
Abstract
Tracheal agenesis (TA) is a severe congenital disorder with often an unexpected emergency presentation. There is complete or partial absence of the trachea below the larynx, with presence or absence of a tracheoesophageal fistula (TOF). A neonate with TA is described, and another 48 cases found in literature are reviewed. Due to absence of a TOF, five cases were diagnosed prenatally because of congenital high airway obstruction syndrome (CHAOS). When a TOF is present, polyhydramnion and several other congenital malformations seen on the ultrasound examination should alert clinicians of potential tracheal problems. Prenatal magnetic resonance imaging (MRI) may provide a definitive diagnosis. Postnatal diagnosis is based on recognition of specific clinical signs in the newborn with TA: respiratory distress with breathing movement without appropriate air entry, no audible cry, and failed endotracheal intubation. Despite progress in surgical interventions, mortality remains high. Prenatal diagnosis of TA is possible, but only if a TOF is absent resulting in CHAOS. Prenatal diagnosis of polyhydramnion and other congenital malformation should alert clinicians of potential tracheal problems. Prenatal MRI may provide a definitive diagnosis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tracheal agenesis (TA) is a rare congenital defect that confronts obstetricians, neonatologists, and anesthesiologists with an unexpected emergency presentation. This usually lethal defect has an incidence of 1 per 50,000 newborns and consists of complete or partial absence of the trachea below the larynx, with or without a concomitant tracheoesophageal fistula (TOF). In the third week of gestation, the layngotracheal groove in the proximal foregut progresses caudally, where it develops two primordial buds, which will develop into the bronchi and lungs. Complete separation of the trachea and esophagus occurs by the sixth week. These relatively separate processes allow for malformations of the trachea in the presence of a normal larynx, bronchi, and lungs. Different theories have been suggested to explain the cause of tracheoesophageal anomalies [12, 13, 25, 27, 34, 39, 41]. Besides environmental factors, animal models of different genetic defects, such as (conditional) inactivation of Gli2, Gli3, Shh, Foxf1, and b-catenin, show TA or incorrect septation of the foregut. A major role for the BMP type I receptor genes in mouse models is suggested [12]. However, no causal gene has been identified in human TA patients yet. TA was first described in 1900 by Payne and was classified based upon the presence and length of the tracheal remnant. Floyd’s classification of TA [19] is used more often than the classification by Faro et al. (Fig. 1) [16]. We describe a case of TA Floyd Type II with a TOF in a male newborn with dysmorphic features and additional congenital malformations. Furthermore, we review the recent literature on cases of TA published between January 2005 until December 2010 taking into account the prenatal diagnosis, clinical presentation, management, and outcome of TA.

Classification system of tracheal agenesis (TA). (1) Faro type A: total pulmonary agenesis. (2) Faro type B/Floyd type III: complete TA with a separate origin of the main bronchi from the esophagus. (3) Faro type C: total TA with normal main bronchi fusing in the midline at the carine; a trachea-esophageal fistula (TOF) is present. Floyd type II: total TA with normal main bronchi fusing in the midline at the carine; a TOF may or may not (not shown) be present. (4) Faro type D: larynx joined by atresic strand to distal trachea with a TOF present. (5) Faro type E/Floyd type I: agenesis of the proximal trachea with a normal caudal segment of the distal trachea and a TOF. Faro type F: agenesis of the proximal trachea with a normal caudal segment of the distal trachea and no TOF is present (not shown). (6) Faro type G: TA in a short segment
Case report
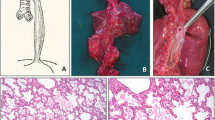
A healthy 34-year-old woman, gravida 2, para 1, was referred to our hospital at 22 + 2 of 7 weeks of gestation because of a double bubble sign on the standard anomaly scan. Third level ultrasonography revealed furthermore a single umbilical artery, postaxial polydactyly of the left hand combined with a polyhydramnion. Amniocentesis showed a normal male karyotype (46,XY), and no subtelomeric and interstitial deletions and/or duplications were found with multiplex ligation-dependent probe amplification (MLPA) analysis. At 34 + 5 of 7 weeks of gestation, an emergency delivery by cesarean section was performed because of fetal distress. A hypotonic boy was born with no audible cry. The birth weight was 2,255 g (weight for gestational age, p25–50). Apgar scores were 5, 5, and 4 after 1, 5, and 10 min, respectively. His heart rate was 70 bpm. Spontaneous breathing movements were absent, and bag and mask ventilation was started. Several attempts for intubation failed, although there was clear vision of the vocal cords. Laryngoscopy by the ear–nose–throat surgeon showed no tracheal opening. Esophageal intubation improved oxygen saturation to 100%, suggesting the presence of a TOF. Tracheotomy was attempted, but no trachea could be identified. The tube was found in the esophagus, confirming TA. The boy died shortly after attempted tracheotomy. Clinical examination showed a premature boy with mild dysmorphic craniofacial features consisting of low implantation of both ears, a small folded right ear, flat face with small forehead, a relatively small head circumference of 23.7 cm (head circumference for gestational age, <p5), and microretrognathy. Furthermore, he had hypertrichosis, unilateral postaxial polydactyly type 1, hypoplastic toenails, and deep-seated hyperconvex finger nails (Fig. 2). At autopsy, the boy had a TA Floyd type II with a TOF (Fig. 3). Both lungs were bilobed with no other signs of left atrial isomerism. Total body X-ray showed fusion of the ninth till 12th rib and hemivertebra of the ninth thoracic vertebra. Furthermore, a duodenal atresia, an arteria lusoria, and a single umbilical artery were diagnosed. Postnatal array-based comparative genomic hybridization (array-CGH) showed no microdeletion or microduplication in the genome.

Postaxial polydactyly type 1 of the left hand of the current case

Autopsy specimen of the current case. Showing a tracheoesophageal fistula (arrow) and bifurcation to the left (L) and right lung (R). Es esophagus
Methods
A Medline (PubMed) literature search was performed for all articles published between January 2005 until December 2010 with MeSH terms and key words including “tracheal/abnormalities,” “tracheal agenesis,” “tracheal aplasia,” “tracheal atresia,” “prenatal diagnosis,” “fetus,” and “newborn.” Titles and abstracts were examined to select potentially relevant studies specific about TA. Statistical analysis was performed using SPSS 17.0 (SPSS, Inc., Chicago, IL, USA). If data relevant for this review was missing, additional information was requested from the authors by e-mail.
Results
Forty-nine cases of TA, including our case, were found [1, 2, 4–7, 9–11, 14, 17, 18, 20, 23, 24, 26, 29–33, 35–38, 40, 42]. The perinatal characteristics, diagnostic investigations, and management are summarized (Table 1). In this review, diagnosis of TA was presumed prenatally in five cases based on congenital high airway obstruction syndrome (CHAOS). In four cases, CHAOS was seen on prenatal ultrasonography [7, 26, 38, 42]. Pregnancy was selectively terminated in two cases. Two newborns died shortly after birth. In one case, initially congenital cystic adenomatoid malformation (CCAM) was suspected on ultrasound; however, after fetal magnetic resonance imaging (MRI), TA was suspected, and an ex utero intrapartum therapy (EXIT) procedure was performed. The child died at the age of 3 months [38]. In another case, CCAM was also suspected, but fetal MRI was not carried out because of a non-reassuring cardiotocogram. The child died shortly after birth [9]. All five prenatal diagnosed cases of TA had no TOF; therefore, they presented as a classic CHAOS. In the cases without prenatal diagnosis, prenatal ultrasonography showed altered amniotic fluid status, mainly polyhydramnion. In 11 of 28 (39%) cases of polyhydramnion, no other malformations besides TA were found. Commonly, clinical symptoms were respiratory distress with breathing movements without appropriate air entry, no audible cry, and failed intubation. In 18 of 31 cases (58.0%), chromosomal analysis was done, but in only two cases, an abnormal karyotype was found: mos 47,XY + mar (43,3)/46,XY (56,5) and 5q11.2 deletion [7, 32]. Associated anomalies were found in 32 (80%) of the cases (Table 2). Most frequent anomalies were cardiac septum defect, laryngeal defect, duodenal atresia, imperforate anus, and kidney and finger malformations. In the remaining eight (20%) cases, no associated malformations were identified by autopsy or computed tomography (CT scan). Mortality was high, as 34 of 40 (85%) children died within 2 days. After surgery, two children are still alive at 10 months and 4 years, respectively [37, 40].
Discussion
Prenatal diagnosis
TA is rarely suspected before birth; prenatal findings leading to the diagnosis of TA are only found when no TOF is present, consistent with the definition of CHAOS. CHAOS is characterized by enlarged hyperechogenic lungs, fluid filled dilated trachea, and bronchi demonstrating an absent flow in the trachea during fetal breathing, and in some cases, even the visualization of the obstruction, compression of the heart, with or without cardiac dysfunction, flattened diaphragm, and/or massive ascites. The classical signs of CHAOS are absent if a TOF is present because lung fluid can pass through the TOF to the stomach or amniotic sac. Therefore, the pressure in the lungs is normal, and the lungs will have a normal ultrasonographic appearance. In the present study, five of 49 cases were diagnosed prenatally because of symptoms related to CHAOS. Prenatal diagnosis in these five cases of TA was confirmed by fetal MRI or detailed ultrasound examination. Detailed ultrasound examination should be focused on abnormal fluid accumulations in the larynx or trachea. If uncertainty remains, fetal MRI can provide additional information [28]. If CHAOS is suspected, absence of amniotic phospholipids in amniotic fluid may support the diagnosis of TA because normally phospholipids are secreted into the amniotic fluid by the respiratory tract [8]. If TA is prenatally suspected, an EXIT procedure can be planned in a tertiary hospital as described by Vaikunth et al. [38]. They also suggested that infants with TA and a TOF may also benefit from the EXIT procedure by preventing hypoxia, but this has not been described as prenatal diagnosis of TA, and a TOF is still not yet possible. In this review, polyhydramnion was reported in 72% of the cases. The presence of TA and a TOF may interfere with fetal swallowing because in 39% of the cases with polyhydramnion, no other congenital abnormalities interfering with fetal swallowing and/or absorption of fluid, such as gastrointestinal obstruction, were present. Development of polyhydramnion, particularly in combination with other frequent associated anomalies with TA, should alert clinician of the possibility of TA. Prenatal MRI may provide a definitive diagnosis.
Clinical presentation and diagnosis
Postnatal diagnosis can be established by recognition of the specific combination of clinical signs in the newborn: respiratory distress with breathing movements without appropriate air entry, no audible cry, and failed endotracheal intubation. Preterm labor and low birth weight (<2,500 g) and an Apgar score below 7 at 5 min are also common characteristics. If TA is accompanied by a TOF, bag and mask ventilation and (accidental) esophageal intubation may temporarily improve the respiratory status. After esophageal intubation, marked abdominal distension develops, and air passes through the fistula into the lungs that will give cycles of stabilization and deterioration. Laryngo-bronchoscopy can diagnose TA and a possible TOF. Radiographic findings may include posterior location of the endotracheal tube (esophageal intubation) and absence of the tracheal shadow. A CT scan is the procedure of choice for delineating the anatomy in newborns with suspected TA [36]. Contrast material may demonstrate a blind laryngeal sac and a TOF but are difficult to perform and unreliable due to potential airway compromise.
Management and outcome
In this review, tracheotomy was attempted in 38% of the cases to explore the options of surgical reconstruction. Often, no surgery could be performed because of severe asphyxia or death of the infant. Surgical management consists of esophageal ligation at the site distal to the TOF. This results in more effective ventilation because the esophagus can be used as a pseudo-trachea. The proximal part of the esophagus has to be taken out as a double barrel esophagostomy, with the distal end for intubation and the proximal end for salivary drainage. An external esophageal stent can prevent the esophagus from collapsing. Gastrostomy for feeding should be performed next. Reconstruction with small intestine, colon interposition, or gastric pull-up has been demonstrated to be feasible and compatible with survival [24]. Despite progress in surgical management, only two children are still alive and have a normal neurodevelopment after surgery [37, 40].
Classification of tracheal agenesis
Both the classifications of Floyd and Faro are inappropriate for all types of TA. This is due to the fact that a TOF, crucial for decision making, can not always be correctly classified, e.g., a Floyd type I without a TOF described in this review does not exist in the Floyd classification. Likewise, Floyd type II without a TOF is missing in the Faro’s classification. We propose to use the Faro’s classification system and add a Faro Type C without a TOF to it.
In the present review, Floyd type II was identified as the most common type of TA in line with the literature. The male:female ratio in this review was 2:1, similar to the literature [22, 39]. Further research is necessary to explain this male predominance of TA.
Associated malformations
Associated malformations can be part of the VACTERL association (Vertebral Defects, Anal Atresia, Cardiovascular Defects, Tracheoesophageal Fistula and/or Esophageal Atresia, Renal Defects, and Limb Defects) or TARCD association (Tracheal Agenesis/Atresia, Radial Ray Defects, Complex Congenital Cardiac Abnormalities, and Duodenal Atresia)15 [15]. Although TA and duodenal atresia are not part of the acronym of VACTERL, tracheal stenosis and tracheal atresia have been described in fetuses with features of VACTERL [3], and duodenal atresia was found in 6.3% of children diagnosed with VACTERL association [21]. So far, no causal genes for these associations have been identified yet. TA can also be a part of Fraser syndrome, which is caused by mutations in FRAS1 and FREM2 genes [7]. Abnormal development of the larynx in combination with or without a TOF or other tracheal problems is also observed in several rare syndromes and in cases of chromosomal aneuploidy or interstitial (micro)deletions or (micro)duplications [5]. In one case, a de novo abnormal karyotype was found (mos 47,XY + mar (43,3)/46,XY (56,5)) as both parents had a normal karyotype. The marker chromosome was identified by FISH as a derivate from chromosome 22 and 15 [32]. This abnormal karyotype could contribute to the TA and associated malformations and may help to unravel the origin of the genetic cause of TA. A deletion on 5q11.2 region was found in another case, whose mother had normal karyotype while no paternal material was available [7]. Because TA is associated with other congenital anomalies, it is important to perform standard karyotyping as well as array-based comparative genomic hybridization (array-CGH) to detect microdeletions or duplications in the genome. Autopsy provides more details especially on malformations that cannot be detected by fetal ultrasound.
Conclusion
TA is a severe congenital defect with an unexpected emergency presentation. Prenatal diagnosis of TA is only possible in case of CHAOS. If a TOF is present, polyhydramnion and other congenital malformations on ultrasound should alert clinicians of potential tracheal problems. Prenatal MRI may provide a definitive diagnosis. Postnatal symptoms suggestive of the diagnosis are respiratory distress with strong respiratory efforts without good air entry, no audible cry, and failed endotracheal intubation. Associated malformations are often found and include cardiac septum defects, laryngeal defects, duodenal atresia, imperforate anus, kidney defects, and finger malformations. The diagnosis TA is often confirmed by laryngo-bronchoscopy, CT scan, and tracheotomy. Despite progress of surgical developments, the prognosis is poor. CGH array or genome wide assays, in addition to routine cytogenetic analysis, and autopsy are important to detect associated malformations and elucidate the cause(s) of TA.
References
Agarwal S, John S, Latta S, Sahni M, Kumar N (2006) Tracheal agenesis: two case reports. Int J Pediatr Otorhinolaryngol 70(4):737–739
Ahmad R, Abdullah K, Mokhtar L, Fadzil A (2009) Tracheal agenesis as a rare cause of difficult intubation in a newborn with respiratory distress: a case report. J Med Case Reports 3:105
Bercker S, Kornak U, Bührer C, Henrich W, Kerner T (2006) Tracheal atresia as part of an exceptional combination of malformations. Int J Pediatr Otorhinolaryngol 70(6):1137–1139
Bhattarai B, Pratap A, Yadav R, Pokharel K (2008) Anaesthetic management of tracheal agenesis. J Nepal Med Assoc 47(171):142–144
Bober K, Musialik-Swietlińska E, Adamiec-Poniewierka E, Pajak J, Bielecki I, Koszutski T, Więcek-Włodarska D, Goc B, Szydłowski L, Swietliński (2008) Tracheal agenesis: perioperative management of one stage correction. Pediatr Anesth 18(10):982–983
Chawla S, Natarajan G, Chouthai N, Kamat D (2009) CHAOS in the delivery room: a dire emergency. J Pediatr 155(5):756
de Jong EM, Douben H, Eussen BH, Felix JF, Wessels MW, Poddighe PJ, Nikkels PGJ, de Krijger RR, Tibboel D, de Klein A (2010) 5q11.2 deletion in a patient with tracheal agenesis. Eur J Hum Genet 18(11):1265–1268
de José MB, Drudis R, Monclús E, Silva A, Santander S, Cusí V (2000) Management of tracheal agenesis. Pediatr Anesth 10(4):441–444
de Luca D, de Carolis MP, Capelli A, Gallini F, Draisci G, Pinto R, Arena V (2008) Tracheal agenesis without esophageal fistula: genetic, resuscitative, and pathological issues. J Pediatr Surg 43(1):e29–e32
Demircan M, Aksoy T, Ceran C, Kafkasli A (2008) Tracheal agensis and esophageal atresia with proximal and distal bronchoesophageal fistulas. J Pediatr Surg 43(8):e1–e3
Dijkman KP, Andriessen P, van Lijnschoten G, Halbertsma FJ (2009) Failed resuscitation of a newborn due to congenital tracheal agenesis; a case report. Cases J 2:7212
Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X (2011) Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 138(5):971–981
Effman EL, Spackman TJ, Berdon WE, Kuhn JP, Leonidas JC (1975) Tracheal agenesis. Am J Roentgenol Radium Ther Nucl Med 125(4):767–781
Erler T, Wetzel U, Biolik HB, Eichhorn T, Gurski A (2008) Trachealaplasie-eine besonders seltene und dramatische fehlbildung. Klin Pädiatr 220(4):271–274
Evans JA, Greenberg CR, Erdile L (1999) Tracheal agenesis revisited: analysis of associated anomalies. Am J Med Gen 82(5):415–422
Faro RS, Goodwin CD, Organ CH Jr, Hall RT, Holder TM, Ashcraft KW, Amoury RA (1979) Tracheal agenesis. Ann Thorac Surg 28(3):295–299
Felix JF, van Looij MA, Pruijsten RV, de Krijger RR, de Klein A, Tibboel D, Hoeve HLJ (2006) Agenesis of the trachea: phenotypic expression of a rare cause of fatal neonatal respiratory insufficiency in six patients. Int J Pediatr Otorhinolaryngol 70(2):365–370
Fischer D, Schloesser R, Veldman A (2007) Management of congenital tracheal agenesis. Eur J Pediatr 166(8):885
Floyd J, Campbell DC Jr, Dominy DE (1962) Agenesis of the trachea. Am Rev Respir Dis 86:557–560
Fraser N, Stewart RJ, Grant J, Martin P, Gibbin KP, Padfield CJH (2005) Tracheal agenesis with unique anatomy. J Pediatr Surg 40(10):e7–e10
Fujishiro E, Suzuki Y, Sato T, Kondo S, Miyachi M, Suzumori K (2004) Characteristic findings for diagnosis of baby complicated with both the VACTERL association and duodenal atresia. Fetal Diagn Ther 19(2):134–137
Haben CM, Rappaport JM, Clarke KD (2002) Tracheal agenesis. J Am Coll Surg 194(2):217–222
Heimann K, Bartz C, Naami A, Peschgens T, Merz U, Hörnchen H (2007) Three new cases of congenital agenesis of the trachea. Eur J Pediatr 166(1):79–82
Kanoija RP, Parikh M, Bhagat H, Bansal S, Menon P, Rao KLN (2009) Tracheal agenesis: case report with review of the current scenario and management reported over the past two decades. Eur J Pediatr Surg 19:1–3
Kluth D, Steding G, Seidl W (1987) The embryology of foregut malformations. J Pediatr Surg 22(5):389–393
Lupi M, Bonetti LR, Trani N, Maccio L, Maiorana A (2009) Congenital tracheal atresia in newborn: case report and review of the literature. Pathologica 101(6):235–239
Merei JM, Hutson JM (2002) Embryogenesis of trachea esophageal anomalies: a review. Pediatr Surg Int 18(5–6):319–326
Mong A, Johnson AM, Kramer SS, Coleman BG, Hedrick HL, Kreiger P, Flake A, Johnson M, Wilson RD, Adzick NS, Jaramillo D (2008) Congenital high airway obstruction syndrome: MR/US findings, effect on management, and outcome. Pediatr Radiol 38(11):1171–1179
Panthagani ID, Santos MC, D’Ángio CT (2009) Use of computed tomography to categorize the type of tracheal agenesis. J Pediatr Surg 44(5):1044–1046
Pragati S, Manisha K, Ruchi N (2007) Tracheal agenesis. Indian J Pathol Microbiol 50(4):848–849
Pratap A, Saha GS, Bhattarai BK, Rohit PY, Nepal A, Bajracharya A, Kumar A, Adhikary S (2007) Tracheal agenesis type B: further evidence to a lethal congenital tracheal malformation. J Pediatr Surg 42:1284–1287
Rupérez Lucas M, Bonet Serra B, Martínez Orgado JA, Guerrero Márquez C (2007) Agenesia traqueal asociada a malformaciones laríngeas como causa inusual de fracaso de la vía aérea. An Pediatr 67(3):236–239
Saccardi C, Ludwig K, Cosmi E, D’Antona D, Salmaso R, Fassina A (2010) Tracheal agenesis with bifurcating common airway arising from midesophagus. Pediatr Dev Pathol 13(3):252–254
Sandu K, Monnier P (2007) Congenital tracheal anomalies. Otolaryngol Clin North Am 40(1):193–217
Schummer W, Schummer C, Klemm P, Brodhun M, Neumann R, Bondartschuk M, Koscielny HA (2006) Trachealagenesie. Eine seltene Ursache der respiratorischen Insuffizienz des Neugeborenen. Anaesthesist 55(12):1259–1265
Strouse PJ, Newman B, Hernandez RJ, Afshani E, Bommaraju M (2006) CT of tracheal agenesis. Pediatr Radiol 36(9):920–926
Usui N, Kamiyama M, Tani G, Takama Y, Soh H, Uehara S, Fukuzawa M (2010) Three-stage reconstruction of the airway and alimentary tract in a case of tracheal agenesis. Ann Thorac Surg 89(6):2019–2022
Vaikunth SS, Morris LM, Polzin W, Gottliebson W, Lim FY, Kline-Faith B, Crombleholme TM (2009) Congenital high airway obstruction syndrome, due to complete tracheal agenesis: an accident of nature with clues for tracheal development and lessons in management. Fetal Diagn Ther 26(2):93–97
van Veenendaal MB, Liem KD, Marres HA (2000) Congenital absence of the trachea. Eur J Pediatr 159(1–2):8–13
Watanabe T, Okuyama H, Kubota A, Kawahara H, Hasegawa T, Ueno T, Saka R, Morishita Y (2008) A case of tracheal agenesis surviving without mechanical ventilation after external esophageal stenting. J Pediatr Surg 43(10):1906–1908
Zaw-Tun HA (1982) The trachea-esophageal septum—fact of fantasy? Origin and development of the respiratory primordium and esophagus. Acta Anat (Basel) 114(1):1–21
Zhou Q, Shi W, Zhang T, Tian L, Zeng S (2009) Prenatal diagnosis of type 1 tracheal agenesis without tracheoesophageal fistula. J Obstet Gynaecol Res 35(2):364–367
Conflicts of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
de Groot-van der Mooren, M.D., Haak, M.C., Lakeman, P. et al. Tracheal agenesis: approach towards this severe diagnosis. Case report and review of the literature. Eur J Pediatr 171, 425–431 (2012). https://doi.org/10.1007/s00431-011-1563-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-011-1563-x