
Abstract
The purpose of this study was to analyze the association between next-generation sequencing (NGS) genotypic profiles and conventional clinicopathologic characteristics in patients with acute myeloid leukemia (AML) with NPM1 mutation (NPM1mut). We selected 238 NPM1mut patients with available NGS information on 112 genes related to blood diseases using the χ2 and Mann-Whitney U tests and a multivariable logistic model to analyze the correlation between genomic alterations and clinicopathologic parameters. Compared with the NPM1mut/FLT3-ITD(−) group, the NPM1mut/FLT3-ITD(+) group presented borderline frequent M5 morphology [78/143 (54.5%) vs. 64/95 (67.4%); P = 0.048], higher CD34- and CD7-positive rates (CD34: 20.6% vs. 47.9%, P < 0.001; CD7: 29.9% vs. 61.5%, P < 0.001) and a lack of favorable−/adverse-risk karyotypes (6.4% vs. 0%; P = 0.031). In the entire NPM1mut cohort, 240 NPM1 mutants were identified, of which 10 (10/240, 4.2%) were missense types. When confining the analysis to the 205 cases with NPM1mut insertions/deletions-type and normal karyotype, multivariable logistic analysis showed that FLT3-ITD was positively correlated with CD34 and CD7 expressions (OR = 5.29 [95% CI 2.64–10.60], P < 0.001; OR = 3.47 [95% CI 1.79–6.73], P < 0.001, respectively). Ras-pathway mutations were positively correlated with HLA-DR expression (OR = 4.05 [95% CI 1.70–9.63], P = 0.002), and KRAS mutations were negatively correlated with MPO expression (OR = 0.18 [95% CI 0.05–0.62], P = 0.007). DNMT3A-R882 was positively correlated with CD7 and HLA-DR expressions (OR = 3.59 [95% CI 1.80–7.16], P < 0.001; OR = 13.41 [95% CI 4.56–39.45], P < 0.001, respectively). DNMT3A mutation was negatively correlated with MPO expression (OR = 0.35 [95% CI 1.48–8.38], P = 0.004). TET2/IDH1 mutations were negatively correlated with CD34 and CD7 expressions (OR = 0.26 [95% CI 0.11–0.62], P = 0.002; OR = 0.30 [95% CI 0.14–0.62], P = 0.001, respectively) and positively correlated with MPO expression (OR = 3.52 [95% CI 1.48–8.38], P = 0.004). In conclusion, NPM1mut coexisting mutations in signaling pathways (FLT3-ITD and Ras-signaling pathways) and methylation modifiers (DNMT3A and TET2/IDH1) are linked with the expressions of CD34, CD7, HLA-DR and MPO, thereby providing a mechanistic explanation for the immunophenotypic heterogeneity of this AML entity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
The human NPM1 gene, located on chromosome 5q35.1 and containing 12 exons, encodes a nucleolar phosphoprotein that possesses multiple functions, including chromatin remodeling, ribosome biogenesis, genomic stability, and regulation of tumor suppressors and transcription factors [1,2,3]. Given its important role in biological significance, the functional category of NPM1 belongs to a separate category according to The Cancer Genome Atlas (TCGA) data [4].
NPM1 gene abnormalities are involved in fusion [5], deletion [2] and mutation, among which the mutation is the most largely studied. The incidence of NPM1 mutation (NPM1mut) accounts for approximately one-third of the cases of de novo acute myeloid leukemia (AML) and up to ~ 50% of normal karyotype (NK) AML [6, 7]. The initial presentations of NPM1mut AML are characterized by multiple clinicopathologic aspects. For instance, its French-American-British (FAB) morphologies commonly have monocytic differentiation (M4 or M5) [8, 9] and are likely to have cup-like nuclei [10]. Immunophenotypically, most NPM1mut cases show CD34 negativity [11]. According to the analysis of myeloid blast population, nearly half of NPM1mut patients show an acute promyelocytic leukemia (APL)-like antigen expression feature represented by CD34(−)/HLA-DR(−)/MPO(str+) [12]. NPM1mut AML mainly arises in an NK situation and is mutually exclusive with recurrent cytogenetic abnormalities [7, 13]. NPM1mut AML has unique gene expression profiles, especially the overexpression of HOX family members [14].
The immunophenotype is not only used in the differential diagnosis of AML but also has prognostic relevance. CD34(+) [11, 15], leukemic stem cell (LSC) phenotype CD34(+)/CD38(−)/CD123(+) [16], APL-like phenotype CD34(−)/HLA-DR(−)/MPO(str+) [12] and clustered type-II phenotype CD34(+)/HLA-DR(+)/CD7(+) [17] have been reported to convey prognostic effects on NPM1mut AML. However, data on genetic information were less integrated into the analysis in those relatively earlier studies. Over recent years, studies regarding prognostic heterogeneity in NPM1mut AML have mainly focused on cytogenetic and gene mutations. Comutations in DNMT3A [18], TET2 [19, 20] or IDH1/2 [21, 22] have been shown to be adverse predictors, and NRAS [23], FLT3-TKD [24] or moCEBPA [25] have been shown to be favorable predictors of clinical outcome in NPM1mut/FLT3-ITD(−)/low AML.
Because NPM1mut AML is mainly seen in intermediate-risk cytogenetics, especially in the NK background, we hypothesize that the diversity of leukemic phenotypes depends to a certain extent on the heterogeneity of coexisting gene mutations in this subtype of AML. Whole genome or exosome sequencing revealed an average of 13 mutations in AML [7], indicating the interplay between mutations as an important pathomechanism of leukemic development and overt onset.
In addition, NPM1mut in association with prognostication is generally described as insertions and/or deletions (indel), which are predominantly characterized by a 4 base-pair insertion in the C-terminus within exon 12 and a resultant frameshift consequence. However, data involving other types of NPM1mut have scarcely been reported. Moreover, types of NPM1mut were not specifically designated in AML classification and treatment guidelines [26, 27]. The development of large-scale parallel sequencing technology, with its enlargement of higher throughput and wider coverage, is bound to detect more diversified mutational loci and types within the NPM1 gene as well as more concurrent mutations.
In this study, we selected newly diagnosed patients with de novo NPM1mut AML and evaluated the correlations of clinicopathologic features with next-generation sequencing (NGS)-based genetic alterations in 112 genes related to blood diseases, aiming profoundly to understand the clinicopathological heterogeneity of this AML subtype.
Methods
Patient selection and clinicopathologic workup
We performed a retrospective review of newly diagnosed de novo AML patients in our institute and Shengjing Hospital of China Medical University from October 2014 to September 2019. AML diagnosis fulfilled World Health Organization (WHO) criteria [28], according to which the clinicopathologic workup included cytomorphology, immunophenotyping, chromosome karyotyping and fluorescence in situ hybridization (FISH), molecular biology and gene mutation analysis (see below). The cytomorphological subtype was based on the FAB classification. Immunophenotyping was performed on freshly EDTA-anticoagulated or heparinized bone marrow (BM) or peripheral blood (PB) samples obtained at the time of initial diagnosis. Four-color analysis was conducted on a FACSCalibur Colorflow Cytometer (Becton-Dickinson, USA) using the following sets of FITC (fluorescein-isothiocyanate), PE (phycoerythrin), PerCP (peridinin-chlorophyII-protein) and APC (allophycocyanin)-labeled mouse anti-human fluorescent monoclonal antibodies: 1) CD34/CD10/CD45/CD19; 2) CD7/CD117/CD45/CD33; 3) CD9/CD2/CD45/CD56; 4) CD15/CD38/CD45/HLA-DR; 5) CD16/CD13/CD45/CD11b; 6) CD4/CD64/CD45/CD14; 7) cMPO/cCD79a/CD45/cCD3; and 8) TdT/CD123/CD45/HLA-DR. G-band karyotyping analysis was conducted using BM aspirate samples. When obtaining BM samples was difficult, PB was used instead. A total of 20 metaphase cells were analyzed for each patient, and chromosomal abnormalities were described according to the International System for Human Cytogenetic Nomenclature [29]. Additionally, KMT2A (MLL) rearrangements (11q23 abnormality) were verified by FISH using Dual-Color, Break-Apart Rearrangement Probe (Vysis, USA), and TP53 deletions (17p-) by locus-specific probe (Vysis, USA). The study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional review board (IRB) of all the participating institutions. All patients provided written informed consent for using their records.
Detection of mutations by NGS and conventional methods
Genomic DNA extraction (Qiagen, Germany), quality control and quantification measurement (Nanodrop Technologies, USA), ultrasonic fragmentation (Covaris, USA), library construction and target enrichment (SureSelect, Agilent Technologies, USA; Illumina, USA) were conducted according to the manufacturer protocols. High-throughput targeted measurement of gene mutations was performed on an Ion torrent PGM™ (Life Technologies) or MiSeq/HiSeq (Illumina) sequencer platform with an average sequencing depth of 800×. The custom-designed panel consisted of 112 potentially mutated genes which are involved in hematological disorders and are related to the following functional categories: signaling pathways, epigenetic regulators, transcription factors, spliceosomes, cohesin complex, tumor suppressors, NPM1 and others. Single nucleotide variants (SNVs) and short fragment indels in protein coding sequences (CDSs) were analyzed by using Ion Reporter™ and Variant Reporter pipelines and annotated referencing the dbSNP, 1000 Genomes, Polyphen-2 and COSMIC databases. NPM1 (exon 12), FLT3-ITD, and potential complex indels in CEBPA (TAD and bZIP domains) were additionally examined by PCR followed by direct sequencing as previously reported [30,31,32].
Statistical analysis
Descriptive statistics are presented as median (range) for non-normally distributed variables and frequency (incidence) for categorical variables. The χ2 test and Mann-Whitney U test were used to calculate the significance of associations between coexisting mutations and clinicopathologic features. To extract independent factors, those with a P-value < 0.15 were included as covariates in the multivariate logistic model using the forward stepwise selection procedure. The results are expressed as odds ratios (ORs) together with 95% confidence intervals (CIs). All calculations were performed applying IBM SPSS v26.0 for Windows. In all analyses, P-values < 0.05 were considered significant. GraphPad Prism 8.4.2, Circos-0.69-9 and R version 4.0.4 were also used for figure plotting.
Results
FAB subtypes of NPM1 mut AML
In this study, we selected 238 patients with NPM1mut AML for our purposive analysis. The study cohort consisted of 105 males and 133 females, with a median age of 49 (range 15–81) years. The most common FAB subtype of NPM1mut AML was AML-M5 (59.7%), followed by M2 (17.6%) and M4 (15.5%), similar to other findings [8, 9]. According to FLT3-ITD, M2 was more common in the NPM1mut/FLT3-ITD(−) group [34/143 (23.8%) vs. 8/95 (8.4%); P = 0.002], while M5 was slightly more common in the NPM1mutFLT3-ITD(+) group [64/95 (67.4%) vs. 78/143 (54.5%); P = 0.048] as shown in Fig. 1a.

Clinicopathologic characteristics in 238 patients with NPM1mut AML (by FLT3-ITD). a Composition ratio of morphologic FAB subtypes. b Positive expression rate of immunophenotypic antigens; *, P < 0.05. c Conventional G-banding karyotype (by FLT3-ITD and cytogenetic risk)
The expression incidence of CD34 and CD7 in the NPM1 mut/FLT3-ITD(+) group was higher than that in the NPM1 mut/FLT3-ITD(−) group
As per the literature [33], leukemic blasts at the initial diagnosis could be divided into leukemic myeloid blasts and leukemic immature monocyte populations, with the latter detected in approximately 50% of cases and mostly in the M4 or M5 morphologic subtypes. Leukemic myeloid blasts recurred when AML relapsed, while leukemic immature monocyte populations often disappeared, indicating that leukemic myeloid blasts may enrich more LSCs, which serve as a source of disease relapse. Consequently, in the description of baseline FCM characteristics, we only analyzed the antigen expression aspects of leukemic myeloid cells. In the entire NPM1mut cohort, the antigens positively expressed at an incidence of 80% or more were CD117 (211/235, 89.8%), CD13 (207/233, 88.8%), CD33 (233/233, 100%), CD123 (230/232, 99.1%) and CD38 (209/233, 89.7%). The positive incidences of CD34 and TDT were 31.5% (74/235) and 6.9% (16/231), respectively. The positive incidence of HLA-DR was 67.4% (157/233) and that of MPO was 74.9% (170/227). CD7 was positively expressed in 43.1% (94/218), CD19 in 3.5% (8/228) and CD79a in 0.4% (1/227) of cases as shown in Fig. 1b. According to FLT3-ITD, the positive expression incidence of CD34 and CD7 in the NPM1mut/FLT3-ITD(+) group was significantly higher than that in the NPM1mut/FLT3-ITD(−) group (CD34: 47.9% vs. 20.6%, P < 0.001; CD7: 61.5% vs. 29.9%, P < 0.001), while the incidence of other antigens was not different between the two genotypic groups.
Chromosomal karyotypes in NPM1 mut AML
Of all 238 patients with NPM1mut, 234 patients had evaluable metaphases, of whom 208 (88.9%) were NKs and 26 (11.1%) were abnormal karyotypes. Among 143 cases with NPM1mut/FLT3-ITD(−), 140 had evaluable metaphases, with 131 cases in the intermediate-risk layer (including 121 cases NK; 10 cases intermediate-risk abnormal karyotype, Fig. 1c), 5 cases in the favorable-risk layer [including 4 cases t (8;21)(q22;q22); 1 case inv (16) (p13q22)] and 4 cases in the adverse-risk layer [including 1 case complex karyotype, monosomy karyotype, t (6;9)(p23;q34), t (8;9;22)(q24;q34;q11.2) for each]. Among 95 cases with NPM1mut/FLT3-ITD(+), 94 had evaluable metaphases, with all of them in the intermediate-risk layer (including 87 cases NK; 7 cases intermediate-risk abnormal karyotype, Fig. 1c) and none in the favorable- or adverse-risk layer. There was no difference in the distribution of intermediate-risk karyotypes (NK plus abnormal) between the NPM1mut/FLT3-ITD(−) and NPM1mut/FLT3-ITD(+) groups (P = 0.144 and 0.930, respectively), while the favorable- plus adverse-risk karyotypes were only enriched in the NPM1mut/FLT3-ITD(−) group and not in the NPM1mut/FLT3-ITD(+) group (6.4% vs. 0%; P = 0.031). No correlation was found between other NPM1mut coexisting gene mutations and abnormal karyotypes (all P > 0.05, data not shown). None of the KMT2A (MLL) translocations or TP53 deletions were identified in 206 NPM1mut patients with available FISH data.
NPM1 mut loci, types and comutation patterns
In the entire NPM1mut cohort, 240 NPM1 mutants were identified, among which 230 (230/240, 95.8%) were out-of-frame indels and 10 (10/240, 4.2%) were missense events (i.e., 3 with c.578A > G → p. K193R, 2 with c.676G > A → p. E226K and 5 with c.733G > C → p.E245Q). All these missense codons did not disrupt any of the tryptophan residues W288 and W290, which are indispensable for the nucleolar localization signal (NoLS). Furthermore, all but one of these missense mutations (9/10, 90.0%) was accompanied by an AML subtype-defining recurrent genetic abnormality, with 7 cases at favorable risk and 2 at adverse risk (Supplementary Table S1). When the analysis was restricted to the NPM1mut indel-types, there was no difference in the incidence of favorable- plus adverse-risk karyotypes between the NPM1mut/FLT3-ITD(−) and NPM1mut/FLT3-ITD(+) groups (3.0% vs. 0%; P = 0.234).
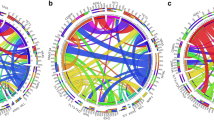
At least one comutation was detected in all 238 NPM1mut cases. Including NPM1mut, the median number of mutated genes per individual was 4.5 (2-14), with 4.0 (2-14) in the NPM1mut/FLT3-ITD(−) group, which was not significantly different from the 5.0 (2-10) in the NPM1mut/FLT3-ITD(+) group (P = 0.378, Fig. 2a). According to gene function categories, the order of incidence was as follows: signaling pathways (72.7%), epigenetic regulators (71.4%), tumor suppressors (31.9%) and myeloid transcription factors (8.8%), spliceosomes (7.1%) and cohesion complex (3.4%, Fig. 2b). DNMT3A (104, 43.7%), FLT3-ITD (95, 39.9%) and FAT1 (57, 23.9%) represented the top three most frequently mutated genes (more details on relatively common genes mutated in > 5% of the entire NPM1mut cohort are displayed in Fig. 3). It was worth mentioning that spliceosomes members SF3A1, ZRSR2, SF3B1, SRSF2, U2AF1 and U2AF2, were uncommonly mutated in 4 (1.7%), 4 (1.7%), 3 (1.3%), 3 (1.3%), 3 (1.3%) and 1 (0.4%) of the 238 cases of entire NPM1mut cohort, respectively. As for cohesion complex members RAD21, STAG2, SMC3 and SMC1A, they were also rarely mutated in 3 (1.3%), 2 (0.8%), 2 (0.8%) and 1 (0.4%) of the entire NPM1mut cohort, respectively. The analysis of gene-gene relationship across NPM1mut coexisting mutations showed a significant accompaniment of FLT3-ITD with DNMT3A (P = 0.005), while FLT3-ITD was mutually exclusive to FLT3-nonITD (P < 0.001), NRAS (P < 0.001), PTPN11 (P = 0.017) and IDH1 (P = 0.005, Fig. 4).

a Number of mutated genes per patient and b the incidence of gene functional categories in NPM1mut AML (by FLT3-ITD)

Mutational landscape for the relatively common genes mutated in > 5% of the entire NPM1mut cohort, as well as for the members of spliceosomes and cohesion complex. Each row represents a different gene, and each column represents an individual patient; A colored cell indicates the presence of mutation, and a blank cell indicates wild type; Mutational types are grouped into five classifications as labeled by varying colors; The 27 individual genes are grouped into seven functional categories as listed in Fig. 2b, and the mutational incidence of each gene is listed on the left panel; Clinical data on cytogenetic risk are accordingly displayed on the bottom panel

A circos plot illustrating pairwise relationships across the relatively common mutated genes in NPM1mut AML. The red ribbon indicates a significant coexistence, and the black ribbon indicates mutual exclusivity; The white ribbon indicates a non-significant association; The width of the ribbon corresponds to the number of cases who have simultaneous presence of a first and a second gene in parallel
Association between NPM1 mut coexisting mutations and immunophenotypic markers
Our results showed that the expressions of CD34 and CD7 were significantly associated with FLT3-ITD. Because NPM1mut AML mostly occurs in the NK context, we hypothesized that diversities in antigen expression in leukemia cells to a certain extent are determined by the heterogeneity of coexisting mutations. To rule out the influence of abnormal karyotypes on the immunophenotype, as well as in view of the deductively insufficient pathogenicity of NPM1mut missense mutations, only patients with NK and NPM1mut indel-types were included for subsequent analysis. A total of 205 NPM1mut patients fulfilling the above conditions were available for distributional crosstabulation between immunophenotypic markers and coexisting mutations. The significant results from the χ2 test and multivariate analysis are shown in Table 1.
Logistic analysis showed that in the entire NPM1mut cohort, FLT3-ITD was positively correlated with the expressions of CD34 and CD7 (OR = 5.29 [95% CI 2.64–10.60], P < 0.001; OR = 3.47 [95% CI 1.79–6.73], P < 0.001). Ras-pathway mutations were positively correlated with HLA-DR expression (OR = 4.05 [95% CI 1.70–9.63], P = 0.002) and negatively correlated with MPO expression (OR = 0.18 [95% CI 0.05–0.62], P = 0.007) in the entire NPM1mut cohort. Stratified analysis according to FLT3-ITD status indicated that this effect was only seen in the NPM1mut/FLT3-ITD(−) group (OR and P values are detailed in Table 1) but not in the NPM1mut/FLT3-ITD(+) group.
DNMT3A-R882 was positively correlated with CD7 and HLA-DR expressions (OR = 3.59 [95% CI 1.80–7.16], P < 0.001; OR = 13.41 [95% CI 4.56–39.45], P < 0.001), and DNMT3A mutation was negatively correlated with MPO expression (OR = 0.35 [95% CI 1.48–8.38], P = 0.004). Stratified analysis indicated that the independent effect of DNMT3A mutations (especially DNMT3A-R882) correlated with CD7 and HLA-DR expressions was significant in both the NPM1mut/FLT3-ITD(+) group and the NPM1mut/FLT3-ITD(−) group (OR and P values are detailed in Table 1). TET2/IDH1 mutations were negatively correlated with CD34 and CD7 expressions (OR = 0.26 [95% CI 0.11–0.62], P = 0.002; OR = 0.30 [95% CI 0.14–0.62], P = 0.001) and positively correlated with MPO expression (OR = 3.52 [95% CI 1.48–8.38], P = 0.004). Stratified analysis indicated the above effects to be prominent only in the NPM1mut/FLT3-ITD(+) group (OR and P values are detailed in Table 1) and not in the NPM1mut/FLT3-ITD(−) group. There were no significant correlations between NPM1mut coexisting mutations and the expression of other antigens.
We finally analyzed the association of NPM1mut coexisting mutations with the APL-like phenotype CD34(−)/HLA-DR(−)/MPO(str+), which has been reported to predict the presence of TET2/IDH1 mutations [12]. In the entire NPM1mut cohort, mutations of the Ras-pathway, DNMT3A-R882 and TET2/IDH1 were each significantly linked with the APL-like phenotype. When stratified by FLT3-ITD, in the NPM1mut/FLT3-ITD(−) group, only Ras-pathway mutations presented an association with the APL-like phenotype (OR = 0.32 [95% CI 0.11–0.96], P = 0.041). Comparatively, a negative correlation of DNMT3A-R882 (OR = 0.04 [95% CI 0.01–0.36], P = 0.004) and a positive correlation of TET2/IDH1 mutation (OR = 6.73 [95% CI 1.83–24.78], P = 0.004) with this phenotype were both seen in the NPM1mut/FLT3-ITD(+) group but not in the NPM1mut/FLT3-ITD(−) group (Table 1).
Discussion
Previous studies regarding the prognostication of NPM1mut often depicted its mutational type as indels, and there was little information about other types of NPM1mut. In our cohort of 238 NPM1mut patients, 240 NPM1 mutant events were identified, among which the vast majority (232, 99.1%) were indel-types. All of these indels derange the tryptophan residues W288 and W290, which are indispensably responsible for NoLS [2]. Ten NPM1mut missense mutations were clustered in the NPM1mut/FLT3-ITD(−) group, and none of them disrupted the two loci of NoLS, nor were they involved in NPM1 posttranslational modification sites [3]. Moreover, all except one (9, 90.0%) missense mutation were accompanied by an AML subtype-defining favorable- or adverse-risk genetic abnormality, indicating that NPM1mut missense mutation may be insufficient to drive leukemogenesis and necessitate other well-characterized pathomechanisms. Consequently, the theme of prognosis concerning NPM1mut AML should be in the context of its indel-types with emphasis or by default, instead of including missense-types of relative rarity and possibly inadequate pathogenicity.
In the present study, NK reached ~ 90% in the entire NPM1mut cohort with analyzable metaphases and accounted for 84.6% in the NPM1mut/FLT3-ITD(−) group, similar to the finding of 82.4% in a large sample survey [13]. Moreover, recurrent cytogenetic translocations were uncommon, and FISH did not detect any KMT2A (MLL) translocation or TP53 deletion, implying that the leukemogenesis of frameshift NPM1mut does not rely on chromosomal abnormalities. Nonetheless, all NPM1mut indels arose together with coexisting mutations, especially those affecting epigenetic regulators and signaling pathways, which points to the necessity of interactivity of NPM1mut with other genetic lesions to promote leukemic overt occurrence. The favorable- and adverse-risk abnormal karyotypes were only aggregated in the NPM1mut/FLT3-ITD(−) group, implying possibly pathogenic independence between FLT3-ITD and those karyotypes in NPM1mut AML.
Compared with the NPM1mut/FLT3-ITD(−) group, the NPM1mut/FLT3-ITD(+) group had higher incidences of CD34 and CD7 expression, similar to other reports [34]. FCM immunophenotyping is not only used in the differential diagnosis of AML but also has prognostic relevance. In terms of an individual immunomarker, CD34(+) in NPM1mut AML was associated with a poor prognosis [11, 15]. CD123 was only expressed in leukemia and other neoplastic cells but hardly in normal hematopoietic cells [35]. A percentage of CD123(+) cells in NPM1mut patients divided by a cutoff of 52% was also reported to predict prognosis [36]. Going forward, the combination of multiple aspects of antigen expression could more potently predict survival. In particular, CD34(+)/CD38(−)/CD123(+), which represents an LSC phenotype, showed inferior prognosis [16]. In addition, most LSC phenotypes also present cross-lineage, antigen overexpression or asynchronous expression phenomena [16]. In our study, the positive incidences of stem cell antigen CD34 and cross-lineage antigen CD7 expression were higher in the NPM1mut/FLT3-ITD(+) subset, which may be implied to encompass more LSCs at initial presentation. LSCs are in the relatively silent cell cycle G0 phase and highly express the drug-resistant efflux transporter P-glycoprotein (PGP) or multidrug-resistant protein (MDR1) [11, 37]. Chen CY et al. [17] clustered immunophenotyping in 94 NPM1mut patients and divided them into two categories according to CD34, CD7 and HLA-DR expressions, showing that the prognosis of type-II class characterized by CD34(+)/HLA-DR(+)/CD7(+) was significantly poorer versus the type-I class CD34(−)/CD7(−). However, their results might be affected by the biased distribution of concurrent FLT3-ITD, which has a positive correlation with CD34 and CD7 expressions. Because of the limited number of cases, it was not clear whether the differential effect of class I and II features on prognosis was independent of FLT3-ITD, although a stratified analysis had been carried out.
We investigated the relationship between NPM1mut coexisting mutations and immunophenotypic markers. In general, there was a distributional association of signaling and methylating mutations with CD34, CD7, HLA-DR and MPO expressions. The regulatory effect of Ras-pathway mutations on the expression of these antigens was only found in the NPM1mut/FLT3-ITD(−) group but not in the NPM1mut/FLT3-ITD(+) group, partly owing to the reciprocal exclusivity of FLT3-ITD with Ras-pathway mutations. DNMT3A mutation was positively correlated with the expressions of CD34, CD7 and HLA-DR in both genotypic groups, while TET2/IDH1 mutations were negatively correlated with those antigens specifically in the NPM1mut/FLT3-ITD(+) group. In contrast, DNMT3A mutation was negatively correlated with MPO expression, while TET2/IDH1 mutations were positively correlated with MPO expression. These results suggested that DNMT3A and TET2/IDH1 mutations might play different roles in regulating the expression of these immunophenotypic markers.
In the NPM1mut/FLT3-ITD(−) group, Ras-pathway mutations and DNMT3A-R882 were positively correlated with the expression of the monocyte marker HLA-DR and negatively correlated with the myeloid marker MPO, which is linked to the FAB morphology of monocytic differentiation (M4/M5) or granulocytic differentiation (M2). Comparatively, in the NPM1mut/FLT3-ITD(+) group, although TET2/IDH1 mutations were negatively correlated with HLA-DR expression, the more commonly coexisting DNMT3A-R882, which was positively correlated with HLA-DR expression, might take precedence and be accountable for a more frequent M4/M5 morphology in this genotypic group.
Mason EF et al. [12] analyzed myeloid blast populations excluding monocytic differentiation in NPM1mut patients. Nearly half of the cases (48%) had an APL-like phenotype represented by CD34(−)/HLA-DR(−)/MPO(str+), which could predict the presence of TET2 or IDH1/2 mutations, a result in line with our findings. Moreover, the authors demonstrated the APL-like phenotype beneficially impacted RFS and OS, and its combination with coexisting TET2 or IDH1/2 mutations was more explicit to refine prognostic subgroups. Our present study extended those findings. We additionally showed an independent negative association of Ras-pathway mutations with the APL-like phenotype only in the NPM1mut/FLT3-ITD(−) group. Additionally, we showed a negative association of DNMT3A-R882 with this phenotype only in the NPM1mut/FLT3-ITD(+) genotypic background. These results suggested that the interplay of NPM1mut coexisting genetic lesions might jointly determine the trend of antigen expression, partly explaining the immunophenotypic heterogeneity in NPM1mut AML.
Conclusions
In summary, NPM1mut missense mutations may be of leukemogenic insufficiency and largely rely on other well-defined pathomechanisms in the development of overt leukemia. The correlation of coexisting mutations in signaling pathways and methylation modifiers with antigen expression (represented by CD34, CD7, HLA-DR and MPO) may partly explain the immunophenotypic diversity in NPM1mut AML. Comprehensively evaluating the FCM immunophenotype and NGS landscape of genetic lesions allows us to gain insight into the clinicopathological heterogeneity of this distinct AML entity.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- BM:
-
Bone marrow
- CDS:
-
Coding sequence
- CI:
-
Confidence interval
- FAB:
-
French-American-British
- FISH:
-
Fluorescence in situ hybridization
- IRB:
-
Institutional review board
- LSCs:
-
Leukemic stem cells
- NGS:
-
Next-generation sequencing
- NK:
-
Normal karyotype
- NPM1 mut :
-
NPM1 mutation
- OR:
-
Odds ratio
- PB:
-
Peripheral blood
- SNVs:
-
Single nucleotide variants
- TCGA:
-
The Cancer Genome Atlas
- WHO:
-
World Health Organization
References
Kunchala P, Kuravi S, Jensen R, McGuirk J, Balusu R. When the good go bad: mutant NPM1 in acute myeloid leukemia. Blood Rev. 2018;32(3):167–83. https://doi.org/10.1016/j.blre.2017.11.001.10.1016/j.blre.2017.11.001.
Brodska B, Sasinkova M, Kuzelova K. Nucleophosmin in leukemia: consequences of anchor loss. Int J Biochem Cell Biol. 2019;111:52–62. https://doi.org/10.1016/j.biocel.2019.04.007.10.1016/j.biocel.2019.04.007.
Heath EM, Chan SM, Minden MD, Murphy T, Shlush LI, Schimmer AD. Biological and clinical consequences of NPM1 mutations in AML. Leukemia. 2017;31(4):798–807. https://doi.org/10.1038/leu.2017.30.10.1038/leu.2017.30.
Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35(9):934–46. https://doi.org/10.1200/JCO.2016.71.2208.10.1200/JCO.2016.71.2208.
Falini B, Nicoletti I, Bolli N, Martelli MP, Liso A, Gorello P, et al. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica. 2007;92(4):519–32. https://doi.org/10.3324/haematol.11007.10.3324/haematol.11007.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–66. https://doi.org/10.1056/NEJMoa041974.10.1056/NEJMoa041974.
Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. https://doi.org/10.1056/NEJMoa1301689.10.1056/NEJMoa1301689.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106(12):3733–9. https://doi.org/10.1182/blood-2005-06-2248.10.1182/blood-2005-06-2248.
Wilson CS, Davidson GS, Martin SB, Andries E, Potter J, Harvey R, et al. Gene expression profiling of adult acute myeloid leukemia identifies novel biologic clusters for risk classification and outcome prediction. Blood. 2006;108(2):685–96. https://doi.org/10.1182/blood-2004-12-4633.10.1182/blood-2004-12-4633.
Chen W, Konoplev S, Medeiros LJ, Koeppen H, Leventaki V, Vadhan-Raj S, et al. Cuplike nuclei (prominent nuclear invaginations) in acute myeloid leukemia are highly associated with FLT3 internal tandem duplication and NPM1 mutation. Cancer. 2009;115(23):5481–9. https://doi.org/10.1002/cncr.24610.10.1002/cncr.24610.
Zeijlemaker W, Kelder A, Wouters R, Valk PJM, Witte BI, Cloos J, et al. Absence of leukaemic CD34(+) cells in acute myeloid leukaemia is of high prognostic value: a longstanding controversy deciphered. Br J Haematol. 2015;171(2):227–38. https://doi.org/10.1111/bjh.13572.10.1111/bjh.13572.
Mason EF, Kuo FC, Hasserjian RP, Seegmiller AC, Pozdnyakova O. A distinct immunophenotype identifies a subset of NPM1-mutated AML with TET2 or IDH1/2 mutations and improved outcome. Am J Hematol. 2018;93(4):504–10. https://doi.org/10.1002/ajh.25018.10.1002/ajh.25018.
Angenendt L, Rollig C, Montesinos P, et al. Chromosomal abnormalities and prognosis in NPM1-mutated acute myeloid leukemia: a pooled analysis of individual patient data from nine international cohorts. J Clin Oncol. 2019;37(29):2632–42. https://doi.org/10.1200/JCO.19.00416.10.1200/JCO.19.00416.
Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43(5):470–5. https://doi.org/10.1038/ng.796.10.1038/ng.796.
Dang H, Chen Y, Kamel-Reid S, Brandwein J, Chang H. CD34 expression predicts an adverse outcome in patients with NPM1-positive acute myeloid leukemia. Hum Pathol. 2013;44(10):2038–46. https://doi.org/10.1016/j.humpath.2013.03.007.10.1016/j.humpath.2013.03.007.
van Rhenen A, Moshaver B, Kelder A, Feller N, Nieuwint AWM, Zweegman S, et al. Aberrant marker expression patterns on the CD34+CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21(8):1700–7. https://doi.org/10.1038/sj.leu.2404754.10.1038/sj.leu.2404754.
Chen CY, Chou WC, Tsay W, Tang JL, Yao M, Huang SY, et al. Hierarchical cluster analysis of immunophenotype classify AML patients with NPM1 gene mutation into two groups with distinct prognosis. BMC Cancer. 2013;13(1):107. https://doi.org/10.1186/1471-2407-13-107.10.1186/1471-2407-13-107.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–98. https://doi.org/10.1182/blood-2016-01-693879.10.1182/blood-2016-01-693879.
Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and leukemia group B study. J Clin Oncol. 2011;29(10):1373–81. https://doi.org/10.1200/JCO.2010.32.7742.10.1200/JCO.2010.32.7742.
Tian X, Xu Y, Yin J, Tian H, Chen S, Wu D, et al. TET2 gene mutation is unfavorable prognostic factor in cytogenetically normal acute myeloid leukemia patients with NPM1+ and FLT3-ITD - mutations. Int J Hematol. 2014;100(1):96–104. https://doi.org/10.1007/s12185-014-1595-x.10.1007/s12185-014-1595-x.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and leukemia group B study. J Clin Oncol. 2010;28(14):2348–55. https://doi.org/10.1200/JCO.2009.27.3730.10.1200/JCO.2009.27.3730.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636–43. https://doi.org/10.1200/JCO.2010.28.3762.10.1200/JCO.2010.28.3762.
Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 2006;107(10):3847–53. https://doi.org/10.1182/blood-2005-08-3522.10.1182/blood-2005-08-3522.
Boddu P, Kantarjian H, Borthakur G, Kadia T, Daver N, Pierce S, et al. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv. 2017;1(19):1546–50. https://doi.org/10.1182/bloodadvances.2017009019.10.1182/bloodadvances.2017009019.
Dufour A, Schneider F, Hoster E, et al. Monoallelic CEBPA mutations in normal karyotype acute myeloid leukemia: independent favorable prognostic factor within NPM1 mutated patients. Ann Hematol. 2012;91(7):1051–63. https://doi.org/10.1007/s00277-012-1423-4.10.1007/s00277-012-1423-4.
Tallman MS, Wang ES, Altman JK, Appelbaum FR, Bhatt VR, Bixby D, et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2019;17(6):721–49. https://doi.org/10.6004/jnccn.2019.0028.10.6004/jnccn.2019.0028.
Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47. https://doi.org/10.1182/blood-2016-08-733196.10.1182/blood-2016-08-733196.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405. https://doi.org/10.1182/blood-2016-03-643544.10.1182/blood-2016-03-643544.
International Standing Committee on Human Cytogenetic Nomenclature, Shaffer LG, McGowan-Jordan J, et al. ISCN 2013 : an international system for human cytogenetic nomenclature (2013). Basel: Karger; 2013. p. 140. 1 folded sheet p
Lin LI, Chen CY, Lin DT, Tsay W, Tang JL, Yeh YC, et al. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005;11(4):1372–9. https://doi.org/10.1158/1078-0432.CCR-04-1816.10.1158/1078-0432.CCR-04-1816.
Rau R, Brown P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol. 2009;27(4):171–81. https://doi.org/10.1002/hon.904.10.1002/hon.904.
Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93(9):3074–80 https://www.ncbi.nlm.nih.gov/pubmed/10216104.
Zhou Y, Moon A, Hoyle E, Fromm JR, Chen X, Soma L, et al. Pattern associated leukemia immunophenotypes and measurable disease detection in acute myeloid leukemia or myelodysplastic syndrome with mutated NPM1. Cytometry B Clin Cytom. 2019;96(1):67–72. https://doi.org/10.1002/cyto.b.21744.10.1002/cyto.b.21744.
Kumar D, Mehta A, Panigrahi MK, Nath S, Saikia KK. DNMT3A (R882) mutation features and prognostic effect in acute myeloid leukemia in coexistent with NPM1 and FLT3 mutations. Hematol Oncol Stem Cell Ther. 2018;11(2):82–9. https://doi.org/10.1016/j.hemonc.2017.09.004.10.1016/j.hemonc.2017.09.004.
Al-Mawali A, Gillis D, Lewis I. Immunoprofiling of leukemic stem cells CD34+/CD38−/CD123+ delineate FLT3/ITD-positive clones. J Hematol Oncol. 2016;9(1):61. https://doi.org/10.1186/s13045-016-0292-z.10.1186/s13045-016-0292-z.
Nomdedeu J, Bussaglia E, Villamor N, Martinez C, Esteve J, Tormo M, et al. Immunophenotype of acute myeloid leukemia with NPM mutations: prognostic impact of the leukemic compartment size. Leuk Res. 2011;35(2):163–8. https://doi.org/10.1016/j.leukres.2010.05.015.10.1016/j.leukres.2010.05.015.
Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–15. https://doi.org/10.1001/jama.2010.1862.10.1001/jama.2010.1862.
Acknowledgments
We thank all coworkers in our laboratory and collaborating centers for their excellent technical assistance and for providing the data.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
B.W. collected clinical-laboratory data and wrote the article; B.Y. and W.W. performed the statistical analysis; X.L. performed sequencing and interpreted mutational data; Hq.L. was responsible for study conception and design. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study conformed to the ethical guidelines of the World Medical Association Declaration of Helsinki. Ethics approval was obtained from the ethics committees of Changzhou First People’s Hospital and Shengjing Hospital of China Medical University. Written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
AML subtype-defining cytogenetic or molecular abnormalities accompanied by NPM1mut missense mutations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, B., Yang, B., Wu, W. et al. The correlation of next-generation sequencing-based genotypic profiles with clinicopathologic characteristics in NPM1-mutated acute myeloid leukemia. BMC Cancer 21, 788 (2021). https://doi.org/10.1186/s12885-021-08455-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08455-7