
Abstract
We report on a 10-year-old patient with developmental delay, craniofacial dysmorphism, digital and genital abnormalities. In addition, muscular hypotonia, strabism, and splenomegaly were observed; inguinal and umbilical hernias were surgically corrected. Mucopolysaccharidoses and CDG syndromes could not be found. Chromosome analysis revealed a normal male karyotype (46,XY). A more detailed investigation of the patient’s genomic DNA by microarray-based comparative genomic hybridization (array CGH) detected an interstitial 3.7 Mb deletion ranging from 15q24.1 to 15q24.3 which was shown to be de novo. Interstitial deletions involving 15q24 are rare. Sharp et al. (Hum Mol Genet 16:567–572, 2007) recently characterized a recurrent 15q24 microdeletion syndrome with breakpoints in regions of segmental duplications. The de novo microdeletion described here colocalizes with the minimal deletion region of the 15q24 microdeletion syndrome. The distinct clinical phenotype associated with this novel microdeletion syndrome is similar to the phenotype of our patient with respect to specific facial features, developmental delay, microcephaly, digital abnormalities, and genital abnormalities in males. We present a genotype–phenotype correlation and comparison with patients from the literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microdeletion syndromes are recurrent deletions associated with a distinct phenotype. These microdeletions often occur between low copy repeats and are commonly due to liability to unequal crossing over. It was recently shown that the well known classical microdeletion syndromes such as DiGeorge and Williams syndromes account for about 5% of all patients with mental retardation [18]. Besides the detection of known microdeletion syndromes, microarray-based comparative genomic hybridization (array CGH) or molecular karyotyping recently led to the discovery of novel recurrent microdeletion syndromes such as 17q21.31 and 15q24 [12, 20, 21]. To date 14 patients have been reported who have interstitial deletions at 15q22-q24 [1, 3, 4, 7, 13, 20, 22], but only three of them are restricted to chromosomal band 15q24 [4, 20]. So far, only a few of these cases have been molecularly characterized in order to determine the size of the affected regions. Despite considerable phenotypic variability, patients with 15q24 microdeletion syndrome share common features including global developmental delay, hypotonia, and genital abnormalities in males. In order to further delineate the 15q24 genotype–phenotype correlation, a novel patient with 15q24 microdeletion detected by array CGH is presented.
Clinical report
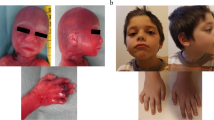
We present a boy who is the second child of healthy Caucasian parents. The mother underwent spine CT scan in the 8th week of gestation. The further course of pregnancy was uneventful and delivery by Caesarean section was at 38 weeks of gestation after preterm hydrorrhoea. Physical examination at the age of 10 years revealed a height of 132.5 cm (P50), weight of 35.7 kg (P50–75) and head circumference of 53.5 cm (P75). Our patient presented with developmental retardation, craniofacial dysmorphism, truncal obesity, and micropenis (Fig. 1a,b). In addition, general muscular hypotonia and splenomegaly were observed. He showed lumbar hyperlordosis, mild genua valga, and joint hypermobility. At the age of 6 months coordination disturbances were noted. Unaided walking was possible at 18 months.

a, b Patient at the age of 10 years. Note truncal obesity, widely spaced, inverted nipples, as well as coarse face with high forehead, broad medial eyebrows, periorbital fullness, slight antimongoloid slant, deep set eyes, hypoplastic nostrils, long philtrum and full cheeks. c, d Hands show mild brachydactyly, clinodactyly of fourth finger (left hand) and broad thumbs. e Note hypoplasia of the distal phalanges, short mesophalanges, delayed carpal ossification (corresponding to the state of a 3 year old child), irregular epiphyses of the second and fifth mesophalanges and of the distal phalanx of the thumb
Speech development was retarded and a mild mental retardation (IQ ∼65) was observed. Nasal speech and a hoarse voice were apparent. Prominent veins were evident on the trunk. Skin laxity was noted especially in the face and hands. We observed a convergent squint, astigmatism, and amblyopia. Inguinal and umbilical hernias as well as unilateral retentio testis inguinalis were surgically corrected at age 4 years. Patient’s hands show mild brachydactyly, clinodactyly of fourth finger (left hand), and broad thumbs (Fig. 1c,d). Radiological investigations including cranial CT were normal except for hand radiographs disclosing hypoplasia of distal phalanges, short mesophalanges, delay of carpal ossification (corresponding to the state of a 3 year old child), irregular epiphyses of the second and fifth mesophalanges and of the distal phalanx of the thumb (Fig. 1e).
Echocardiography as well as renal ultrasound and neurophysiological investigations (i.e. nerve velocity, EEG, ECG) did not reveal any pathological findings. In repeated metabolic screenings triglycerides and the LDL/HDL quotient were always elevated. He was only able to walk up to 100 m. Clinically, a metabolic defect was suspected. However, examinations concerning mucopolysaccharidoses and CDG syndrome disclosed no abnormalities. All serum parameters including growth related factors (IGF1, IGFBP3) were normal. FSH (18 U/l) and testosterone (1.4 ng/ml) values were normal for the patient’s age. This study was approved by the ethics committee of the Charité, Universitätsmedizin Berlin, and all individuals gave their informed consent prior to their inclusion in the study.
Materials and methods
Cytogenetics
Karyotyping of GTG-banded chromosomes from lymphocytes at 450 bands resolution was performed according to standard procedures.
Fluorescence in situ hybridization (FISH)
BAC clones RP11–414J4 (genomic position 72.7–72.9 Mb) and RP11–94P14 (genomic position 75.2–75.3 Mb) were obtained from the RZPD (Deutsches Ressourcenzentrum für Genomforschung, Berlin, Germany). Genomic positions are according to the Ensembl Genome Browser v44, April 2007. BAC DNA was fluorescently labelled using nick translation and hybridized to metaphase spreads of the patient’s and parents’ lymphocytes using standard procedures. BAC clones RP11–414J4 and RP11–94P14 were labelled with spectrum orange. Centromere probe, Cep15 (D15Z1; Vysis, Downers Grove, IL), labelled with spectrum green was used as control probe. Chromosomes were counterstained with DAPI (4′,6-diamidino-2-phenylindole). Hybridization of the commercial probes for SNRPN (Prader-Willi/Angelman region) and TUPLE1 (DiGeorge/VCFS region) were performed on metaphase spreads of the patient as recommended by the manufacturer (Vysis, Downers Grove, IL).
Microarray-based comparative genomic hybridization (array CGH)
Array CGH was carried out using a submegabase whole human genome tiling path BAC array consisting of the human 32k Re-Array set (http://bacpac.chori.org/pHumanMinSet.htm) [14, 18], the 1Mb Sanger Clone set (Wellcome Trust Sanger Institute) [6], and a set of 390 subtelomeric clones (generated in the course of the EU initiative COSTB19: Molecular cytogenetics of solid tumours). Detailed protocols are available at the following webpage: http://www.molgen.mpg.de/~abt_rop/molecular_cytogenetics/Protocols.html. Array CGH was performed as described previously [11] and 33028 BACs were included in the analysis. The log2ratio of test to reference was calculated and plotted according to chromosomal position of the clones. Copy number gains and losses were determined by a conservative threshold of 0.3 and −0.3, respectively. Profile deviations consisting of three or more neighbouring BAC clones were considered as genomic aberrations and were further evaluated by FISH, unless they coincided with a published variant as listed in the Database of Genomic Variants (http://projects.tcag.ca/variation/; version Oct. 11, 2006). For visualizing the content of low copy repeats in the ratio plots (Fig. 2a,b), each BAC clone was classified into one out of seven categories and colour-coded as described previously [2].

a Array CGH profile of chromosome 15 visualized using CGHPRO software. Each spot represents one BAC clone on the array. The red and green lines indicate the log2ratio thresholds −0.3 (loss) and 0.3 (gain), respectively. Note: the aberration close to the centromere on 15q11.2 constitutes a known CNV indicated by the turquoise colour of the spots. b Detailed view of the interstitial deletion on chromosome 15q24. The microdeletion is flanked by low-copy repeats indicated by the turquoise colour of the spots. Horizontal bars represent DNA copy number variants as listed in the Database of Genomic Variants (Dec. 2005)
Results
Standard karyotyping of G-banded chromosomes did not reveal any chromosomal abnormalities. According to the FISH analyses there was no evidence for a deletion of the Prader-Willi syndrome region or a microdeletion 22q11.2. Therefore, we performed array CGH analysis with the patient’s genomic DNA using a submegabase resolution whole genome tiling path array to detect microdeletions or duplications. A submicroscopic interstitial deletion on chromosome 15q24.1-q24.3 represented by 33 BAC clones was detected (Fig. 2). The size of the deletion is approximately 3.7 Mb extending from 72.2 Mb to 75.9 Mb (Ensembl release 43; Feb. 2007). According to the array CGH data the two breakpoints are located between RP11–247C02 and RP11–672A20 (proximal breakpoint) and RP11–758J16 and RP11–745I11 (distal breakpoint). Both breakpoints were found to be enriched for low copy repeats, which, according to the Segmental Duplication Database (http://humanparalogy.gs.washington.edu/), share a 49.9 kb segment with 95% sequence similarity (no .9417), and the proximal breakpoint coincided with published DNA copy number variants. Given this high sequence similarity the deletion most likely emerged through nonallelic homologous recombination (NAHR) of the two breakpoint flanking low copy repeats (LCRs), a mechanism associated with genomic rearrangements [16]. The deleted region comprises 39 annotated genes (based on v39, June 2006, Ensembl genome browser; (http://www.ensembl.org). In addition, a deletion on 15q11.2 was observed which represents a CNV listed in the Database of Genomic Variants and was therefore considered as a benign variant without pathological relevance.
For verification of array CGH data by FISH we used a set of BAC clones mapping to 15q24. As expected from the array data (log2ratio < −0.3) only one signal on the patient’s metaphases was detected for BAC clones RP11–414J4 and RP11–94P14 (data not shown). FISH on metaphase chromosomes of the patient’s parents and his brother detected two signals on chromosome 15 for all probes investigated and karyotypes were normal (data not shown). Thus, the aberration observed in our patient occurred de novo.
Discussion
Recently, Sharp et al. described a recurrent microdeletion of 15q24 in four cases ranging in size from 1.7–3.9 Mb characterised by molecular cytogenetic techniques [21]. The minimal deletion region was delineated to be 1.7 Mb in size (72.15–73.85 Mb). The patient presented here constitutes an interstitial microdeletion at 15q24 which shares common proximal (BP1) and distal breakpoints (BP3) with cases IMR349 and C45/06 of the publication by Sharp et al. [21] (Fig. 3). Since all deletions occurred in the maternal lineage the possibility of an underlying imprinting effect was pointed out [21]. The parental origin of the deletion was not analysed in our patient. A clinical comparison of our patient to these patients from the literature is shown in Table 1. The phenotype between patients with microdeletion 15q24 is variable; however, our patient shares all of the major features described for 15q24 microdeletion syndrome by Sharp et al. [21], i.e. unusual facial features (high frontal hairline, broad medial eyebrows, downslanted palpebral fissures, and long philtrum), developmental delay, digital abnormalities, genital abnormalities, and loose connective tissue (manifestation: joint laxity, inguinal hernia). However, our patient does not present with microcephaly (3/4), prenatal and postnatal growth deficiency (3/4), hearing problems (2/4), or bowel atresia (2/4).

Schematic representation of 15q24 region. The three recurrent breakpoints as delineated by Sharp et al. [21] as well as the deletion sizes of the presented case and published cases are indicated. The critical region is located between the recurrent breakpoints BP1 and BP2
Among the deleted genes are several coding for enzymes, e.g. mannose phosphate isomerase (MPI), alpha-mannosidase 2C1 (MAN2C1), and alpha-subunit of electron transfer flavoprotein (ETFA), as well as the cytochrome P450 side-chain cleavage enzyme (P450scc) which catalyzes the first step in steroidogenesis. Several of these enzymes are involved in the synthesis, export, and degradation of glycoproteins. Mutations in the MPI gene (MIM 154550) cause congenital disorder of glycosylation type Ib (CDG Ib) [9, 19]. CDG Ib is an autosomal recessive disorder showing abnormalities in the glycosylation of glycoproteins. Therefore, the haploinsufficiency is asymptomatic. MAN2C1 (MIM 154580) is involved in the degradation of oligomannosides derived from dolichol intermediates, the degradation of newly synthesized glycoproteins, and in the processing of free oligosaccharides that are formed in the cytosol [23]. Secretory carrier membrane proteins (SCAMP2, SCAMP5) are a family of post-Golgi and Golgi membrane proteins which have been implicated in vesicular trafficking. SCAMPs interact with NHE7 (Na+/H+ exchanger) and participate in the shuttling and retrieval of NHE7 from peripheral recycling endosomes to the trans-Golgi network [15]. Mucopolysaccharidoses and CDG syndrome were ruled out biochemically. However, the second allele was not analysed for point mutations. Thus, we cannot exclude uncovering of a recessive condition which could result in a homozygous loss of alleles which leads to deficiency of one of the other enzymes in this region.
The cholesterol side chain cleavage enzyme P450scc is responsible for the conversion of cholesterol to pregnenolone in mitochondria [17]. This is the first and rate-limiting step in steroidogenesis [17]. So far, only one patient with a heterozygous mutation in P450scc has been described [24]. This patient had a late-onset form of congenital lipoid adrenal hyperplasia without the characteristic enlargement of the adrenals. A complete absence of P450scc activity causes congenital adrenal insufficiency, complete 46,XY sex reversal, and severe adrenal failure [8, 10]. Congenital adrenal hyperplasia is a severe disorder with an impairment of adrenal and gonadal steroid synthesis. Although, we did not observe such a severe phenotype, and current steroid levels are normal in our patient. The haploinsufficiency of P450scc might contribute to the genital abnormalities in our patient and other affected male patients (microphallus with small scrotum, undescended testes, hypospadias) with deletions spanning the P450scc locus on 15q24.1 [4, 20].
In summary, the patient presented here constitutes an interstitial microdeletion at 15q24 and represents another case of the recently described 15q24 microdeletion syndrome. This is another example of the impact of array CGH as a diagnostic tool in clinical medicine [5].
Abbreviations
- Array CGH:
-
microarray-based comparative genomic hybridization
- BAC:
-
bacterial artificial chromosome
- CNV:
-
copy number variation
- FISH:
-
fluorescence in situ hybridization
- P:
-
percentile
References
Bettelheim D, Hengstschlager M, Drahonsky R, Eppel W, Bernaschek G (1998) Two cases of prenatally diagnosed diaphragmatic hernia accompanied by the same undescribed chromosomal deletion (15q24 de novo). Clin Genet 53(4):319–320
Chen W, Erdogan F, Ropers H, Lenzner S, Ullmann R (2005) CGHPRO - a comprehensive data analysis tool for array CGH. BMC Bioinformatics 6:85
Clark RD (1984) Del(15)(q22q24) syndrome with Potter sequence. Am J Med Genet 19(4):703–705
Cushman LJ, Torres-Martinez W, Cherry AM, Manning MA, Abdul-Rahman O, Anderson CE, Punnett HH, Thurston VC, Sweeney D, Vance GH (2005) A report of three patients with an interstitial deletion of chromosome 15q24. Am J Med Genet A 137(1):65–71
Denayer E, Legius E (2007) What’s new in the neuro-cardio-facial-cutaneous syndromes? Eur J Pediatr 166(11):1091–1098 DOI 10.1007/s00431-007-0535-7
Fiegler H, Carr P, Douglas EJ, Burford DC, Hunt S, Scott CE, Smith J, Vetrie D, Gorman P, Tomlinson IP, Carter NP (2003) DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer 36(4):361–374
Formiga LD, Poenaru L, Couronne F, Flori E, Eibel JL, Deminatti MM, Savary JB, Lai JL, Gilgenkrantz S, Pierson M (1988) Interstitial deletion of chromosome 15: two cases. Hum Genet 80(4):401–404
Hiort O, Holterhus PM, Werner R, Marschke C, Hoppe U, Partsch CJ, Riepe FG, Achermann JC, Struve D (2005) Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY sex reversal, and severe adrenal failure. J Clin Endocrinol Metab 90(1):538–541
Jaeken J, Matthijs G (2001) Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2:129–151
Katsumata N, Ohtake M, Hojo T, Ogawa E, Hara T, Sato N, Tanaka T (2002) Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J Clin Endocrinol Metab 87(8):3808–3813
Klopocki E, Neumann LM, Tonnies H, Ropers HH, Mundlos S, Ullmann R (2006) Ulnar-mammary syndrome with dysmorphic facies and mental retardation caused by a novel 1.28 Mb deletion encompassing the TBX3 gene. Eur J Hum Genet 14:1274–1279
Koolen D, Vissers L, Pfundt R, de Leeuw N, Knight S, Regan R, Kooy R, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid B, Schoumans J, Knoers N, van Kessel A, Sistermans E, Veltman J, Brunner H, de Vries B (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38(9):999–1001
Kristoffersson U, Heim S, Mandahl N, Sundkvist L, Szelest J, Hagerstrand I (1987) Monosomy and trisomy of 15q24–qter in a family with a translocation t(6;15)(p25;q24). Clin Genet 32(3):169–171
Krzywinski M, Bosdet I, Smailus D, Chiu R, Mathewson C, Wye N, Barber S, Brown-John M, Chan S, Chand S, Cloutier A, Girn N, Lee D, Masson A, Mayo M, Olson T, Pandoh P, Prabhu AL, Schoenmakers E, Tsai M, Albertson D, Lam W, Choy CO, Osoegawa K, Zhao S, de Jong PJ, Schein J, Jones S, Marra MA (2004) A set of BAC clones spanning the human genome. Nucleic Acids Res 32(12):3651–3660
Lin P, Williams W, Luu Y, Molday R, Orlowski J, Numata M (2005) Secretory carrier membrane proteins interact and regulate trafficking of the organellar (Na+, K+)/H+ exchanger NHE7. J Cell Sci 118:1885–1897
Lupski JR, Stankiewicz P (2005) Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 1(6):e49
Miller W (1988) Molecular biology of steroid hormone synthesis. Endocrinology Reviews 9:295–318
Osoegawa K, Mammoser AG, Wu C, Frengen E, Zeng C, Catanese JJ, de Jong PJ (2001) A bacterial artificial chromosome library for sequencing the complete human genome. Genome Research 11:483–496
Schollen E, Dorland L, de Koning TJ, Van Diggelen OP, Huijmans JG, Marquardt T, Babovic-Vuksanovic D, Patterson M, Imtiaz F, Winchester B, Adamowicz M, Pronicka E, Freeze H, Matthijs G (2000) Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Hum Mutat 16(3):247–252
Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE (2006) Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38(9):1038–1042
Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, Striano P, Coppola A, Regan R, Price SM, Knoers NV, Eis PS, Brunner HG, Hennekam RC, Knight SJ, de Vries BBA, Zuffardi O, Eichler EE (2007) Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet 16(5):567–572
Spruijt L, Engelen JJ, Bruinen-Smeijsters IP, Albrechts JC, Schrander J, Schrander-Stumpel CT (2004) A patient with a de novo 15q24q26.1 interstitial deletion, developmental delay, mild dysmorphism, and very blue irises. Am J Med Genet A 129(3):312–315
Suzuki T, Hara I, Nakano M, Shigeta M, Nakagawa T, Kondo A, Funakoshi Y, Taniguchi N (2006) Man2C1, an alpha-mannosidase, is involved in the trimming of free oligosaccharides in the cytosol. Biochem J 400(1):33–41
Tajima T, Fujieda K, Kouda N, Nakae J, Miller W (2001) Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocrinol Metab 86:3820–3825
Acknowlegdements
The authors thank Fabienne Trotier, Karen Stout-Weider, and Milena Vetter for their excellent technical assistance in the array CGH and FISH experiments. We are also grateful to the patient and his family for their collaboration in this study. Additionally, we thank Pieter de Jong and the BACPAC Resources Centre for providing the DNA of the human 32k Re-array set, the COST B19 Action for the clones of the subtelomeric set, Nigel Carter and the Mapping Core and Map Finishing groups of the Wellcome Trust Sanger Institute for initial clone supply and verification of the 1Mb array, as well as Claus Hultschig for spotting the arrays.
Author information
Authors and Affiliations
Corresponding author
Additional information
This project was supported by the European Fund for Regional Development.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Klopocki, E., Graul-Neumann, L.M., Grieben, U. et al. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH. Eur J Pediatr 167, 903–908 (2008). https://doi.org/10.1007/s00431-007-0616-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-007-0616-7