes were stored at − 80 °C, as recommended by the manufacturer.
Collection of metadata
Both clinical (during admission) and post-discharge metadata were also collected (Table 1). For the clinical data, this included both maternal — antenatal antibiotics, chorioamnionitis (clinically diagnosed), preeclampsia (clinically diagnosed), and diabetes (type 1 & 2), and infant data — mode of delivery (vaginal birth versus caesarean section), diet, gestation, NEC (stage 2 or greater), sepsis (confirmed through culture), neonatal antibiotics and retinopathy of prematurity (ROP) (stage 1 or greater). The post-discharge information was collected through the previously mentioned brief questionnaire.
16S rRNA short amplicon sequencing
We used the Bioline ISOLATE Fecal DNA Kit for DNA extraction [20]. Modifications were made in consultation with the manufacturer to optimise DNA yield and included increased beta-mercaptoethanol from 0.5 to 1% (increasing DNA solubility and reducing secondary structure formation), addition of an extra wash step (improving purity) and decreased elution buffer volume from 100 to 50 μl (increasing final DNA concentration). After consultation with the manufacturer, 150 µl was chosen for the initial sample volume, in place of the usual 150 µg required by the kit, for compatibility with the OMNIgene® GUT kit. The Illumina metagenomics library preparation protocol was used for library preparation [21], using the Index Kit v2 C [22] and Platinum™ SuperFi™ PCR Master Mix [23]. Sequencing was performed on the Illumina MiSeq system using the MiSeq Reagent Kit V3 [22], targeting the V3 and V4 regions with the S-D-Bact-0431-b-S-17/S-D-Bact-0785-a-A-21primer combination [21]. Both the pre-analytical bioinformatics and statistical analyses were conducted in R Studio Version 3.6.1 [24] with a pipeline adapted from our previous work [25], which can be found in the Supplementary material. DADA2 [26] was used for quality filtering and trimming, demultiplexing, denoising and taxonomic assignment (SILVA Database). In addition, microDecon [27] was used to remove homogenous contamination from samples using extraction blanks.
Admission and discharge samples for longitudinal analyses
Data for a subset of individuals that had samples collected at both admission and just prior to discharge were obtained from previous work. As we recruited from the same hospital, a small subset (n = 6) had samples collected at previous time points, allowing us to make comparisons across these three time points, within the probiotic-supplemented group. However, it should be noted that for one infant, we did not receive an admission sample. The recruitment, collection and sequencing protocols are as previously described [25].
Shotgun metagenomics
The shotgun metagenomics was performed by Microba Life Sciences [28]. Once samples had DNA extracted for 16S rRNA gene amplicon sequencing, the samples were then again stored at − 80 °C, and, soon after, shipped to Microba on dry ice. Sequencing was conducted on the Illumina NovaSeq6000 system with 300 bp, paired-end reads. This workflow was completed using Microba’s patented metagenomics analysis platform (MAP), which includes the Microba Genome Database, the Microba Community Profiler and the Microba Gene and Pathway Profiler [28]. The MAP produces taxonomic and functional profiles.
Statistical analysis
For both the 16S rRNA short amplicon sequencing and shotgun metagenomics, we assessed beta diversity, alpha diversity and taxonomic abundance using mixed effects models. For beta diversity comparisons, we performed both PERMANOVA and EnvFit analyses from the Vegan package [29], which compare the differences in the centroids relative to total variation. Both analyses were applied to Bray–Curtis dissimilarity matrices [30] based on data normalised through total sum scaling (TSS) [31]. The significance was based on 10,000 permutations and was transformed based on the Benjamini-Hochberg (BH) procedure [32].
For alpha diversity comparisons, we performed generalised linear mixed effects models. The generalised linear mixed effects regression models were created using the package lme4 [33]. Shannon diversity was calculated at the ASV level, on normalised data (TSS), and continuous predictors were scaled and centred. Multicollinearity was assessed using the AED package [34], and significance was using an analysis of deviance (type II Wald chi-square test) from the car package [35]. This was followed by subsequent post-hoc pairwise Tukey comparisons, to correct for multiple comparisons, using the emmeans package [36].
DESeq2 [37], which uses a negative binomial generalised linear model and variance stabilising transformation, was used for comparing taxonomic abundances between groups. For the 16S rRNA short amplicon sequencing, taxa were agglomerated at the genus level, due to the limited taxonomic depth of 16S-target technologies. A Wald test with the BH multiple inference correction was performed to obtain taxa that were significantly differentially abundant. The pre-analytical bioinformatics and statistical analyses can be found in the GitHub link in the Supplementary material. The results for each of the statistical analyses can be found in the Supplementary File 1.
Results
Changes in the gut microbiome of probiotic-supplemented infants over time
The data from the 16S amplicon sequencing revealed that the microbiome composition of probiotic-supplemented infants changed dramatically over time, stabilising at discharge (Fig. 1A). Samples clustered significantly by the sampling time based on their taxonomic composition (Fig. 1A, p < 0.01), coupled with a significant increase in alpha diversity post-discharge (Fig. 1B, admission and post-discharge: p < 0.0001, and discharge and post-discharge: p < 0.0001), as taxa continued to colonise. The composition is dominated early on by the phylum Firmicutes, followed by Proteobacteria and Actinobacteriota at discharge, and then Bacteroidota and Firmicutes post-discharge (Fig. 2A). At the genus level, we see Streptococcus, a facultative anaerobe, dominating at admission, followed by Bifidobacterium at discharge, and then maturation to a more diverse ecosystem post-discharge. The changes in Streptococcus over time were significant (p < 0.01), with it being in significantly greater abundance early on, compared with both discharge (p < 0.001) and post-discharge (p < 0.01) samples. A similar pattern was seen for Bifidobacterium, which was in significantly greater abundance at both admission (p < 0.001) and discharge (p < 0.001), relative to post-discharge samples.

A Principal coordinate analysis plot based on Bray–Curtis distances using ASV level taxonomy obtained through 16S rRNA short amplicons sequencing demonstrating the changes in gut microbial composition for the six infants tracked over time, with significant (p < 0.01) clustering of samples. B Dot plot representing the time-based increases in alpha diversity metrics for the same six infants tracked over time and based on transformed ASV level taxa (16S amplicon sequencing), both observed (richness) and the Shannon Index, where pairwise comparisons found significant differences between admission and discharge samples (p = 0.01), admission and post-discharge (p < 0.0001) and discharge and post-discharge (p < 0.0001)

A Changes in the proportions of taxa for the six infants tracked over time at both the phylum and genus levels (16S amplicon sequencing) across admission, just prior to discharge and post discharge, describing the significant (p < 0.0001) reduction in Bifidobacterium abundance post-discharge relative to the first two time points. B Changes in the proportions of both Bifidobacterium and Lactobacillus for the six infants tracked (16S amplicon sequencing) across admission, just prior to discharge, and post discharge, using 16S amplicon sequencing
Persistence of probiotic species present at discharge up to 2 years of age
Bifidobacterium was present at admission, discharge and post-discharge; however, Lactobacillus was sparse at all time points (Fig. 2B). Bifidobacterium was at its greatest abundance at discharge, and thus, towards the end of supplementation. However, despite being present across all infants, there was a significant reduction in its abundance post-discharge. Using shotgun metagenomics, we observed that what remains of the genus post-discharge is mostly other Bifidobacterium species, with B. bifidum only present in 4/14 infants (Table 2). The other Bifidobacterium species present were B. adolescentis, B. animalis, B. breve and B. longum, with B. longum as the most common and B. breve as having the greatest mean relative abundance. L. acidophilus was also not present post-discharge (Table 2). The only two species from this genus present post-discharge were L. paracsei and L. rhmanosus. The lack of long-term colonisation with L. acidophilus is consistent with previous work; however, B. bifidum has been observed to persist post-discharge at ~ 58 weeks [14]. The scarcity of probiotic species present in our cohort of infants suggests transient colonisation.
Comparison of probiotic-supplemented infants and non-supplemented infants
Previously identified positive modulation of the gut microbiome associated with probiotic prophylaxis during hospital admission does not persist at 18 months to 2 years post-supplementation; however, several other associations were observed. Overall community composition did not differ significantly between those who received probiotic prophylaxis and those who did not (Fig. 3A, PERMANOVA: p = 0.4, envfit: p = 0.88). However, differences in several taxa were oberved (Fig. 4). Specifically, we observed greater abundance of Clostridium_M sp001517625 (p < 0.01) and Flavinofractor plauti (p < 0.01), in combination with lower abundances of Alistipes finegoldi (p < 0.01), in those that received probiotic prophylaxis. Clostridium_M sp001517625 (11/14 infants) and Flavonifractor plauti (13/14 infants) were only observed in the probiotic group. In contrast, Alistipes finegoldi was only found in half of those supplemented, but all of those who did not receive probiotics. Lastly, counter to what was expected, alpha diversity, both richness (p < 0.05) and the Shannon Index (p < 0.05) were significantly lower in those infants supplemented with probiotics (Fig. 3B). However, it is unclear whether this associated modulation is a result of probiotic prophylaxis or evidence of an inability of probiotics to exhibit lasting modulation beyond the supplementation period.

A principal coordinate analysis plot based on Bray–Curtis distances exploring the clustering of samples post-discharge by probiotic-supplementation (coloured) using species level taxonomy obtained through shotgun metagenomics. B Dot plots describing the significant difference in alpha diversity metrics post-discharge, both observed (richness) (p < 0.05) and the Shannon Index (p < 0.05), between probiotic supplementation groups and obtained through shotgun metagenomics

A Bar plots comparing the relative distribution of the top 30 most abundant species identified through shotgun metagenomics, and across individuals and between probiotic-supplementation status. B results of Wald-test on the probiotic-supplementation comparison from DESeq2 mixed effects modelling, that also accounted for diet, on species level taxonomy obtained through shotgun metagenomics
Discussion
We have previously described the short-term positive modulatory impact that probiotic supplementation with Infloran® can have on the developing gut microbiome of preterm infants during their NICU admission [17], supporting an increased diversity and colonisation with beneficial taxa, such as Bifidobacterium. This study sets out to investigate whether colonisation by these probiotic species affected the development of a healthy microbiome at 18 months to 2 years post discharge. We observed significant changes in the microbiome of supplemented infants over time, with increases in alpha diversity and dynamic changes in taxonomic abundance, culminating in a reduction in heterogeneity between samples and stabilisation of the microbiome. However, these dynamic changes were coupled with a significant reduction in probiotic species. Several studies report persistence of the probiotic species in the faeces of preterm infants up to the time of hospital discharge [38,39,40,41], yet evidence for long-term colonisation with probiotic species is limited. The inability of probiotic species to colonise the infant gut may mean that probiotic-associated modulation is short-lived. However, the reported benefits of probiotic prophylaxis may extend beyond colonisation and include competitive pathogen exclusion, changes to intestinal barrier function and immune modulation [8]. These positive effects on the developing microbiome may prove important for pre-term infants.
Changes in probiotic-supplemented infants over time
Despite high levels of heterogeneity between individuals early in life, the infant microbiome generally follows a standardised colonisation process. As previously described, the choregraphed progression leads to typical microbial communities at different stages of the developmental process [42], as well as a reduction in heterogeneity and increased stabilisation with time [43]. Typically, aerobes dominate early on, defined by Dogra et al. as Cluster 1. Cluster 2 is characterised by higher levels of facultative anaerobes, especially those from Enterobacteriaceae, and Cluster 3 has higher abundances of strict anaerobes, particularly Bifidobacterium, in combination with lower abundances of aerobes like Streptococcus [42]. This previous work describes Cluster 3 as an end point, as once Cluster 3 is reached, subsequent samples from the same infant stay within this cluster. However, our cohort of infants had a significant reduction in Bifidobacterium after it rose to dominance.
The drop in Bifidobacterium and the lack of persistence of B. bifidum may be the result of confounding factors. Delayed colonisation or reduced counts of Bifidobacterium have previously been linked to caesarean section [42], and greater colonisation to breastfeeding [25]. The link to breastfeeding largely stems from the presence of human milk oligosaccharides in breastmilk, which are complex glycans that selectively nourish specific microbes [44, 45]. Without nourishment, microbes like B. bifidum may not persist. This may also explain why our work does not align with that of Abdulkadir et al. who observed persistence of B. bifidum following supplementation with Infloran® and post discharge [14]. An important distinction between this study and theirs is that their entire cohort was breastfed, contrasting with only four infants in this study. Additive to this is that formula was introduced to all our infants’ post-discharge (Supplementary Table 1). Thus, diet may be an important factor for sustaining colonisation, and a modifiable factor that could encourage the long-term persistence of probiotic species in preterm infants.
L. acidophilus was not present post-discharge. Previous work has also struggled to isolate both the species and genus in supplemented infants [14, 15, 46]. However, unlike B. bifidum, this may not be a result of diet. Yousuf et al. who also observed an inverse relationship of Lactobacillus and antibiotic exposure, suggests that this is due to Lactobacillus being a coloniser of the small intestine and less likely to be found in faecal samples [15]. In addition, in our longitudinal analysis, we observed that the genus does not consistently establish itself in the gut during admission/prophylaxis, which is also supported by our previous work [17]. In this previous work, we suggest that this lack of colonisation could be the result of poor probiotic integrity. However, more work still needs to be done to provide conclusive evidence. Taken together, it is likely that the persistence of probiotic species and even bacterial community succession over the long term is determined by multiple environmental factors. Development of the gut microbiome in early life appears to mimic ecological primary succession, involving pioneer organisms which colonise the newly developed and relatively sterile habitat, changing the environmental conditions and thereby dictating succession through provision of niche conditions.
Comparison of probiotic-supplemented infants and non-supplemented infants
Positive probiotic-associated modulation may not persist beyond discharge. Previous work suggests probiotic prophylaxis contributes to acute increases in bacterial diversity and abundance of known commensals, as well as a reduction in potential pathogens. This positive modulation may explain why probiotics have been demonstrated to significantly reduce the incidence of stage 2 or more NEC and LOS, albeit with some level of heterogeneity [47]. However, the modulation contributing to disease reduction appears to be temporary and may result from the limited persistence of probiotic species as previously discussed. Durack et al. observed that probiotics can temporarily correct for the delayed diversification associated with preterm infants, but that the inability of the probiotic species to engraft meant that these benefits are lost when probiotic prophylaxis is complete [48]. Additionally, Yousuf et al. demonstrated that probiotic exposure in preterm infants resulted in increased relative abundance of Bifidobacterium, but not Lactobacillus, potentially due to the colonisation in the small bowel of the latter genus [15]. However, this effect reduced over time, particularly at 5-month follow-up [15]. The cessation of probiotic prophylaxis may be why we do not see previously observed modulation persists.
Although lower diversity is associated with probiotic supplementation, it is unclear if probiotics are a driver of this low diversity. This is especially true when one considers the inability of the probiotic species to persist. Rather, these results may suggest that probiotics cannot correct for the lower diversity common to the most premature of infants. However, if probiotics are the causative factor, the drop in diversity may be a result of the restructuring of the microbial ecology, where the probiotic supports growth of a few specific taxa. Either way, whether this lower diversity will have significant consequences for these infants is not known, and beyond providing stability, greater diversity may also have limited benefits. Many of the benefits afforded by probiotic prophylaxis are likely to come through support of key taxa that possess invaluable functionality, or functional benefits provided through the probiotic species themselves during the supplementation period. This may be through the ability to combat pathogens by preventing adhesion to the mucosa [49] or through the production of short chain fatty acids [50]. This is not to say that diversity is not beneficial, just that the presence of key taxa may provide more benefits to infant health.
There were significant differences in the abundance of three species between the probiotic groups. Although it has been implicated in fatty liver disease, Clostridium_M sp001517625 is a relatively un-studied species [51]. Clostridium species are of particular interest in maintenance of gut health and an imbalance in two of the species clusters (XIVa and IV) has been implicated in the development of ulcerative colitis and overall gut health, due to its involvement in metabolism of bile acids and generation of short-chain fatty acids [50]. Flavonifractor plauti on the other hand is considered a common inhabitant of the human gut microbiome, but its role/significance is unclear. The species has been implicated in both beneficial and pathogenic roles, being linked to reduced Th2 immune responses in mice [52], potentially through catechin metabolism, and to colorectal cancer [53], potentially acting through its capacity to degrade beneficial flavonoids. Lastly, Alistipes finegoldi is also thought to be a common inhabitant of the gut microbiome, albeit at lower levels relative to other Bacteroidetes. In terms of the species pathogenicity, there is contrasting evidence. The species has been suggested to be protective against some diseases, such as liver fibrosis and colitis, but it has also been implicated as a pathogen in colorectal cancer and depression [54]. Thus, although significant differences in taxa exist, the consequences are again indeterminant.
Despite the apparent limited long-term benefits in microbial modulation, the acute modulation observed previously during supplementation may provide lasting benefits. Microbial perturbations, including lower diversity, have been consistently associated with disease. This includes obesity [55], metabolic syndrome [56], Crohn’s disease and ulcerative colitis [57, 58], multiple sclerosis [59] and more. However, equally important to note is the effect on the development of both innate and adaptive immune function [60], as perturbations in the gut microbiome have also been shown to have long-lasting metabolic and immunological dysregulation [61]. Delayed gut microbial diversification over the first year of life, along with altered composition and metabolic function, is significantly associated with a greater risk of atopy and asthma development in childhood [48]. Thus, the early-life gut microbiome, colonising a relatively sterile habitat, influencing the developing ecosystem, and in turn, immune and physiological conditions, may have the greatest impact on long-term health.
Limitations
This work has limited statistical power and was unable to account for all known microbial covariates due to its small sample size. As stated in the methods, the recruitment and collection protocol involved contacting parents/guardians at home and relying on their involvement for the collection and postage of stool samples. This proved too much of a burden for a demographic of people who have limited incentive to be involved in the project and are dealing with the stresses of being a new parent. We recommend that future studies take this into consideration during study design and either have greater involvement in the collection process or target a larger group to ensure adequate sample size.
Conclusion
Probiotic-supplemented preterm infants are protected from infectious diseases during their stay in the NICU; however, this study suggests that the prophylactic probiotic species from Infloran® do not persist 18 months–2 years post discharge. The implications of this are unclear. While probiotic-supplemented infants showed a healthier microbiome at discharge compared to other infants who did not receive probiotic supplementation, probiotic-supplemented infants had lower diversity in their gut microbiome at 18 months to 2 years of age. The small sample size reduces the certainty of this result. Nonetheless, with the emergence of a significant body of literature implicating the early gut microbiome in immune system development, it is unclear if lower diversity at this age would have significant implications.
Availability of data and material
The sequencing dataset generated and/or analysed during the current study is available through the International Nucleotide Sequence Database Collaboration at the National Center for Biotechnology Information (NCBI) repository; BioProject IDs: PRJNA751712, PRJNA687291 & PRJNA805057.
Code availability
The analysis workflow, both the bioinformatics pipeline and statistical analyses, can be found at JacobAFW/Long_term_effects_of_probiotics (github.com).
Abbreviations
- ASV:
-
Amplicon sequence variant
- B:
-
Bifidobacterium
- BH:
-
Benjamini-Hochberg
- DNA:
-
Deoxyribonucleic acid
- EC:
-
Enzyme commission
- EC:
-
Enzyme commission number
- HMO:
-
Human milk oligosaccharide
- L:
-
Lactobacillus
- LoS:
-
Late-onset sepsis
- MetaCyc:
-
Metabolic pathway database
- MPA:
-
Metagenomics analysis platform
- NEC:
-
Necrotising enterocolitis
- NICU:
-
Neonatal intensive care unit
- NMDS:
-
Non-metric multidimensional scaling
- NQLD:
-
North Queensland
- PCoA:
-
Principal coordinate analysis
- PCR:
-
Polymerase chain reaction
- PERMANOVA:
-
Permutational analysis of variance
- ROP:
-
Etinopathy of prematurity
- rRNA:
-
Ribosomal ribonucleic acid
- SCN:
-
Special care nursery
- TCDB:
-
Membrane transport proteins
- TSS:
-
Total sum scaling
- THHS:
-
Townsville Hospital and Health Service
References
World Health Organisation (2018) Preterm Birth. Available from: https://www.who.int/news-room/fact-sheets/detail/preterm-birth#:~:text=Preterm%20is%20defined%20as%20babies%20born%20alive%20before,planned%20before%2039%20completed%20weeks%20unless%20medically%20indicated. Accessed 2 May 2022
Forsgren M, Isolauri E, Salminen S, Rautava S (2017) Late preterm birth has direct and indirect effects on infant gut microbiota development during the first six months of life. Acta Paediatr 106(7):1103–1109
Jacquot A, Neveu D, Aujoulat F, Mercier G, Marchandin H, Jumas-Bilak E et al (2011) Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J Pediatr 158(3):390–396
Chang JY, Shin SM, Chun J, Lee JH, Seo JK (2011) Pyrosequencing-based molecular monitoring of the intestinal bacterial colonization in preterm infants. J Pediatr Gastroenterol Nutr 53(5):512–519
Tamburini S, Shen N, Wu HC, Clemente JC (2016) The microbiome in early life: implications for health outcomes. Nat Med 22(7):713–722
Berrington JE, Stewart CJ, Embleton ND, Cummings SP (2013) Gut microbiota in preterm infants: assessment and relevance to health and disease. Arch Dis Child Fetal Neonatal Ed 98(4):286–290
Thomas JP, Raine T, Reddy S, Belteki G (2017) Probiotics for the prevention of necrotising enterocolitis in very low-birth-weight infants: a meta-analysis and systematic review. Acta Paediatr 106(11):1729–1741
Raheem A, Liang L, Zhang G, Cui S (2021) Modulatory effects of probiotics During pathogenic infections with emphasis on immune regulation. Front Immunol 12
Zuo F, Appaswamy A, Gebremariam HG, Jonsson A-B (2019) Role of sortase A in Lactobacillus gasseri Kx110A1 adhesion to gastric epithelial cells and competitive exclusion of helicobacter pylori. Front Microbiol 10
Saitoh T, Yamamoto M, Miyagishi M, Taira K, Nakanishi M, Fujita T et al (2005) A20 Is a negative regulator of IFN regulatory factor 3 signaling. J Immunol 174(3):1507
Pei R, Martin DA, DiMarco DM, Bolling BW (2017) Evidence for the effects of yogurt on gut health and obesity. Crit Rev Food Sci Nutr 57(8):1569–1583
Saliganti V, Kapila R, Kapila S (2016) Consumption of probiotic Lactobacillus rhamnosus (MTCC: 5897) containing fermented milk plays a key role in development of the immune system in newborn mice during the suckling–weaning transition. Microbiol Immunol 60(4):261–267
van Zyl WF, Deane SM, Dicks LMT (2020) Molecular insights into probiotic mechanisms of action employed against intestinal pathogenic bacteria. Gut microbes 12(1):1831339
Abdulkadir B, Nelson A, Skeath T, Marrs EC, Perry JD, Cummings SP et al (2016) Routine Use of probiotics in preterm infants: longitudinal impact on the microbiome and metabolome. Neonatology 109(4):239–247
Yousuf EI, Carvalho M, Dizzell SE, Kim S, Gunn E, Twiss J et al (2020) Persistence of suspected probiotic organisms in preterm infant gut microbiota weeks after probiotic supplementation in the NICU. Front Microbiol 11:574137
Taft DH, Ambalavanan N, Schibler KR, Yu Z, Newburg DS, Ward DV et al (2014) Intestinal microbiota of preterm infants differ over time and between hospitals. Microbiome 2(1)
Westaway JAF, Huerlimann R, Kandasamy Y, Miller CM, Norton R, Watson D et al (2022) To probiotic or not to probiotic: a metagenomic comparison of the discharge gut microbiome of infants supplemented with probiotics in NICU and those who are not. Front Pediatr 10
Health Q (2018) The health of Queenslanders 2018. Published by the State of Queensland (Queensland Health), Brisbane
Infloran (2019) Evidence based probiotics. Available from: https://www.infloran.com.au/?gclid=CjwKCAiA-_L9BRBQEiwA-bm5fjBoxiUHkDF7r40k4SgIjF7M_MDTTVue4HDOB6QFbsX1XD_WgJICshoCPY8QAvD_BwE (Cited 25 Nov 2020)
Meridian (2020) Meridian Bioscience. Available from: https://www.bioline.com/ (Cited 5 Nov 2020)
Illumina Inc (2018) 16S Metagenomic Sequencing Library Preparation. Available from: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (Cited 24 Dec 2020)
Illumina Inc (2020) Illumina. Available from: https://www.illumina.com/index-d.html (Cited 25 Nov 2020)
ThermoFisher Scientific (2020) ThermoFisher Scientific. Available from: https://www.google.com/search?q=platinum+superfi+pcr+master+mix&oq=platinum+superfi&aqs=chrome.1.69i57j0l7.3863j0j4&sourceid=chrome&ie=UTF-8 (Cited 25 Nov 2020)
RStudio Team (2020) RStudio: Integrated Development for R. RStudio, PBC, Boston, MA. http://www.rstudio.com/
Westaway JAF, Huerlimann R, Kandasamy Y, Miller CM, Norton R, Staunton KM et al (2021) The bacterial gut microbiome of probiotic-treated very-preterm infants: changes from admission to discharge. Pediatr Res
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13(7):581–583
McKnight D, Huerlimann R, Bower D, Schwarzkopf L, Alford R, Zenger K (2019) microDecon: a highly accurate read‐subtraction tool for the post‐sequencing removal of contamination in metabarcoding studies. Environmental DNA (1):14:25
Parks DH, Rigato F, Krause L, Tousignant K, Pribyl AL, Hugenholtz P, Tyson GW, Wood DL (2020) Microba’s community profiler enables precise measurement of the gut microbiome. Microba Life Sci
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14(6):927–930
Jari Oksanen FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MH, Szoecs E, Wagner H (2020) Vegan: community ecology package. Community Ecol 2.5–7:719
McKnight DT, Huerlimann R, Bower DS, Schwarzkopf L, Alford RA, Zenger KR (2019) Methods for normalizing microbiome data: an ecological perspective. Methods Ecol Evol 10(3):389–400
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol) 57(1):289–300
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4 2015;67(1):48
Luštrik R, Stachelek J (2009) AED: package accompanying ‘mixed effects models and extensions in ecology with R’. p. R package version 1.0
Fox J, Weisberg S (2019) An R companion to applied regression. Third ed. Sage, Thousand Oaks CA
Searle SR, Speed FM, Milliken GA (1980) Population marginal means in the linear model: an alternative to least squares means. Am Stat 34(4):216–221
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550
Li Y, Shimizu T, Hosaka A, Kaneko N, Ohtsuka Y, Yamashiro Y (2004) Effects of bifidobacterium breve supplementation on intestinal flora of low birth weight infants. Pediatr Int 46(5):509–515
Mohan R, Koebnick C, Schildt J, Schmidt S, Mueller M, Possner M et al (2006) Effects of Bifidobacterium lactis Bb12 supplementation on intestinal microbiota of preterm infants: a double-blind, placebo-controlled, randomized study. J Clin Microbiol 44(11):4025–4031
Plummer EL, Bulach DM, Murray GL, Jacobs SE, Tabrizi SN, Garland SM et al (2018) Gut microbiota of preterm infants supplemented with probiotics: sub-study of the ProPrems trial. BMC Microbiol 18(1):184
Horigome A, Hisata K, Odamaki T, Iwabuchi N, Xiao J-Z, Shimizu T (2021) Colonization of supplemented bifidobacterium breve M-16V in low birth weight infants and its effects on their gut microbiota weeks post-administration. Front Microbiol 12
Dogra S, Sakwinska O, Soh S-E, Ngom-Bru C, Brück WM, Berger B et al (2015) Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. Mbio [Internet] 6(1). Available from: http://europepmc.org/abstract/MED/25650398. Accessed 2 May 2022
Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC et al (2018) Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562(7728):583–588
Su P, Henriksson A, Mitchell H (2007) Prebiotics enhance survival and prolong the retention period of specific probiotic inocula in an in vivo murine model. J Appl Microbiol 103(6):2392–2400
Panigrahi P, Parida S, Pradhan L, Mohapatra SS, Misra PR, Johnson JA et al (2008) Long-term colonization of a Lactobacillus plantarum synbiotic preparation in the neonatal gut. J Pediatr Gastroenterol Nutr 47(1):45–53
Alcon-Giner C, Dalby MJ, Caim S, Ketskemety J, Shaw A, Sim K et al (2020) Microbiota supplementation with Bifidobacterium and Lactobacillus modifies the preterm infant gut microbiota and metabolome: an observational study. Cell Rep Med 1(5):100077
Meyer MP, Chow SSW, Alsweiler J, Bourchier D, Broadbent R, Knight D et al (2020) Probiotics for prevention of severe necrotizing enterocolitis: experience of New Zealand neonatal intensive care units. Front Pediatr 8:119
Durack J, Kimes NE, Lin DL, Rauch M, McKean M, McCauley K et al (2018) Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by Lactobacillus supplementation. Nat Commun 9(1):707
Adlerberth I, Wold AE (2009) Establishment of the gut microbiota in Western infants. Acta Paediatr 98(2):229–238
Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A (2013) Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 5(1):23
Ruuskanen MO, Åberg F, Männistö V, Havulinna AS, Méric G, Liu Y et al (2021) Links between gut microbiome composition and fatty liver disease in a large population sample. Gut Microbes 13(1):1–22
Ogita T, Yamamoto Y, Mikami A, Shigemori S, Sato T, Shimosato T (2020) Oral administration of flavonifractor plautii strongly suppresses Th2 immune responses in mice. Front Immunol 11:379
Gupta A, Dhakan DB, Maji A, Saxena R, P KV, Mahajan S et al (2019) Association of Flavonifractor plautii, a flavonoid-degrading bacterium, with the gut microbiome of colorectal cancer patients in India. mSystems 4(6)
Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A (2020) The genus Alistipes: gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol 11:906
Winer DA, Luck H, Tsai S, Winer S (2016) The intestinal immune system in obesity and insulin resistance. Cell Metab 23(3):413–426
Richard C, Wadowski M, Goruk S, Cameron L, Sharma AM, Field CJ (2017) Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res Care 5(1):000379
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ et al (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105(43):16731–16736
Kudelka MR, Hinrichs BH, Darby T, Moreno CS, Nishio H, Cutler CE et al (2016) Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk. Proc Natl Acad Sci USA 113(51):14787–14792
Kallaur AP, Reiche EMV, Oliveira SR, Simão ANC, Pereira W, Alfieri DF et al (2017) Genetic, immune-inflammatory, and oxidative stress biomarkers as predictors for disability and disease progression in multiple sclerosis. Mol Neurobiol 54(1):31–44
Gensollen T, Iyer SS, Kasper DL, Blumberg RS (2016) How colonization by microbiota in early life shapes the immune system. Science 352(6285):539–544
Ruiz VE, Battaglia T, Kurtz ZD, Bijnens L, Ou A, Engstrand I et al (2017) A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Commun 8(1):518
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was funded through a Study, Education and Research Trust Account (SERTA) research grant (Townsville Hospital and Health Service). The funding body played no role in the design of the study or collection, nor the interpretation of data and writing.
Author information
Authors and Affiliations
Contributions
Substantial contributions to conception and design, acquisition of data or analysis and interpretation of data: JW, DR, YK, RH, RN, SV and DW. Drafting the article or revising it critically for important intellectual content: JW, DR, CM, YK, RH and RN. Final approval of the version to be published: DR, CM, YK, RH, RN, JW, SV and DW.
Corresponding author
Ethics declarations
Ethics approval
The research was performed in accordance with the Declaration of Helsinki and ethics approval was obtained from the Human Research Ethics Committee from the Townsville Hospital and Health Service (HREC/QTHS/65181).
Consent to participate
Informed consent was obtained from parents/legal guardians of all subjects through the signing of a Parental Information Sheet and Consent Form (PICF).
Consent for publication
Informed consent was obtained from parents/legal guardians of all subjects through the signing of a Parental Information Sheet and Consent Form (PICF).
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Daniele De Luca
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Westaway, J.A.F., Huerlimann, R., Kandasamy, Y. et al. Exploring the long-term colonisation and persistence of probiotic-prophylaxis species on the gut microbiome of preterm infants: a pilot study. Eur J Pediatr 181, 3389–3400 (2022). https://doi.org/10.1007/s00431-022-04548-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-022-04548-y