
Abstract
Juvenile myelomonocytic leukemia (JMML) is a heterogeneous childhood leukemia. The management of patients with JMML requires accurate assessment of genetic and clinical features to help in patient risk stratification. This study aimed to investigate the association between genomic alterations and prognosis in children with JMML. Genomic DNA was extracted from a total of 93 patients with JMML for targeted sequencing. Univariable and multivariable analysis were used to evaluate the correlation between gene mutations and prognosis of the patients. Patients with PTPN11 mutation exhibited significantly lower event-free survival (EFS) compared with non-PTPN11 mutations (P = 0.005). Patients without or with one somatic alteration at diagnosis showed significantly better prognosis in comparison with those with more than two alterations (P = 0.009). PTPN11 mutation with additional alterations showed significantly the poorest outcome in comparison with those with only one non-PTPN11 mutation, only one PTPN11 mutation, and combined mutations without PTPN11, respectively (P < 0.0001).
Conclusion: Both PTPN11 mutation and the number of somatic alterations detected at diagnosis are likely to be the major determinant of outcome in JMML. The subgroup of patients with PTPN11 mutation showed the shortest survival which was even worsened when a secondary mutation was present.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare, early childhood neoplasm with features characteristic of both myelodysplastic and myeloproliferative disorders. Most JMML cases are severe, with the only curative treatment being hematopoietic stem cell transplantation (HSCT) [1, 4]. However, clinical evolution is heterogeneous, with occasional transformation to acute myeloid leukemia (AML) and frequent relapses after HSCT, whereas some rare “long-term survivors” experience spontaneous remission and survive without treatment [3, 4, 16]. The management of patients with JMML requires accurate assessment of genetic and clinical features to help in patient risk stratification [2, 5]. We hypothesized that complete genomic characterization of JMML would aid in distinguishing these cases.
Driver mutations converge on the RAS-signaling pathway. Mutations in NF1, NRAS, KRAS, PTPN11, and CBL currently allow for a molecular diagnosis in 85% of patients [14, 15, 23]. Recently, secondary mutations, such as SETBP1 and JAK3, were identified in a number of patients with JMML at diagnosis [20]. We therefore sequenced a series of genes involved in signal transduction, splicing, transcription, and epigenetic modification, in addition to the canonical RAS genes. These findings uncovered a level of genetic complexity in JMML.
Methods
Patient samples
This study included 93 patients with JMML, diagnosed from 2009 to 2018 at the Department of Hematology and Oncology, Shanghai Children’s Medical Center (SCMC), China. All patients harboring gene alterations were diagnosed according to WHO criteria for the diagnosis of JMML.
Targeted NGS
Genomic DNA was extracted from total peripheral blood (PB) in 68 cases or bone marrow (BM) aspirates in 25 cases at diagnosis, and 500 ng of DNA were fragmented on the Covaris M220 Focused-ultrasonicator and purified to yield fragments of 450–550 bp. Fragmented DNA was modified by end-repair, 3′ end adenylylation and Illumina adaptor ligation. The adapter-ligated DNA fragments were captured by a customized panel of biotinylated oligoprobes (Roche NimbleGen) and amplified. Amplified material was validated and quantified using an Agilent 2100 Bioanalyzer. Each DNA library was then sequenced on an Illumina X Ten for paired-end reads at 150 bp by the targeted gene panel designed (Table 1), which was designed to cover the RAS-signaling gene regions and the possible synergistic gene regions involved in signal transduction, splicing, transcription, and epigenetic modification. Genetic analysis in non-hematopoietic tissues (such as fingernails, hair follicles, or buccal swab samples) of patients or PB of the patients’ parents was of critical importance in addition to the screening in leukemic cells to determine whether the mutation was somatic or germline, considering the milder clinical course described in germline mutations. When potential leukocyte contamination in hair follicles or buccal swab samples existed, fingernails or the parents’ samples were done.
Bioinformatics
The raw FASTQ data were trimmed, filtering the low quality or undefined bases, and deleting the reads that has a length less than 50 bp. The clean reads were then aligned to the hg19 reference genome using the BWA-MEM (BWA-0.7.10). Duplicates were removed using Picard. BAM files were further processed according to Genome Analysis Toolkit best practices (https://www.broadinstitute.org/gatk/guide/bp_step.php?p=1), by performing Indel Realignment and Base Quality Recalibration. Variants were called using VarScan 2 and the variants were annotated using ANNOVAR. The final output positive mutations were confirmed by using the Sanger sequencing method.
Statistical analysis
Patients’ outcome data updated on December 31, 2018, were used. Event-free survival (EFS) time was calculated from diagnosis to the first failure, including death, relapse, or treatment abandonment due to disease progression. Overall survival (OS) time was considered from the time of diagnosis to death. When no events occurred, the observation was censored at the time of last follow-up. EFS and OS curves were estimated by the Kaplan-Meier method and compared with the log-rank test. Univariable and multivariable analyses were performed using the Cox model. The chi-square test was used to assess the associations and distribution characteristics between categorical variables. All P values were 2-sided, with values of < 0.05 indicating statistical significance. For statistical analysis, we used the SPSS Mac23.0.0 (IBM Corp.) software package.
Results
Clinical and biological features at diagnosis
In the 93 cases, the median age of diagnosis was 32.6 (range, 3.3–168.0) months, 50 cases were less than 24 months, 61 cases were male, and 32 were female. White blood cells’ number at diagnosis was 29.2 (2.7–127.1) × 103/μL, platelet count was 73.3 (4.0–357.0) × 103/μL, and monocytes’ number was 5.2 (1.1–48.1) × 103/μL. Percentage of myeloid and erythroid precursors on PB smear was 5.0 (0–24.0)%. Median percentage of BM blasts was 7.8 (0.4–20.0)%.
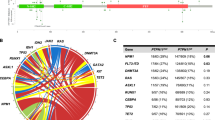
There were 30 cases with PTPN11 mutation, 20 with NF1 mutation, 16 with NRAS mutation, 10 with KRAS mutation, 4 with CBL mutation, and 13 with other mutations unable to be simply categorized to the five classical RAS-signaling mutations aforementioned (Table 2). In these cases, there were 89 somatic mutations and only 4 germline mutations, including 2 NF1, 1 PTPN11, and 1 KRAS mutation.
After diagnosis, we recommended swift HSCT for all children with NF1, somatic PTPN11, or KRAS mutations, and for the vast majority of children with somatic NRAS mutations. Children with CBL mutations were followed closely and were not offered HSCT immediately. Transplantation was considered, however, if disease progressed. Yet some patients did not receive HSCT due to fee or some other reasons. Forty-five patients received transplantation in 2–3 months after diagnosis. Thirty-two patients received mild chemotherapy before transplantation, with 15 receiving hydroxycarbamide, 8 receiving 6-mercaptopurine, 3 receiving 13-cis retinoic acid, and 6 receiving combined chemotherapy of these drugs. And 4 patients received courses of intensive chemotherapy for AML, 2 with daunorubicin-cytarabine-etoposide, and 2 with homoharringtonine-cytarabine. Five patients received decitabine before HSCT. The other 48 patients did not receive HSCT. Among these cases, only 2 patients of these cases received recorded chemotherapy, 1 with 13-cis retinoic acid, and 1 with a course of daunorubicin-cytarabine-etoposide before death.
After a median follow-up of 25 (0–125) months, 51 children were alive; By the Kaplan-Meier method, the 5 -year
OS was estimated at 50.7%. In the untransplanted patients, 9 cases died of primary disease due to lack of treatment and 1 died of transformation to AML. In the HSCT patients,7 cases died of relapse, 4 died of transplantation complications, and 2 died of AML. Relapse remained the major cause of death in JMML after HSCT. Some other studies have assigned poorer prognostic significance to several clinical and laboratory characteristics in patients with JMML, such as age > 24 months, male sex, lower platelet count, and monosomy 7 [12, 13]. However, in our cohort, no clinical characteristics, including age, sex, platelet count, higher hemoglobin F (HbF) concentration for age, abnormal G-band karyotype of chromosome, and monosomy 7 detected by fluorescence in situ hybridization, reached significance in univariate Cox analysis (Table 3).
Moreover, when the patients were divided into group with HSCT and group without HSCT, no relationship was found between these characteristics and disease outcome except age (P = 0.008) (Table 4). Patients with HSCT showed no differences in sex, platelet count, elevated HbF concentration for age, karyotype, monosomy 7 status, mutation genes, and the number of alterations in comparison with patients without HSCT (P > 0.05). But the HSCT group was enriched with patients older than 24 months in comparison with the no-HSCT group (P = 0.031).
Expanding the spectrum of RAS-pathway mutations
Although RAS-pathway lesions have traditionally been thought to represent largely mutually exclusive events [12], coexisting mutations in NRAS, KRAS, PTPN11, CBL, and NF1 were found in 10 of 93 (10.8%) patients, 3 with PTPN11 and NF1, 2 with PTPN11 and CBL, 1 with PTPN11 and NRAS, 1 with NF1 and NRAS, 1 with NF1 and KRAS, 1 with PTPN11 and NRAS and KRAS, and 1 with PTPN11 and NRAS and CBL. Acquisition of NF1 haploinsufficiency with PTPN11 was the most frequent subclone event. No significant difference in EFS was noted among the five RAS-signaling genes (P = 0.123) (Fig. 1a). No difference was found between germline and somatic mutation either (EFS 66.7% vs. 44.0%, hazard ratio (HR) = 1.53, confidence interval (CI) = 0.21–11.15, P = 0.675) (Table 3).

The EFS and genotype in JMML. a EFS based on the type of the five classical RAS-signaling genes. No significant difference was noted. b EFS based on status of PTPN11. Patients with PTPN11 mutation exhibited significantly lower EFS compared with non-PTPN11 mutations. c EFS based on the number of somatic events. Patients without or with one somatic alteration at diagnosis showed significantly better prognosis in comparison to those with more than two alterations. d EFS based on the four subgroups stratified by the mutational gene and number. PTPN11 mutation with additional alteration exhibited poorer outcome when compared with other three subtypes, only one non-PTPN11 mutation, only one PTPN11 mutation, and combined mutations without PTPN11.
However, patients with PTPN11 mutation exhibited significantly lower EFS compared with non-PTPN11 mutations (EFS 27.2% vs. 57.4%, HR = 2.25, CI = 1.27–3.99, P = 0.005) (Fig. 1b). There were no differences in age, sex, distribution of clinical parameters, and HSCT treatment in patients with PTPN11 or non-PTPN11 mutations (P > 0.05).
The secondary mutational signature correlating with disease aggressiveness
In addition to the classical JMML-associated mutations affecting RAS-pathway genes (PTPN11, NF1, KRAS, NRAS, and CBL), gene sequencing detected secondary mutations in ASXL1 (7/93, 7.5%), JAK3 (6/93, 6.4%), SETBP1 (4/93, 4.3%), EZH2 (2/93, 2.2%), JAK2 (1/93, 1.1%), SRSF2 (1/93, 1.1%), GATA2 (1/93, 1.1%), and NOTCH1 (1/93, 1.1%), many of which were known epigenetic modifiers including members of the polycomb repressive complex 2 (PRC2) or genes implicated in RAS-RAF-MEK-ERK pathway activation.
Five of the six patients with JAK3 mutations at diagnosis co-occurred with PTPN11 mutations and all six patients went on to poor outcome, uncovering activated JAK-STAT corresponding with clinical outcome.
We identified a mutation in GATA2, a transcription factor broadly involved in hematopoiesis [18] and a mutation in SRSF2, a member of the spliceosome complex. Recent work has shown that germline GATA2 mutations are responsible for several syndromes, leading to a predisposition to myeloid malignancies [7, 9].
In contrast to previous reports that emphasized the rarity of genetic mutations in epigenetic modifier genes in JMML [8, 19, 22], we identified components of PRC2, including EZH2 and ASXL1, were mutated at diagnosis (9/93, 9.68%). Mutations in epigenetic modifiers are frequent in JMML.
Somatic alterations at diagnosis predicting outcome
Using the number of somatic events at diagnosis, we evaluated differences in prognosis. In our 93-patient cohort, patients without or only with one somatic alteration at diagnosis showed significantly better prognosis in comparison with those with more than two alterations (EFS 56.4% vs. 25.0%, HR = 2.13, CI = 1.21–3.76, P = 0.009) (Fig. 1c).
HSCT could improve the EFS and OS significantly (EFS 54.1% vs. 33.6%, HR = 1.95, CI = 1.09–3.47, P = 0.024 and OS 66.8% vs. 33.6%, HR = 3.09, CI = 1.67–5.85, P < 0.0001) compared with patients without HSCT (Fig. 2). Although most of patients received chemotherapy before HSCT, we thought it was not beneficial to the final outcome because intensity of chemotherapy (non-, mild, or AML-like) prior to HSCT did not induce remission and showed no impact on HSCT outcome (P = 0.767), being not in favor of intense chemotherapy before transplantation, to avoid the adverse reactions associated with intense chemotherapy.

HSCT can improve the EFS and OS in JMML significantly
The OS of no-HSCT patients in our study was better than some other reports, maybe because this group was enriched with more KRAS (11/43, 25.6%) and CBL (4/43, 9.3%) mutations, 1 germline mutation in PTPN11, and 1 germline mutation in NF1.
Furthermore, when a Cox multivariate regression model was applied, PTPN11 mutation remained independently prognostic of poor outcome (EFS HR = 2.57, CI = 1.41–4.69, P = 0.002) after adjusting for the number of alterations and HSCT treatment. Two or more somatic events also remained independently prognostic of poor prognosis (EFS HR = 2.05, CI = 1.15–3.67, P = 0.015) after adjusting for PTPN11 status and HSCT (Table 5). Not only PTPN11 mutation but also the number of somatic alterations at diagnosis retained statistical significance for EFS.
On account of the relevance between PTPN11 status and the number of mutations (P = 0.029) and some patients harboring both characteristics, 93 cases were then subdivided into four groups stratified by their mutational gene and number, only one non-PTPN11 mutation (41/93, 44.0%), only one PTPN11 mutation (20/93, 21.5%), combined mutations but without PTPN11 abnormity (14/93, 15.1%), and PTPN11 mutation with additional somatic alteration (18/93, 19.4%). The PTPN11-mutated subgroup of patients showed the shortest survival which worsened when a secondary mutation was present (EFS 5.6%, HR = 3.73, CI = 1.83–7.62, P < 0.0001). The subgroup of only one PTPN11 mutation (EFS 47.8%, HR = 1.45, CI = 0.64–3.27, P = 0.369) and the subgroup holding combined mutations, but excluding PTPN11 abnormity (EFS 50.0%, HR = 1.27, CI = 0.51–3.14, P = 0.610), exhibited a little more hazard compared with the subtype of only one non-PTPN11 mutation (EFS 60.9%), albeit no statistically significant difference (Fig. 1d).
Discussion
JMML is characterized by the presence of mutations activating the RAS-signaling pathway in about 90% of cases [1, 6], in which the mutation of PTPN11 is a marker of poor prognosis. Recent studies have shown that oncogenic RAS signaling can mediate genomic DNA methylation [6, 11, 21]. RAS-activating mutations in different genes might have distinct effects on epigenome remodeling. Analyzing 167 cases, the EWOG-MDS demonstrated that the low-methylation cluster comprised patients with CBL and NRAS mutations known to have a favorable prognosis. KRAS mutation was associated with the intermediate cluster. The high methylation group was dominated by cases with PTPN11 mutation, resulting in poor outcome, suggesting RAS-pathway mutation patterns define epigenetic subclasses in JMML [10]. In addition, hinting at possible functional links between Ras activation and methylation classes, some research also reported that DNA hypermethylation was more pronounced when additional mutations in Ras-pathway genes or epigenetic modifier genes were present [10, 17].
Furthermore, we have shown that secondary mutations providing additional activation of RAS-signaling and other signaling pathways were frequent in poor-outcome JMML. It seemed that these secondary mutations, as second hits targeting the RAS pathway, contribute to further augment the extent of epigenetic remodeling. Pre-existing epigenetic alterations might provide a fertile ground for malignant transformation following single or few genetic hits. This sequence of events has been shown in lung cancer where hypermethylation of PRC2 target genes sensitizes epithelial cells to single-step transformation by mutant KRAS [24]. No matter whether the RAS-signaling genes or the so-called secondary mutations are primary to drive disease, it implies that each mutation in a series of mutations has biological activity, more mutations, more hits, thus maybe more aggressive.
In our cohort, DNA was extracted from PB or BM. The mutation frequencies in PB or BM were still comparable because leukemia cells could distribute in both PB and BM with almost the same proportion in JMML other than the predominant rate in BM in acute leukemia. Thus, the outcome determined by the mutation patterns was not biased regardless of sample sources.
In conclusion, both PTPN11 mutation and the number of somatic alterations detected at diagnosis are likely to be the major determinant of outcome in JMML. Notably, in addition to identifying patients with aggressive disease, our data provides patient selection criteria for therapeutic options in this heterogeneous childhood leukemia. Yet, the effects of RAS-signaling genes on epigenome remodeling and the interactions of these secondary events in RAS-signaling and other signaling pathways remain to be explored.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BM:
-
Bone marrow
- CI:
-
Confidence interval
- EFS:
-
Effect-free survival
- HbF:
-
Hemoglobin F
- HR:
-
Hazard ratio
- HSCT:
-
Hematopoietic stem cell transplantation
- JMML:
-
Juvenile myelomonocytic leukemia
- OS:
-
Overall survival
- PB:
-
Peripheral blood
- PRC2:
-
Polycomb repressive complex 2
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le BM et al (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405
Bresolin S, Zecca M, Flotho C, Trentin L, Zangrando A, Sainati L, Stary J, de Moerloose B, Hasle H, Niemeyer CM, te Kronnie G, Locatelli F, Basso G (2010) Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28:1919–1927
Bretz CL, Langohr IM, Lee S, Kim J (2015) Epigenetic instability at imprinting control regions in a Kras(G12D)-induced T-cell neoplasm. Epigenetics. 10:1111–1120
Chang TY, Dvorak CC, Loh ML (2014) Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 124:2487–2497
Flotho C, Kratz CP, Bergsträsser E, Hasle H, Stary J, Trebo M et al (2008) Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood. 111:966–967 author reply 967–8
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa S (2014) Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 28:241–247
Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, Frucht DM, Vinh DC, Auth RD, Freeman AF, Olivier KN, Uzel G, Zerbe CS, Spalding C, Pittaluga S, Raffeld M, Kuhns DB, Ding L, Paulson ML, Marciano BE, Gea-Banacloche JC, Orange JS, Cuellar-Rodriguez J, Hickstein DD, Holland SM (2011) Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 118:2653–2655
Kar SA, Jankowska A, Makishima H, Visconte V, Jerez A, Sugimoto Y, Muramatsu H, Traina F, Afable M, Guinta K, Tiu RV, Przychodzen B, Sakaguchi H, Kojima S, Sekeres MA, List AF, McDevitt MA, Maciejewski JP (2013) Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica. 98:107–113
Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar- Rodriguez J et al (2012) Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 119:1283–1291
Lipka DB, Witte T, Toth R, Yang J, Wiesenfarth M, Nöllke P, Fischer A, Brocks D, Gu Z, Park J, Strahm B, Wlodarski M, Yoshimi A, Claus R, Lübbert M, Busch H, Boerries M, Hartmann M, Schönung M, Kilik U, Langstein J, Wierzbinska JA, Pabst C, Garg S, Catalá A, de Moerloose B, Dworzak M, Hasle H, Locatelli F, Masetti R, Schmugge M, Smith O, Stary J, Ussowicz M, van den Heuvel-Eibrink MM, Assenov Y, Schlesner M, Niemeyer C, Flotho C, Plass C (2017) RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat Commun 8:2126
Liu W, Yu WM, Zhang J, Chan RJ, Loh ML, Zhang Z, Bunting KD, Qu CK (2017) Inhibition of the Gab2/PI3K /mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia. 31:1415–1422
Locatelli F, Niemeyer CM (2015) How I treat juvenile myelomonocytic leukemia. Blood. 125:1083–1090
Locatelli F, Crotta A, Ruggeri A, Eapen M, Wagner JE, Macmilan ML et al (2013) Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood. 122:2135–2141
Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH et al (2004) Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 103:2325–2331
Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, Emanuel PD, Hasle H, Issa JP, van den Heuvel-Eibrink MM, Locatelli F, Stary J, Trebo M, Wlodarski M, Zecca M, Shannon KM, Niemeyer CM (2009) Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 114:1859–1863
Molognoni F, de Melo FH, da Silva CT, Jasiulionis MG (2013) Ras and Rac1, frequently mutated in melanomas, are activated by superoxide anion, modulate Dnmt1 level and are causally related to melanocyte malignant transformation. PLoS One 8:e81937
Murakami N, Okuno Y, Yoshida K, Shiraishi Y, Nagae G, Suzuki K, Narita A, Sakaguchi H, Kawashima N, Wang X, Xu Y, Chiba K, Tanaka H, Hama A, Sanada M, Ito M, Hirayama M, Watanabe A, Ueno T, Kojima S, Aburatani H, Mano H, Miyano S, Ogawa S, Takahashi Y, Muramatsu H (2018) Integrated molecular profiling of juvenilemyelomonocytic leukemia. Blood. 131:1576–1586
Pasquet M, Bellanné-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O et al (2013) High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 121:822–829
Pérez B, Kosmider O, Cassinat B, Renneville A, Lachenaud J, Kaltenbach S, Bertrand Y, Baruchel A, Chomienne C, Fontenay M, Preudhomme C, Cavé H (2010) Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol 151:460–468
Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shirashi Y, Takahashi M et al (2013) Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45:937–941
Serra RW, Fang M, Park SM, Hutchison L, Green MR (2014) A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife. 3:e02313
Sugimoto Y, Muramatsu H, Makishima H, Prince C, Jankowska AM, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H, Takahashi Y, Kato K, Manabe A, Kojima S, Maciejewski JP (2010) Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br J Haematol 150:83–87
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD, Gelb BD (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148–150
Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O'Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, Baylin SB (2017) Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell 32:360–376
Acknowledgments
We would like to express our sincere gratitude to Dr. Jiaoyang Cai who provided statistical advice for the data from the inception of the study and undertook statistical analysis.
Funding
The study was fully supported by a grant from the Science and Technology Commission of Shanghai Municipality, China (Project No. 14411950600).
Author information
Authors and Affiliations
Contributions
Jing Chen conceptualized and designed the study, interpreted the data, and critically revised the manuscript. Yan Miao interpreted the data and drafted the manuscript. Benshang Li contributed to study design, interpreted the data, and reviewed the manuscript. Lixia Ding and Hua Zhu analyzed the data and reviewed the manuscript. Changying Luo, Jianmin Wang, and Chengjuan Luo were in charge of follow-up.
Corresponding author
Ethics declarations
Ethical approval
The study was approved by the Institutional Ethical Review Board at SCMC, Shanghai Jiao Tong University School of Medicine. All the parents signed informed consent forms. Informed written consent was obtained from the parents of each participant. The procedure followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional or regional) and with the Helsinki Declaration of 1964, as revised in 2000.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Peter de Winter
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Authors’ summary: We found that both PTPN11 mutation and the number of somatic alterations detected at diagnosis are likely to be the major determinant of outcome in JMML. Moreover, the subgroup of patients with PTPN11 mutation showed the shortest survival which even worsened when a secondary mutation was present. Notably, in addition to identifying patients with aggressive disease, our data expanded mutational spectrum on the pathogenesis of JMML, providing functional guidance, prognostic markers, and patient selection criteria for therapeutic options in this heterogeneous childhood leukemia.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Miao, Y., Li, B., Ding, L. et al. PTPN11 mutation with additional somatic alteration indicates unfavorable outcome in juvenile myelomonocytic leukemia: a retrospective clinical study from a single center. Eur J Pediatr 179, 463–472 (2020). https://doi.org/10.1007/s00431-019-03468-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-019-03468-8