
Abstract
Our aim was to evaluate the performance of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), routinely used in the microbiology laboratory for bacterial identification, for bacterial typing in the setting of extended spectrum beta-lactamase producing Klebsiella pneumoniae (ESBL-KP) outbreak in the neonatal intensive care unit (NICU). Isolates from a 2011 outbreak in the NICU were retrieved from frozen stocks and analyzed by MALDI-TOF. The MALDI typing was compared with core genome multilocus sequence typing (cg-MLST). MALDI typing divided the 33 outbreak isolates into 2 clones: sequence type (ST)-290 and 405. These results were in complete agreement with cg-MLST results. The differentiation of the outbreak isolates into two clones correlated with the patients’ location in the NICU, but also with their place of residence.
Conclusion: Here, we show that MALDI-TOF MS, which has been integrated into the microbiology laboratory workflow for microbial species identification, can be secondarily used for epidemiological typing at no added cost.
What is Known: • Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is now routinely used in the microbiology laboratory for bacterial identification What is New: • MALDI typing was used for outbreak investigation in the NICU and divided the outbreak isolates into two clones • MALDI-TOF MS may be secondarily used for epidemiological typing at no added cost. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Every neonatal intensive care unit (NICU) faces the challenge of rapid identification and control of outbreaks due to resistant or hypervirulent pathogens. Every increase in infections over a baseline rate may represent an outbreak and requires effective intervention. Outbreak control frequently requires the assessment of clonal relationship among circulating isolates. In a setting of widespread circulation of multidrug resistant isolates in the community, it is challenging to characterize the relative contribution of within-hospital transmission of resistant strains versus recurrent importation of isolates into the NICU.
Genotyping methods, such as pulsed field gel electrophoresis (PFGE) and multilocus sequence typing (MLST), have good discriminatory power, but are costly, have a long turnaround time, and require highly trained personnel. Recently, whole-genome sequencing (WGS) became increasingly available, as a result of improved technology and decreasing cost. However, WGS data analysis still requires significant expertise. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), which has been established as a routine method for the identification of clinically relevant microorganisms [1], is increasingly used for typing of diverse microorganisms (MALDI typing) [2]. This method is based upon the analysis of whole-cell mass spectra and is capable of mapping the microbial proteins to two dimensional coordinates based on their mass-to-charge (m/z) values. The generated spectrum provides a unique profile (“fingerprint”) of the microorganism, which is then compared against a database of reference strains. With MALDI-TOF MS, colonies growing on culture can be identified in minutes, inexpensively and accurately. The same data used for bacterial identification can be also used for isolate typing; therefore, MALDI-TOF MS may be a very efficient tool for on-site monitoring of the spread of pathogens in an outbreak setting. Nevertheless, for some species, or for isolates within the same species, MALDI-TOF may not have enough discriminatory power compared with the available reference-typing methods [3]. So far, MALDI-TOF typing was not introduced routinely to the algorithms of outbreak investigation and control. To determine the utility of MALDI-TOF MS for outbreak investigation in the NICU, we used isolates from a past outbreak, which was investigated with the available typing methods at the time. We compared the discriminatory power of the past analysis with the present MALDI-TOF MS and WGS analysis and characterized the possible implications for outbreak control.
Methods
Isolate collection and epidemiological setting
During the first 3 months of 2011, we experienced an outbreak of extended spectrum beta-lactamase-producing Klebsiella pneumoniae (ESBL-KP) in the NICU of Shaare-Zedek Medical Center (SZMC, Jerusalem, IL), a tertiary care academic hospital. The NICU then was a 35-bed unit that consisted of 20 intensive care beds and 15 step-down/intermediate care beds. As part of the outbreak control efforts, surveillance cultures were performed three times a week, using rectal swabs. At the time, all bacterial isolates were identified as Klebsiella spp. in the clinical laboratory by non-automated biochemical tests [4]. Antibiotic susceptibility testing of all isolates was performed by Kirby-Bauer’s disc diffusion method and interpreted using CLSI criteria [5].
Screening for ESBL isolates in rectal cultures was performed by applying the double disc synergy test, using ceftazidime and cefotaxime both individually and in combination with clavulanic acid. All outbreak isolates were frozen in − 70 °C. Later on, in a research setting, we used Enterobacteriaceae repetitive intergenic consensus sequence-based PCR (ERIC-PCR) typing to characterize the outbreak isolates. Recently, with the integration of MALDI-TOF MS and the increasing availability of WGS, we retrieved the outbreak isolates from frozen stocks, to determine clonality and compare the utility of these methods for outbreak investigation in the NICU.
MALDI-TOF MS typing
Samples were prepared from bacterial strains grown on blood agar (Novamed Ltd, Jerusalem, IL) and incubated for 18 h to 24 h at 37 °C. Isolated colonies of each strain were selected and used for MALDI-TOF MS identification and typing using the Microflex LT spectrometer (Bruker Daltonics, Bremen, Germany), as previously described [6]. The obtained spectra were downloaded into a MALDI Biotyper 3.0 system (Bruker Daltonics). Triplicates of the samples were prepared and measured to ensure the reproducibility of the obtained patterns. To discriminate between different Klebsiella strains, cluster analysis of mass spectra was performed and used to create a single main spectrum for each isolate. For each run, a standard (bacterial test standard, Bruker Daltonics) was included to calibrate the instrument and validate the run. Identification criteria were used as recommended by the manufacturer. Primary cluster analysis of isolates grown on the same day, under the same conditions, was performed using the MALDI Biotyper®.
Molecular analyses
Shortly after the outbreak, we performed fingerprinting of the isolates using ERIC-PCR as described by Versalovic et al. [7]. This method uses PCR amplification of extragenic repetitive sequences to directly fingerprint bacterial genomes. PCR for major genes encoding extended spectrum beta-lactamases—TEM-1, SHV-1, and CTX-M-1,3, CTX-M-2, and CTX-M-9—groups was performed as described by Dallenne et al. [8].
A subgroup of 6 isolates representing the different clones as characterized by the MALDI-TOF were chosen for WGS analysis. Whole-genome sequencing libraries were prepared using the Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer protocol. Indexed libraries were then normalized and pooled, followed by 2 × 150 paired-end whole-genome sequencing using the Illumina-MiSeq platform. Sequences were aligned to a KP reference isolate (Klebsiella pneumoniae subsp. pneumoniae ATCC 700721) using the Snippy software (https://github.com/tseemann/snippy). A minimum spanning tree was constructed based on whole-genome single-nucleotide polymorphisms (wgSNP) using the FastTree software [9].
Results
Outbreak description
Over a course of 3 days, 3 premature infants developed sepsis with ESBL-KP growing in blood cultures. Infection control interventions were promptly initiated and included weekly surveillance rectal cultures of all patients, followed by contact precautions and cohorting of colonized patients. In the initial surveillance, 6 of 26 infants (23%) were found to be colonized with ESBL-KP. Outbreak investigation revealed that one of the colonized infants had a sputum culture positive for ESBL-KP more than 10 weeks prior to this outbreak. Two of the colonized children developed urinary tract infection with ESBL-KP, and one developed bacteremia 4 weeks later. Figure 1 and Table 1 show the outbreak outline and the clinical details of the infants in the first stage of the outbreak. Overall, 37 isolates were collected, 8 of clinical specimens and 29 isolates representing colonization.

Timeline showing the number of infants screened for ESBL-KP and number of infected or colonized infants
Four outbreak isolates were identified by biochemical tests as K. oxytoca strains and therefore were not further analyzed.
Molecular characterization
The remaining 33 KP strains were resistant (in addition to cephalosporins) to amoxicillin/clavulanic acid, gentamicin, tetracycline, and trimethoprim/sulfamethoxazole, and intermediate to piperacillin/tazobactam and ciprofloxacin. Following the outbreak, we typed the strains using ERIC-PCR, in order to examine the utility of the method for clinical decision-making. Using 80% genetic relatedness as a threshold, ERIC-PCR typing divided the outbreak strains to 8 clones (Fig. 2). This method was extremely labor intensive and the results did not support the outbreak control efforts.

Dendrogram illustrating the genetic relationships among ESBL-KP outbreak strains using ERIC-PCR typing. Dashed line indicates 80% genetic relatedness
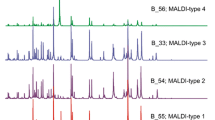
MALDI typing divided the isolates into 2 clones: In comparison with clone A1, clone A2 did not have a peak in 4100 m/z and had a shift in peaks 4705 → 4715 and 4640 → 4660 (Fig. 3). These results were in complete agreement with the cg-MLST results which are considered the gold standard (Supplementary figure S1). MALDI type A1 corresponded to sequence type (ST)-290, while MALDI type A2 corresponded to ST-405. Principal component analysis (PCA) of MALDI results show again the separation into 2 major clones. A 3D visualization of isolate clustering shows that isolates that are hard to separate by mere visualization (e.g., look like outliers or fall in the overlap zone between the clones in one dimension) are clearly separated in 3D visualization (Supplementary figure S2).

Mass spectral fingerprints of ESBL-KP strains representing the two outbreak types. In comparison with clone A1(blue), clone A2 (red) lacks a peak in 4100 m/z (A) and has a shift in peaks 4640 → 4660 (B) and 4705 → 4715 (C)
The agreement of MALDI typing and cg-MLST with the ERIC-PCR, on the other hand, was poor. For example, the 4 isolates of ST-405 had overall < 9% genetic relatedness according to ERIC-PCR typing.
The two outbreak clones shared most antibiotic resistance genes with the exception of blaSHV-145 that was positive in ST-290 isolates (clone A1 in MALDI typing) and negative in ST-405 (clone A2 in MALDI typing), and blaSHV-76 that was positive in ST-405 and negative in ST-290 (supplementary file S3).
The differentiation of the outbreak isolates into two clones correlated with the location of the patients (room 1 versus room 2), but also with their epidemiological characteristics—patients with clone A1/ST-290 lived in Central/Northern Jerusalem, whereas the 2 patients with clone A2/ST-405 lived in southern neighborhoods of Jerusalem (Fig. 4).

The correlation between the two outbreak clones and the area of maternal residence in Jerusalem
Discussion
The present study demonstrates the utility of MALDI-TOF MS in the setting of an ESBL-KP outbreak in the NICU. In real time (e.g., 2011), we could differentiate outbreak clones only based on differences in susceptibility patterns. Later on, ERIC-PCR typing was examined for outbreak investigation; however, this method was cumbersome and hard to interpret. Recently, MALDI-TOF MS became readily available, and we examined whether employing it to the isolates of the past outbreak would aid in outbreak investigation and control. MALDI typing identified two distinct outbreak clones that correlated with the epidemiological characteristics of the patients. Had this information been available in real time, we could screen and cohort patients based on these different clones.
Recently, the MALDI-TOF MS has been integrated into the microbiology laboratory workflow for quick microbial species identification. It was shown that this method might be secondarily used for epidemiological typing at no added cost; however, so far, the incorporation of MALDI typing into routine clinical practice was delayed, likely due to lack of clear guidelines for data interpretation [10]. When comparing mass spectra for typing purposes, three types of differences should be considered: signal intensity, loss of signal, and shift of signal (i.e., “peak shift”). Differences in signal intensity may reflect bacterial adaptation to different growth conditions or post-translational modification. Peaks that are considered type-specific should be recorded consistently with good signal intensities in order to avoid false interpretation [10]. For example, differences in bacterial growth conditions can affect protein expression, thereby changing the resulting “fingerprint” and the microbial assignation to a specific subgroup. Therefore, specific peaks that are expressed differently under different growth conditions should not be used for typing. Moreover, the m/z value for a given protein can change, for example, as a result of post-translational modification, which may result in a change in the peak position [10].
Another consideration in MALDI typing is what would be considered a “significant” difference between strains. This may not be the same for all bacterial species, since a number of peaks and their variability differ among species. For K. pneumoniae, for instance, the average number of peaks per spectrum is 70, and 26 of them are variable in > 70% of spectra [10]. Spinali et al. proposed that an average difference of 15 peaks could be tolerated (i.e., could define related types) in MALDI-TOF MS typing [10]. Our strains were variable in only 3 peaks.
The information provided by the MALDI typing, had it been available in the time of the outbreak, was clinically relevant. Here, we show a correlation between the 2 circulating outbreak clones and distinct geographical areas of Jerusalem. Patients infected with isolate ST-290 resided in Northern/Central Jerusalem, while patients infected with ST-405 reside in the southern neighborhoods of Jerusalem. Nevertheless, patients with clone A1 resided in room 1, while patients with clone A2 resided in room 2. Therefore, in this case, we could not differentiate nosocomial spread in the NICU from importation of isolates from the community (e.g., maternal flora).
In summary, MALDI-TOF MS typing is a quick, low-cost method that can provide timely information regarding the different circulating strains in an outbreak setting. Such information can speed up the implementation of infection control measures, guide outbreak investigation and control efforts, and limit the spread of pathogens in the NICU. Nevertheless, in order for the MALDI typing to be incorporated as a first-line tool in an outbreak setting, its performance in various bacterial species and comparison with other widely accepted typing methods (especially cg-MLST) should be further evaluated, preferably by multicenter validation studies.
Abbreviations
- ERIC-PCR:
-
Enterobacteriaceae repetitive intergenic consensus sequence-based PCR
- ESBL-KP:
-
Extended spectrum beta-lactamase-producing Klebsiella pneumoniae
- MALDI-TOF MS:
-
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MLST:
-
Multilocus sequence typing
- NICU:
-
Neonatal intensive care unit
- PCA:
-
Principal component analysis
- PFGE:
-
Pulsed field gel electrophoresis
- WGS:
-
Whole-genome sequencing
References
Patel R (2013) Matrix-assisted laser desorption ionization-time of flight mass spectrometry in clinical microbiology. Clin Infect Dis 57(4):564–572
Welker M, Moore ER (2011) Applications of whole-cell matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry in systematic microbiology. Syst Appl Microbiol 34(1):2–11
Bernardo K, Pakulat N, Macht M, Krut O, Seifert H, Fleer S, Hünger F, Krönke M (2002) Identification and discrimination of Staphylococcus aureus strains using matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Proteomics 2(6):747–753
Versalovic J, Carroll KC, Funke G, Jorgensen JH, Landry ML, Warnock DW (eds) (2011) Manual of clinical microbiology, 10th edn. American Society of Microbiology
European Committee on Antimicrobial Susceptibility Testing, B.T.f.I.o.M.a.Z.D., version 1.3, 2011 http://www.eucast.org/clinical_breakpoints
Seng P, Rolain JM, Fournier PE, la Scola B, Drancourt M, Raoult D (2010) MALDI-TOF-mass spectrometry applications in clinical microbiology. Future Microbiol 5(11):1733–1754
Versalovic J, Koeuth T, Lupski JR (1991) Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res 19(24):6823–6831
Dallenne C, da Costa A, Decré D, Favier C, Arlet G (2010) Development of a set of multiplex PCR assays for the detection of genes encoding important beta-lactamases in Enterobacteriaceae. J Antimicrob Chemother 65(3):490–495
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26(7):1641–1650
Spinali S, van Belkum A, Goering RV, Girard V, Welker M, van Nuenen M, Pincus DH, Arsac M, Durand G (2015) Microbial typing by matrix-assisted laser desorption ionization-time of flight mass spectrometry: do we need guidance for data interpretation? J Clin Microbiol 53(3):760–765
Author information
Authors and Affiliations
Contributions
Conceptualization, MB, MVA; methodology, LK,DZ, MVA; investigation, EB, LK, DZ, and MB; writing–original draft, MB; writing–review and editing, MB and MVA; supervision, MB and MVA.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Nicole Ritz
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bar-Meir, M., Berliner, E., Kashat, L. et al. The utility of MALDI-TOF MS for outbreak investigation in the neonatal intensive care unit. Eur J Pediatr 179, 1843–1849 (2020). https://doi.org/10.1007/s00431-020-03696-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-020-03696-3