
Abstract
Severe tracheal stenosis, resulting in functional atresia of the trachea is a rare congenital malformation with an estimated occurrence of two in 100,000 newborns. If no esophagotracheal fistula is present to allow for spontaneous breathing, this condition is usually fatal. We report on a male infant born at 32 weeks of gestation. The patient presented with respiratory distress immediately after delivery due to severe congenital tracheal stenosis resulting in functional atresia of the trachea. Endotracheal intubation failed and even emergency tracheotomy did not allow ventilation of the patient lungs. The patient finally succumbed to prolonged hypoxia due to functional tracheal atresia. The etiology of tracheal atresia and tracheal stenosis is still unclear, but both conditions are frequently combined with other anomalies of the VACTERL (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal/radial anomalies and limb defects) and TACRD (tracheal agenesis, cardiac, renal and duodenal malformations) association. Conclusion Successful treatment of severe congenital tracheal stenosis and tracheal atresia depends on either prenatal diagnosis or recognition of this condition immediately after birth to perform tracheotomy without delay. Nevertheless, despite any efforts, the therapeutical results of severe tracheal stenosis and tracheal atresia are still unsatisfactory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe tracheal stenosis, resulting in functional atresia of the trachea is a rare congenital malformation that is frequently combined with other anomalies of the vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal/radial anomalies, and limb defects (VACTERL)—and tracheal agenesis, cardiac, renal, and duodenal malformations (TACRD)—association. Affected neonates present with severe respiratory distress immediately after birth. Without esophagotracheal fistula, severe tracheal stenosis, resulting in functional atresia of the trachea is fatal and usually results in death within the first minutes of life despite all efforts for salvage.
We report on a preterm neonate with severe congenital tracheal stenosis resulting in functional tracheal atresia. It is of note, that no esophagotracheal fistula was present. The child developed respiratory distress immediately after birth and died at the age of 3 h due to respiratory failure. No other malformations, particularly an esophagotracheal fistula, were found on post-mortem examination.
Tracheal stenosis, functionally resulting in atresia of the trachea without esophagotracheal fistula is an always lethal congenital malformation because ventilation of the lungs cannot be achieved. Survival is only possible with either a small tracheal lumen remaining, sufficiently large enough for spontaneous breathing, or an emergency tracheotomy. The results of surgical treatment of high-grade congenital tracheal stenosis and tracheal atresia however are disappointing.
Case report
A male infant was born at 32 weeks of gestation to a 35-year-old gravida II, para I by cesarean section because of maternal hepatitis C infection. Because of prior drug abuse, the mother had been on polamidone, a synthetic opioid used as part of the treatment of dependence on opioid drugs. Pregnancy was uncomplicated until the development of cervical incompetence. The APGAR scores were 6 after 1 min, 7 after 5 min, and 8 after 10 min, respectively. The infant developed respiratory distress and high oxygen demand immediately after birth and was admitted to the neonatal intensive care unit on nasopharyngeal CPAP support. On admission, the patient presented with hypercapnia with a pCO2 of 93 mmHg. Because of persisting respiratory distress and progression of CO2 retention, the decision was made to intubate and ventilate the infant. Direct laryngoscopy revealed a clearly visible glottic plane with unaffected vocal cord mobility, but it was impossible to advance tracheal tubes of various sizes beyond the vocal cords. After several attempts, mask ventilation became impossible as well. Intubation of the esophagus to ventilate the lungs via a potentially existing esophagotracheal fistula was also attempted but was unsuccessful.
Because of progressive bradycardia and poor oxygenation, cardiopulmonary resuscitation was started. Even emergency tracheotomy did not allow ventilation of the lungs since the infant lacked a visible tracheal lumen. After prolonged unsuccessful cardiopulmonary resuscitation, the infant finally died at the age of 3 h.
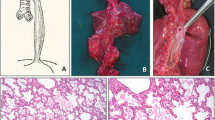
Post-mortem examination revealed congenital high-grade stenosis of the trachea directly distal to the vocal cords over a distance of more than 1 cm with a lumen of less than 1 mm in diameter. Microscopically, the normal architecture of the proximal trachea was replaced by fibrous connective tissue (Fig. 1). The severely stenotic lumen of the distal part of trachea was surrounded by compact cartilage (Fig. 2). Macroscopically, the lungs appeared normal, whereas the microscopic examination revealed only poorly ventilated alveoli (Fig. 3). There were no additional malformations.

In the proximal part of the trachea, normal architecture was replaced by fibrous connective tissue

On microscopic examination, a small lumen of the distal trachea surrounded by compact cartilage was found

Microscopic examination of the lung revealed poorly ventilated alveoli
Discussion
Severe congenital stenosis and atresia of the trachea are rare malformations occurring with an estimated rate of two per 100,000 live births [13]. Less than 100 cases have been reported in literature since the first description by Payne in 1900 [16]. Several years ago, we described a case of tracheal atresia. This term infant was found to have type II tracheal atresia according to Floyd [8]. The tracheal malformation was associated with esophageal atresia proximal to the esophagotracheal fistula [8, 18]. Taking the regional birth rate into consideration, together with the case described in this report, the predicted rate of two per 100,000 live births is closely met. Classifying tracheal abnormalities anatomically, it is important to distinguish between tracheal agenesis and tracheal atresia. Tracheal agenesis describes an entity without any remnant of tracheal organ structure, whereas the latter characterizes an anomaly with no tracheal lumen but remaining organ structure [20]. The etiology of tracheal atresia and tracheal agenesis is still unclear, but abnormal epithelial–mesenchymal interactions may be involved since the respiratory tract develops from the ventral wall of the foregut in the third embryonic week [6]. The lower respiratory tract starts to develop as a median groove in the anterior wall of the pharynx. From this groove, the respiratory tract buds out into the mesenchyme ventral to the foregut, where the lung bud appears paired early in its development. Tracheal agenesis develops when the first outgrowth of the lung bud is aborted early during development and the larynx or the very proximal part of the trachea stays atretic. The paired lung primordium grows into the ventral mesenchyme, forming a bronchial tree without being connected to the larynx [14, 23]. Therefore, tracheal agenesis can be embryologically interpreted as a result of defective foregut differentiation, whereas tracheal atresia represents faulty development of an already differentiated anatomical structure [12]. The development of an esophagotracheal fistula is possibly be caused by a too close proximity of the trachea and the esophagus due to an abnormal localization of both organs. The proximity between both organs might lead to epithelial cellular death and consecutively the formation of an esophagotracheal fistula. Cellular death caused by the too close vicinity of two anatomical structures is a process known to occur physiologically during the normal development of embryos as well [12]. The underlying mechanisms for this phenomenon of cellular death and the factors ultimately leading to the development of esophagotracheal fistula are, however, unidentified. Likewise, the factors directing the development of tracheal atresia are unknown. Neither a chromosomal defect nor a single gene defect has been identified as the underlying cause. In the reported case, genetic studies including a microarray analysis were not performed due to parental refusal. External factors seem to interfere with the normal development of the respiratory tract in patients with tracheal atresia or severe tracheal stenosis [6, 20], but to date no common environmental agents or drugs have been identified to be the underlying cause for tracheal atresia and its associated anomalies. Although opioids are known to be of teratogenic effect to central nervous structures [22], there is no evidence for opioids to be involved in the determination of tracheal malformations. Since it is unknown, whether the mother of the reported baby took any other drugs than polamidone during early pregnancy, a drug effect contributing to the observed malformations cannot be ruled out.
According to Floyd [8], three anatomical types of tracheal atresia can be differentiated. In type I, accounting for approximately 15% of the cases, atresia of the proximal trachea is combined with a normal distal trachea, which is connected to the esophagus via an esophagotracheal fistula. In type II (approximately 50%), the trachea is completely absent, both main bronchi join in the midline and an esophagotracheal fistula arises from the carina. In type III (approximately 30%), the main bronchi arise directly from the esophagus. Five percent of the reported cases could not be categorized according to Floyd [20].
In the majority of the cases, tracheal atresia is associated with other congenital anomalies [6, 7]. These include malformations of the respiratory tract, cardiac malformations, and anomalies of the genitourinary as well as the gastrointestinal tract. Because of the associated malformations, some authors refer to tracheal atresia as part of the VACTERL syndrome [7]. More recently, cases of tracheal atresia have been reported with associated abnormalities described as TACRD association [5–7, 21]. In most of the cases of tracheal atresia, an esophagotracheal fistula was present; only five cases without esophagotracheal fistula have been published [4, 6, 15–17]. Remarkably, the infant, we report on, did neither fit into Floyd’s classification of tracheal atresia nor did we find any associated anomalies. Therefore, we describe one of the few cases, where an isolated tracheal atresia occurred. In contrast to our patient, in none of the reported cases without esophagotracheal fistula, a residual lumen of the trachea was described. Since the term tracheal agenesis is often used synonymously with tracheal atresia, it is difficult to compare our patient with other reported cases, but reviewing the published literature on congenital tracheal malformations, the reported case is unique for two reasons. First, no other malformations could be found on post-mortem examination and second, a small residual lumen of the patient’s trachea was evident.
Tracheal atresia is one of the rarest tracheal abnormalities. It should be suspected in pregnancies with a history of polyhydramnios and newborns with respiratory distress without an audible cry and in cases of difficult intubation [3]. There is a male predominance (2:1) and an association with prematurity [20]. The postnatal management of neonates with tracheal atresia is a challenging problem. If tracheal atresia is associated with an esophagotracheal fistula, the infants are not able to breathe spontaneously, since the esophagus collapses during inspiration. Mask ventilation via the esophagus and the fistula may improve the condition of the neonate. Esophageal intubation and PEEP ventilation are approaches to secure a patent upper airway. In cases without communication between trachea and esophagus and no remaining tracheal lumen, spontaneous breathing as well as mask ventilation are not effective. Emergency tracheotomy or coniotomy can be successful, whenever a patent distal trachea exists. In the reported case, the post-mortem examination revealed a severely stenotic trachea with an endoluminar width of less than 1 mm in diameter that extended from the subglottic region to the bifurcation.
If the diagnosis of tracheal atresia is suspected prenatally, it can be confirmed by fetal ultrasound and fetal magnetic resonance imaging (MRI). From the results of the MRI scan, a decision should be made to employ an ex utero intrapartum tracheotomy (EXIT) to ventilate the lungs of the neonate. EXIT tracheotomy will be only successful in cases with a patent distal trachea. The EXIT procedure has been originally described for the reversal of tracheal occlusion in patients with congenital diaphragmatic hernia [2, 10].
The therapeutical results of tracheal atresia are unsatisfactory. Different surgical reconstructive procedures have been developed, using either the esophagus or synthetic material to create an upper airway. None of them has turned out to be effective concerning long-term survival of the patients. Most of the patients died within a few hours to a few days [11, 13]. Keeping patients on extracorporeal membrane oxygenation after tracheal reconstructive surgery to provide “airway rest” during the initial healing phase could improve the outcome [1, 9]. The longest surviving infant was a 6-year-old female with Floyd type I tracheal aresia and associated congenital anomalies (tetralogy of Fallot, rectovaginal fistula). She died at the age of 6 years and 10 months because of an acute esophageal bleeding [19]. In the future, generating new tracheal tissue by means of tissue engineering might be an approach to repair tracheal defects like tracheal atresia or tracheal agenesis. Animal experiments showed promising results in this direction [20].
In conclusion, tracheal atresia is a rare congenital malformation and the management of neonates with tracheal atresia is difficult. The resuscitative management could be improved with the diagnosis of tracheal atresia being made prenatally. However, none of the surgical procedures employed to date has been proven to be satisfactory. Using tracheal tissue generated by tissue engineering to reconstruct the atretic trachea might be the therapy of choice in the future.
Reference
Angel C, Murillo C, Zwischenberger J, Swischuk L, Graves D, Chernin J (2000) Perioperative extracorporeal membrane oxygenation for tracheal reconstruction in congenital tracheal stenosis. Pediatr Surg Int 16:98–101
Bouchard S, Johnson MP, Flake AW, Howell LJ, Myers LB, Adzick NS, Crombleholme TM (2002) The EXIT procedure: experience and outcome in 31 cases. J Pediatr Surg 37:418–426
De Jose MB, Drudis R, Monclus E, Silva A, Santander S, Cusi V (2000) Management of tracheal agenesis. Paediatr Anaesth 10:441–444
DeLuca D, De Carolis MP, Capelli A, Gallini F, Draisci G, Pinto R, Arena V (2008) Tracheal agenesis without esophageal fistula: genetic, resuscitative, and pathological issues. J Pediatr Surg 43:e29–e32
Diaz EM Jr, Adams JM, Hawkins HK, Smith RJ (1989) Tracheal agenesis. A case report and literature review. Arch Otolaryngol Head Neck Surg 115:741–745
Evans JA, Greenberg CR, Erdile L (1999) Tracheal agenesis revisited: analysis of associated anomalies. Am J Med Genet 82:415–422
Evans JA, Reggin J, Greenberg C (1985) Tracheal agenesis and associated malformations: a comparison with tracheoesophageal fistula and the VACTERL association. Am J Med Genet 21:21–38
Floyd J, Campbell DC Jr, Dominy DE (1962) Agenesis of the trachea. Am Rev Respir Dis 86:557–560
Hines MH, Hansell DR (2003) Elective extracorporeal support for complex tracheal reconstruction in neonates. Ann Thorac Surg 76:175–178
Kanamori Y, Kitano Y, Hashizume K, Sugiyama M, Tomonaga T, Takayasu H, Egami S, Goishi K, Shibuya K, Kawana Y, Marumo G, Kikuchi A, Kozuma S, Taketani Y, Sekiyama Y (2004) A case of laryngeal atresia (congenital high airway obstruction syndrome) with chromosome 5p deletion syndrome rescued by ex utero intrapartum treatment. J Pediatr Surg 39:E25–E28
Kerschner J, Klotch DW (1997) Tracheal agenesis: a case report and review of the literature. Otolaryngol Head Neck Surg 116:123–128
Kluth D, Steding G, Seidl W (1987) The embryology of foregut malformations. J Pediatr Surg 22:389–393
Manschot HJ, van den Anker JN, Tibboel D (1994) Tracheal agenesis. Anaesthesia 49:788–790
Merei JM, Farmer P, Hasthorpe S, Qi BQ, Beasley SW, Myers NA, Hutson JM (1997) Timing and embryology of esophageal atresia and tracheo-esophageal fistula. Anat Rec 249:240–248
Milles G, Dorsey DB (1950) Intra-uterine respiration-like movements in relation to development of the fetal vascular system. A discussion of intra-uterine physiology based upon cases of congenital absence of the trachea, abnormal vascular development, and other anomalies. Am J Pathol 26:411–425
Payne W (1900) Congenital absence of the trachea. Brooklyn Med J 14:568
Sankaran K, Bhagirath CP, Bingham WT, Hjertaas R, Haight K (1983) Tracheal atresia, proximal esophageal atresia, and distal tracheoesophageal fistula: report of two cases and review of literature. Pediatrics 71:821–823
Schiffmann JH, Rehder H, Speer CP (1991) Tracheal agenesis, a rare cause of respiratory insufficiency in newborn infants. Monatsschr Kinderheilkd 139:102–104
Soh H, Kawahawa H, Imura K, Hagi M, Yoneda A, Kubota A, Okada A (1999) Tracheal agenesis in a child who survived for 6 years. J Pediatr Surg 34:1541–1543
van Veenendaal MB, Liem KD, Marres HA (2000) Congenital absence of the trachea. Eur J Pediatr 159:8–13
Wei JL, Rodeberg D, Thompson DM (2003) Tracheal agenesis with anomalies found in both VACTERL and TACRD associations. Int J Pediatr Otorhinolaryngol 67:1013–1017
Yanai J, Ben-Shaanan TL, Haimovitch H, Katz S, Kazma M (2006) Mechanism-based approaches for the reversal of drug neurobehavioral teratogenicity. Ann NY Acad Sc 1074:659–671
Zaw-Tun HA (1982) The tracheo-esophageal septum—fact or fantasy? Origin and development of the respiratory primordium and esophagus. Acta Anat (Basel) 114:1–21
Conflicts of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Krause, U., Rödel, R.M.W. & Paul, T. Isolated congenital tracheal stenosis in a preterm newborn. Eur J Pediatr 170, 1217–1221 (2011). https://doi.org/10.1007/s00431-011-1490-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-011-1490-x