
Abstract
The role of adaptive immune responses in the control of hepatitis B virus (HBV) infection is well accepted. The contribution of innate immune responses to the viral control is recognized but yet not fully understood. Toll-like receptors (TLRs) sense pathogen-associated molecule patterns and activate antiviral mechanisms including the intracellular antiviral pathways and the production of antiviral effectors like interferons (IFNs) and pro-inflammatory cytokines. Activation of the TLR3 pathway and the production of IFN-β represent one of the major mechanisms leading to the suppression of HBV replication in the liver, as shown in different in vitro and in vivo models. TLR4 signaling and TLR2 signaling result in the activation of intracellular pathways including MAPK and PI-3 K/Akt in hepatocytes and reduce HBV replication in an IFN-independent manner. HBV is able to counteract the actions of TLR3 and TLR2/4 through downregulation of TLR expression and attenuation of the cellular signaling pathways. Thus, TLR ligands are promising candidates as immunomodulators and therapeutics for the treatment of chronic HBV infection. Specific antiviral treatment against HBV could recover the TLR functions in chronic HBV infection and increase the effectiveness of therapeutic approaches based on TLR activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the estimation of World Health Organisation, there are about 240 million people who are chronically infected with hepatitis B virus (HBV). The chronic HBV infection is one of the major causes of hepatocellular carcinoma and liver cirrhosis. There is a large body of evidence for the essential role of cell-mediated immune responses for viral clearance in acute HBV infection. Patients with chronic HBV infection fail to develop adequate HBV-specific immune responses [1]. Standard treatment regimens with pegylated interferon (IFN)-α and nucleoside/nucleotide analogs are used for therapy of chronic hepatitis B but are only partially successful. Several recent studies indicated the possibility of stimulating specific immune responses against HBV in chronically infected patients. The recent approaches were summarized in previous reviews of our group and others and in the present issue [2–5]. A number of therapeutic vaccination trials using conventional HBV vaccines failed to demonstrate the effectiveness in terms of the induction of HBV-specific immune responses and suppression of HBV replication in chronic HBV carriers [4, 5]. New approaches based on DNA vaccines or anti-HBs antibody-HBs immune complex are now being tested in clinical trials [6–8]. As a principle recognized on the basis of available information, a combined strategy including antiviral treatment and immunomodulation will be needed to stimulate the full range of immune responses to achieve effective control over HBV infection. Important aspects of the human HBV infection have been studied with a genetically closely related virus of Hepadnaviridae, woodchuck hepatitis virus (WHV), which infects a North American rodent, the woodchuck. In the woodchuck model, combinations of antiviral treatment and therapeutic vaccinations led to the induction of specific T cell and B cell responses to WHV antigens and sustained suppression of WHV replication in some individual animals [2, 4, 9, 10]. Stimulation of innate immune responses may further improve the immunotherapeutic effect of combination strategies against the hepadnaviral infection.
Toll-like receptor (TLR) system
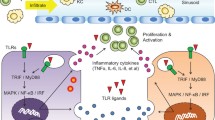
The significance of the innate immune response as a defense against microbial infections and its link to the adaptive immune responses has been recognized during the past years. Toll-like receptors (TLRs) are a group of highly conserved molecules that play a critical role in the recognition of pathogen-associated molecular patterns (PAMPs) and in the activation of innate immune responses to infectious agents [11]. TLRs are structurally characterized by an ectodomain composed of leucine-rich repeats for binding and recognition of PAMPs and a cytoplasmic domain homologous to the cytoplasmic region of the interleukin (IL)-1 receptor, known as the TIR domain, for downstream signaling [12]. TLR ligands are natural macromolecular components derived from pathogens and may be composed of lipids, lipopeptides, proteins, and nucleic acids. Some synthetic small molecules could mimic TLR ligands and activate TLR-mediated cellular signaling. A subgroup within the TLR family including TLR3, TLR7, TLR8, and TLR9 is localized in endosomes and recognizes nucleic acids such as viral DNA or RNA. The other subgroup of surface-expressed TLR1, TLR2, TLR4, TLR5, and TLR6 recognizes extracellular bacterial and fungal cell wall components, as well as some viral proteins [13, 14]. Binding of TLR agonists to their receptors initiates the activation of complex networks of intracellular signal transduction pathways to coordinate the inflammatory response. Conformational changes and dimerization of TLRs occur upon binding with ligands. The important components of these signaling networks are the adaptor proteins and several protein kinases including ERK, JNK, p38 MAP kinase, and PI-3 k kinase, and the transcription factors IRF3/5/7, nuclear factor kappa B (NF-κB), and AP-1. The activation of these transcription factors leads to the induction of type I IFNs, pro-inflammatory cytokines, or co-stimulatory molecules, which are involved in antiviral responses [15, 16]. The crucial adaptor proteins including myeloid differentiation primary-response protein 88 (MyD88), used by nearly all TLRs except TLR3, TIR domain-containing adaptor protein (TIRAP), TIR domain-containing adaptor protein inducing interferon (IFN)-β (TRIF), and TRIF-related adaptor molecule (TRAM) are recruited [17]. TLR4 is unique among TLRs being able to activate two distinct signaling pathways, TIRAP/MyD88 and TRAM/TRIF [17]. The MyD88-dependent pathway leads to the activation of downstream signal transduction involving IL-1R-associated kinases (IRAKs), tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6), transforming growth factor (TGF)-β-activated kinase (TAK1), and the inhibitor of nuclear factor-κB (IκB) kinase complex. Through the NF-κB and activating protein 1 (AP1), the MyD88-dependent TLR activation results in the production of pro-inflammatory cytokines IL-6, IL-10, IL-12, and TNF-α. In contrast, the TRIF-dependent pathway leads to the activation of IFN regulatory factors (IRFs) and production of type I IFNs [15, 17]. Exceptionally, plasmacytoid dendritic cells produce type I IFN after TLR7 and TLR9 activation via the MyD88-IRF7-dependent pathway [18]. Figure 1 schematically depicts human TLR signaling pathways.

TLR signaling. Upon the activation of TLRs by their respective ligands, the adaptor molecules MYD88, TIRAP, TRIF, and TRAM are recruited and further activate the kinases TAK1, MAPKs, TRAF3, TBK1, and IKKs, resulting in nuclear translocation of transcriptions factors AP-1, NF-κB, IRF3, or IRF7, and subsequent transcription of IFNs and pro-inflammatory cytokines
Recognition of HBV by host cells
There is accumulating evidence that the innate branch of the host immune system plays an important role in the control of HBV infection [19–21]. The previous studies in chimpanzees and in patients showed that HBV infection does not lead to a measurable response involving type I IFNs. Wieland et al. [22] investigated the transcriptome of the liver in three chimpanzees during the course of acute HBV infection. Their analysis focused on two diverse groups of cellular genes: Those in the early phase are associated with the innate immune response, and those in the late phase are associated with the adaptive immune response that terminates infection. They demonstrated that HBV does not induce any genes during entry and expansion, leading the authors to suggest that HBV is a “stealth virus” in the early phase of infection. By contrast, a large number of IFN-γ-regulated genes are expressed in the liver during viral clearance. The upregulation of IFN-γ-regulated genes in the liver results from the adaptive T cell response as specific T cells infiltrating the liver are major producers of IFN-γ [22]. Thus, HBV infection strongly differs from other viral infections like HCV infection in the early phase, as HCV induces a strong IFN-α response in chimpanzees [23]. Dunn et al. [24] measured type I IFN production in patients with acute HBV infection and found only a low level of type I IFN not higher than those found in healthy controls. Similarly, IL-15 and IFN-λ1 were not induced during peak viraemia. In contrast, IL-10 is induced at the early stages of acute HBV. The lack of early IFN response in vivo during acute HBV infection does not necessarily exclude the triggering of host IFN responses by HBV. HBV infection may be initiated only with few viral particles and may not induce a host response at the initial phase of infection that is measurable by gene array technology or cytokine detection. In addition, HBV may inhibit host IFN responses by specific mechanisms as described below. Experimental data are available, indicating that HBV interacts with the host innate immune system but is able to inhibit host responses. It has been shown that HBV interacts with hepatic non-parenchymal cells (NPCs) and induces the production of IL-6 [25], though it is not clear how hepatic NPCs sense HBV. Within 3 h, these cells release inflammatory cytokines including IL-1β, IL-6, IL-8, and TNF-α without inducing an IFN response. IL-6 is able in turn to inhibit the expression of hepatocyte nuclear factor (HNF) 1α and HNF 4α, two transcription factors essential for HBV gene expression and replication. However, relatively high doses of HBV particles are usually used in such experiments to induce measurable responses of host cells. Future in vivo experiments are required to compare and verify these findings.
TLR3 and HBV
TLR3 activation results in the production of type I IFNs in different cell types. IFN-β has been identified as a major antiviral factor produced by NPCs in response to TLR3 [26]. Wieland et al. [27] showed that TLR3 ligand poly I:C induces intrahepatic IFN-β production and inhibits HBV replication by non-cytolytic mechanisms that either destabilize pregenomic (pg)RNA-containing capsids or prevent their assembly. Isogawa et al. [28] tested the ability of different TLR ligands to inhibit HBV replication in the HBV transgenic mouse model. Consistently, a single-dose injection of TLR3, TLR4, TLR5, TLR7, and TLR9 ligands suppressed HBV replication in the liver in an IFN-α/β-dependent manner. In a doxycycline (dox)-inducible HBV replication system, IFN-β pretreatment prevents the production of replication-competent pgRNA-containing capsids but does not change the turnover rate of preformed HBV RNA-containing capsids [29]. Apparently, the formation of replication-competent HBV capsids is one of the major targets of IFN-mediated antiviral actions. A great number of cellular IFN-stimulated genes (ISGs) are activated by IFNs and may inhibit the different steps of the HBV life cycle [30]. A recent publication suggests that HBV covalently closed circular (ccc) DNA could be degraded by the action of APOBEC3A and 3B cytidine deaminases [31]. These findings partly explain the therapeutic effect of IFN-α in patients with chronic HBV infection. IFN-α is widely used to treat chronic HBV infection and can lead to sustained decrease of HBsAg and virus clearance in about 30 % of chronically HBV-infected patients. In addition, type I IFNs may modulate specific immune responses to HBV, resulting in HBe seroconversion or complete control of HBV infection.
TLR3 activation of hepatic NPCs could lead to IFN-β production and HBV inhibition in in vitro [26, 32, 33]. Upon poly I:C stimulation, hepatic NPCs such as Kupffer cells (KC) and liver sinusoid endothelial cells (LSEC) release antiviral cytokines which are able to inhibit HBV replication in a co-culture model utilizing HBV-Met cells that contains an integrated HBV genome [34]. Blocking with specific antibodies to type I and II IFNs identified IFN-β as the major anti-HBV factor produced by poly I:C-treated NPCs [26]. While HBV DNA replicative intermediates were efficiently suppressed, viral mRNAs as well as secretion of HBsAg and HBeAg remained largely unchanged. Importantly, screening of different TLR ligands demonstrated that hepatic NPCs show a significant production of IFN-β only in response to TLR3 stimulation (and a lower extent to TLR4 stimulation, see below). Therefore, TLR3-mediated response and IFN-β production in the liver may contribute to the control of pathogens including HBV in a unique way.
Several studies suggest that HBV is able to inhibit pattern recognition receptor (PRR) and IFN signaling. HBV surface and “e” antigen (HBsAg, HBeAg) and HBV particles could inhibit the activation of NPCs by TLR3 ligands [35]. Co-culture of hepatic NPCs with HBV-Met cell supernatants, HBsAg, HBeAg as well as HBV virions results in abrogation of TLR-induced antiviral activity, correlating with decreased activation of IRF3, NF-κB, and ERK1/2 in NPCs. Our most recent data suggest that HBsAg may trigger IL10 production on hepatic cells and thereby attenuates the TLR3-mediated activation of NPCs [33]. This is consistent with the recent publication that HBsAg induces TNF-α and IL-10 production by monocytes which leads to downregulation of TLR9 expression on pDCs, thereby inhibiting IFN-α production by pDCs [36]. At high amounts of HBV, even TLR-induced expression of TNF-α and IL-6 in NPCs was suppressed. In addition, many studies provide evidence that HBV polymerase may counteract the innate responses at two or more steps: (1) HBV polymerase is able to inhibit the IRF3 activation by interacting with RNA helicase DDX3 in hepatoma cells [37, 38]; (2) HBV polymerase is able to block IFN signaling by inhibition of PKC-δ-mediated phosphorylation of stat 1 and importin-dependent translocation of stat 1/stat 2/IRF9 complex [39, 40]. HBx protein was reported to promote the decay of mitochondrial antiviral signaling protein (MAVS), the adaptor of RIG-I, and MDA5 receptors [41, 42]. However, the relevance of these findings for the human HBV infection remains to be defined.
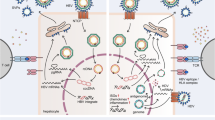
TLR3-mediated functions are impaired in patients with chronic HBV infection and may recover partially under successful antiviral treatment [43]. In the woodchuck model, PBMCs from animals with chronic WHV infection show reduced responses to poly I:C stimulation [44]. Taken together, the interaction of HBV or the molecular components of HBV with the innate immune system is complex, leading both to activation and inhibition of host innate responses. Figure 2 depicts the interaction of TLR3 signaling pathway with HBV in a schematic way.

Interaction of HBV and TLR3. TLR3 activation in hepatic NPCs leads to the production of IFN-β and subsequently the upregulation of ISGs in hepatocytes. Antiviral ISGs like MxA and IFIT1/2 inhibit HBV replication at the transcriptional and posttranscriptional steps. HBV virions and proteins are able to suppress TLR3 signaling and block IFN-β production. HBV polymerase could interfere with IRF3 action and block the nuclear translocation of Stat1/2
TLR2/4 and HBV
Unlike to TLR3, TLR4 activation by lipopolysaccharide (LPS) leads to low IFN-β production only in KCs but not in LSECs and hepatocytes [26]. However, TLR4-activated KCs release other yet undefined antiviral factors, inhibiting HBV replication in HBV-Met cells. In contrast to TLR3 ligands, Zhang et al. [45] demonstrated that activation of cellular pathways by TLR4 ligands leads to inhibition of hepadnaviral replication. In the model of WHV-infected primary hepatocytes (PWHs), LPS stimulation led to a pronounced reduction of WHV replication intermediates without a significant IFN induction, while poly I:C transfection resulted in the IFN production and a highly increased expression of antiviral genes in PWHs, but only slight inhibitory effect on WHV replication. LPS was able to activate NF-κB, MAPK, and PI-3 k/Akt pathways in PWHs. The inhibitors of MAPK-ERK and PI-3 k/Akt pathways, but not those of IFN signaling pathways, block the antiviral effect of LPS, indicating that IFN-independent pathways which activated by LPS are able to downregulate hepadnaviral replication in hepatocytes [45].
TLR2 and TLR4 share the cellular MyD88-dependent signaling pathway in mammalian cells (Fig. 3). Consequently, TLR2 and TLR4 mediate the activation of the same signaling pathways downstream of MyD88, including NF-κB, MAPK, and PI-3 k/Akt pathways. Similarly, TLR2 is able to inhibit HBV or WHV replication in human hepatoma cells or PWHs [46, 47]. Again, the antiviral action of TLR2 is dependent on the presence of adaptor molecules like TAK1, IRAK1/4, and TRAF6 and the downstream MAPK and PI-3 k/Akt pathways [47]. Silencing of the expression of adaptor molecules or blocking the MAPK and PI-3 k/Akt pathways with chemical inhibitors significantly enhanced HBV replication. In the HBV transgenic mouse model, the injection of a single dose of TLR2 ligands reduced HBV replication in the liver but not as effective as IFN-inducing ligands [28]. It was not examined whether TLR2 ligands also activate MAPK and PI-3 k/Akt pathways in vivo and thereby exert the antiviral action.

Interaction of HBV and TLR2/4. TLR2/4 activation in hepatocytes inhibits HBV replication in an IFN-independent manner but requires the participation of intracellular signaling pathways like MAPK pathway. TLR4 stimulation in KCs leads to the production of IFN-β and an unknown antiviral effector which inhibits HBV replication in hepatocytes. HBV downregulates TLR2/4 expression during chronic HBV infection. HBsAg and HBeAg are able to block TLR2 signaling at different steps, preventing the production of pro-inflammatory cytokines
TLR2 activation and TLR4 activation lead to the production of pro-inflammatory cytokines IL6 and TNF-α in hepatic NPCs and hepatocytes [32, 45, 47, 48]. Though the antiviral effect of TLR2 and TLR4 ligands does not directly depend on the production of pro-inflammatory cytokines, IL6 and TNF-α have been shown to inhibit HBV replication in primary hepatocytes [25, 49]. Xu et al. [49] explored the Tupaia model to investigate the effect of TNF-α on HBV infection. Stimulation of HBV-infected primary Tupaia hepatocytes with recombinant Tupaia TNF-α led to viral suppression, while covalently closed circular DNA and viral RNA were still detectable, leading to the conclusion that TNF-α may also contribute to control HBV infection.
Obviously, HBV developed measures to counteract the antiviral functions mediated by TLR2. Using hepatocytes and KCs isolated from liver biopsies of patients with CHB, Visvanathan et al. [50] showed significantly decreased TLR2 expression on hepatocytes, KCs, and peripheral monocytes in patients with HBeAg-positive CHB in comparison with HBeAg-negative CHB and controls. The level of TLR4 expression did not significantly differ between these groups. Hepatic cell lines harboring a recombinant baculovirus encoding HBV significantly reduced TNF-α expression as well as phospho-p38 kinase expression in the presence of HBeAg. In the absence of HBeAg, HBV replication was associated with upregulation of the TLR2 pathway resulting in increased TNF-α expression [50]. HBeAg was found to co-localize with Toll/IL-1 receptor (TIR)-containing proteins TRAM, Mal, and TLR2, interact with TIR proteins Mal and TRAM, and disrupt the homotypic TIR–TIR interaction. Consequently, HBeAg suppressed TIR-mediated activation of the inflammatory transcription factors, NF-κB, and interferon-β promoter activity [51]. Consistently, TLR2 expression was found to be significantly suppressed in PBMCs from chronically HBV-infected patients and in woodchuck liver tissue and PBMCs if chronically infected with WHV [47, 52]. Previously, Wu et al. [39] demonstrated that HBV blocks the MyD88 expression, the central adaptor molecule in TLR-mediated innate immune responses, by an antagonistic activity of the terminal protein (TP) domain of the HBV polymerase. It could be shown that HBV polymerase is able to block the nuclear translocation of stat 1 thus representing a general inhibitor of IFN signaling [40] and IFN-inducible MyD88 expression.
Activation of innate immunity is a prerequisite for proper adaptive immune responses. As an example, TLR2 is expressed widely such on antigen-presenting cells (APC), endothelial and epithelial cells as well as on T-lymphocytes on which it acts as a costimulatory molecule. Wu et al. [32] found previously that TLR2 ligands could trigger the expression of costimulatory molecules on hepatic NPCs. LSECs are unique organ-resident antigen-presenting cells capable of antigen cross-presentation and reported to prime naïve CD8+ T cells to memory cells at non-inflammatory conditions [53]. Under certain conditions, LSECs could also directly promote T cell immunity [54]. Recently, we examined functional maturation of LSECs by TLR ligand stimulation, demonstrating that pretreatment of LSECs with TLR1/2 ligand but not TLR3 and TLR4 ligands reverts their suppressive properties to induce specific T cell immunity [55]. IL-12 was identified to be one essential mediator for LSEC-mediated CD8+ T cell immunity, which was produced at a low level but sustainably after TLR2 stimulation. Our findings suggest that TLR2 activation has a great impact on T cell immunity in the liver and may be used to stimulate specific immune responses to persistent infection of HBV and HCV.
On the other hand, Wang et al. [56] could show that HBsAg inhibits TLR2-mediated stimulation of human PBMCs and IL12 production. In the presence of HBsAg, both Pam3CSK4-triggered IL-12p40 mRNA expression and IL-12 production in phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 macrophages are reduced in a dose-dependent manner, while the production of IL-1β, IL-6, IL-8, IL-10, and TNF-α is not affected. The presence of HBsAg inhibits the TLR2-mediated activation of NF-κB and MAPK signaling, by selective impairment of JNK-1/2 and c-Jun phosphorylation. Thus, HBsAg likely interferes with the initiation of adaptive immune responses by selective inhibition of TLR2-stimulated IL-12 production in monocytes.
Therapeutic approaches
The findings mentioned above suggest that TLR ligands may be used for therapeutic approaches against chronic viral infection. However, only few trials using TLR ligands for therapies against viral infections have been carried out until now [57]. CpG oligonucleotides (ODN), ligands of TLR9, have been considered as promising candidates. A large number of new reagents that are potentially suitable as immunomodulators and therapeutics the sequence could be generated by modifications of CpG ODN [58, 59]. However, a candidate CpG ODN has been tested in clinical trials and the woodchuck model for treatment of chronic hepatitis C and B but failed to show sufficient therapeutic effect if applied alone. TLR3 and TLR4 ligands are not tested in clinical trials yet. Recently, TLR7 ligand GS-9620 has been examined for its antiviral effect in the woodchuck and chimpanzee models. Interestingly, a 4-week treatment with GS-9620 resulted in a sustained, marked reduction of serum WHV DNA and WHV surface antigen (WHsAg) levels and in the induction of anti-WHs antibody response, as well as a markedly decreased incidence of hepatocellular carcinoma in chronic WHV-infected woodchucks [60]. In chimpanzees, GS-9620 induced an increase of serum IFN-α in a dose-dependent manner and triggered the ISG expression in PBMCs and the liver. A reduction of HBV viral load and serum HBsAg was observed in three chronically HBV-infected chimpanzees treated with GS-9620 [61]. TLR7/8 ligands are promising drug candidates if their toxicity could be reduced to a tolerable range.
A potential use of TLR ligands as adjuvants for therapeutic vaccines has been considered for long time. The rational design of specific TLR agonists may increase potency and tolerability of new adjuvants and provides the opportunity to meet the stringent safety criteria for new vaccine formulation [62]. Two improved adult HBV vaccines Fendrix and Supervax using TLR4 agonists as adjuvant are now on the market [63, 64]. Monophosphoryl lipid A (MPLA), a chemically modified derivative of the lipid A moiety of LPS, is considerably less toxic but has similar immunostimulatory activity [65]. MPLA-formulated hepatitis B vaccines elicit protective anti-HBs antibody titers with only two injections instead of three [64]. A class B CpG ODN called 1018 ISS in combination with recombinant HBsAg has been tested in a Phase III clinical trial for persons older than 40 years of age. This vaccine formulation increases seroprotection rates to 100 %, compared with a rate of only 64 % in the alum-adjuvanted rHBsAg group [66]. Such a vaccine formulation may be used for patients with impaired immune system, as it is more effective in the hypo-responsive population than conventional HBV vaccines [67].
Perspectives
Based on the current knowledge, TLR-mediated innate responses may not control HBV infection alone. Though stimulation with TLR ligands reduces HBV replication in hepatocytes, the TLR-mediated antiviral action against HBV is far less efficient than those achieved by nucleoside analogs. The innate immune system is known to respond fast and aimed to slow down viral spread in primary infections; thus, the link from innate to adaptive immune responses may be more important for the control of viral infection. The innate immune system plays a pivotal role for the regulation of adaptive immunity [68]. Recently, it was shown that poly I:C treatment leads to HBV clearance in hydrodynamic injection mouse model [69]. In this model, HBV clearance required IFN-α and IFN-γ, indicating a complex mode of poly I:C-triggered action. CXCR3 was also essential for HBV clearance after poly I:C injection, apparently responsible for the recruitment of T cells. Other studies conducted by several groups have shown that TLR2 is expressed on activated and memory CD4+ and CD8+ T cells and serves as co-stimulatory molecule to enhance their proliferation, survival, and functions [70–73]. TLR2 agonists stimulate activated T cells thus promoting their proliferation and differentiation in vitro and in vivo. Consistently, TLR2 ligand Pam3CSK4 application in vivo with transferred tumor antigen (Ag)-specific CD8+ T cells results in enhanced therapeutic efficacy of these CD8+ T cells in tumor models [74, 75]. Moreover, covalent linkage of TLR2 ligand Pam3CSK4 or Pam2CSK4 with peptides representing CD8+ T cell or B cell epitopes efficiently primed respective specific CD8+ T cell or B cell immune responses in vivo [76–79]. It was shown that TLR2 engagement on CD8+ T cells increased T-bet transcription in a MyD88-Akt-mTOR- and protein kinase C-dependent manner [71]. The molecular mechanisms underlying TLR2-mediated T cell proliferation and functional differentiation in HBV infection need in-depth analysis. Thus, future research should not only investigate the direct antiviral effect of TLR-mediated action but also analyze and optimize the connection of innate and adaptive immune responses. In the specific case of TLR2, it is desirable to identify specific markers expressed on CD8+ T cells after TLR2 stimulation. Such markers may facilitate future analysis of CD8+ T cells in vitro and vivo and understanding the inhibitory action of HBV on TLR2 co-stimulation of CD8+ T cells. These approaches could also be extended to studies about other TLRs.
References
Bertoletti A, Gehring AJ (2006) The immune response during hepatitis B virus infection. J Gen Virol 87:1439–1449
Roggendorf M, Lu M (2005) Woodchuck hepatitis virus. In: Thomas TH, Zuckermann A, Lemon A (eds) Viral hepatitis, 3rd edn. Blackwell Publishing Ltd, Oxford, pp 210–224
Inchauspé G, Michel ML (2007) Vaccines and immunotherapies against hepatitis B and hepatitis C viruses. J Viral Hepat 14(Suppl 1):97–103
Lu M, Menne S, Yang D, Xu Y, Roggendorf M (2007) Immunomodulation as an option for the treatment of chronic hepatitis B virus infection: preclinical studies in the woodchuck model. Expert Opin Investig Drugs 16:787–801
Michel ML, Tiollais P (2010) Hepatitis B vaccines: protective efficacy and therapeutic potential. Pathol Biol (Paris) 58:288–295. doi:10.1016/j.patbio.2010.01.006
Mancini-Bourgine M, Fontaine H, Scott-Algara D, Pol S, Brechot C, Michel ML (2004) Induction or expansion of T-cell responses by a hepatitis B DNA vaccine administered to chronic HBV carriers. Hepatology 40:874–882
Xu DZ, Wang XY, Shen XL, Gong GZ, Ren H, Guo LM, Sun AM, Xu M, Li LJ, Guo XH, Zhen Z, Wang HF, Gong HY, Xu C, Jiang N, Pan C, Gong ZJ, Zhang JM, Shang J, Xu J, Xie Q, Wu TF, Huang WX, Li YG, Xu J, Yuan ZH, Wang B, Zhao K, Wen YM, YIC Efficacy Trial Study Team (2013) Results of a phase III clinical trial with an HBsAg-HBIG immunogenic complex therapeutic vaccine for chronic hepatitis B patients: experiences and findings. J Hepatol 59:450–456
Fontaine H, Kahi S, Chazallon C, Bourgine M, Varaut A, Buffet C, Godon O, Meritet JF, Saïdi Y, Michel ML, Scott-Algara D, Aboulker JP, Pol S, ANRS HB02 study group (2015) Anti-HBV DNA vaccination does not prevent relapse after discontinuation of analogues in the treatment of chronic hepatitis B: a randomised trial–ANRS HB02 VAC-ADN. Gut 64:139–147
Kosinska AD, Zhang E, Johrden L, Liu J, Seiz PL, Zhang X, Ma Z, Kemper T, Fiedler M, Glebe D, Wildner O, Dittmer U, Lu M, Roggendorf M (2013) Combination of DNA prime–adenovirus boost immunization with entecavir elicits sustained control of chronic hepatitis B in the woodchuck model. PLoS Pathog 9:e1003391. doi:10.1371/journal.ppat.1003391
Liu J, Zhang E, Ma Z, Wu W, Kosinska A, Zhang X, Möller I, Seiz P, Glebe D, Wang B, Yang D, Lu M, Roggendorf M (2014) Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog 10:e1003856. doi:10.1371/journal.ppat856.1003856
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Aderem A, Ulevitch RJ (2000) Toll-like receptors in the induction of the innate immune response. Nature 406:782–787
Gay NJ, Gangloff M (2007) Structure and function of Toll receptors and their ligands. Annu Rev Biochem 76:141–165
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Schwabe RF, Seki E, Brenner DA (2006) Toll-like receptor signaling in the liver. Gastroenterology 130:1886–1900
Seki E, Brenner DA (2008) Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48:322–335
Lee MS, Kim YJ (2007) Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem 76:447–480
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Ait-Goughoulte M, Lucifora J, Zoulim F, Durantel D (2010) Innate antiviral immune responses to hepatitis B virus. Viruses 2:1394–1410
Broering R, Lu M, Schlaak JF (2011) Role of Toll-like receptors in liver health and disease. Clin Sci (Lond) 121:415–426
Chang J, Block TM, Guo JT (2012) The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antivir Res 96:405–413
Wieland S, Thimme R, Purcell RH, Chisari FV (2004) Genomic analysis of the hostresponse to hepatitis B virus infection. Proc Natl Acad Sci U S A 101:6669–6674
Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV (2002) Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A99:15669–15674
Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ, Dusheiko GM, Jacobs M, Klenerman P, Maini MK (2009) Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 137:1289–1300
Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, Esser K, Arzberger S, Kirschning CJ, Langenkamp A, Falk C, Büning H, Rose-John S, Protzer U (2009) Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 50:1773–1782
Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, Schlaak JF (2007) Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 46:1769–1778
Wieland SF, Guidotti LG, Chisari FV (2000) Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J Virol 74:4165–4173
Isogawa M, Robek MD, Furuichi Y, Chisari FV (2005) Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 79:7269–7272
Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV (2005) Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A 102:9913–9917
Pei R, Chen X, Lu M (2014) Control of hepatitis B virus replication by interferons and Toll-like receptor signaling pathways. World J Gastroenterol 20:11618–11629
Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Durantel D, Hüser N, Liang TJ, Münk C, Heim MH, Browning JL, Dejardin E, Schindler M, Dandri M, Heikenwalder M, Protzer U (2014) Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228
Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF (2010) Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 129:363–374
Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G, Lu M, Schlaak JF (2014) Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J Viral Hepat. doi:10.1111/jvh.12216
Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV (2002) Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J Virol 76:5646–5653
Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, Bucchi A, Sowa JP, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF (2009) Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 49:1132–1140
Shi B, Ren G, Hu Y, Wang S, Zhang Z, Yuan Z (2012) HBsAg inhibits IFN-α production in plasmacytoid dendritic cells through TNF-α and IL-10 induction in monocytes. PLoS One 7:e44900
Wang H, Ryu WS (2010) Hepatitis B virus polymerase blocks pattern recognitionreceptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 6:e1000986
Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z (2010) Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol 91:2080–2090
Wu M, Xu Y, Lin S, Zhang X, Xiang L, Yuan Z (2007) Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J Gen Virol 88:3260–3269
Chen J, Wu M, Zhang X, Zhang W, Zhang Z, Chen L, He J, Zheng Y, Chen C, Wang F, Hu Y, Zhou X, Wang C, Xu Y, Lu M, Yuan Z (2013) Hepatitis B virus polymerase impairs interferon-α-induced STATs activation through inhibition of importin-α5 and protein kinase C-δ. Hepatology 57:470–482
Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL (2011) Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol 85:987–995
Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, Jia Y, Yuan Y, Guan K, Xu Y, Cheng X, Zhang Y, Yang X, Wang Y, Wen C, Wu Q, Shi W, Zhong H (2010) The hepatitis B virus X protein disrupts innate immunity by down regulating mitochondrial antiviral signaling protein. J Immunol 185:1158–1168
Huang YW, Lin SC, Wei SC, Hu JT, Chang HY, Huang SH, Chen DS, Chen PJ, Hsu PN, Yang SS, Kao JH (2013) Reduced Toll-like receptor 3 expression in chronic hepatitis B patients and its restoration by interferon therapy. Antivir Ther 18:877–884
Lu Y, Xu Y, Yang D, Kemper T, Roggendorf M, Lu M (2008) Molecular characterization of woodchuck type I interferons and their expression by woodchuck peripheral blood lymphocytes. Cytokine 41:127–135
Zhang X, Meng Z, Qiu S, Xu Y, Yang D, Schlaak JF, Roggendorf M, Lu M (2009) Lipopolysaccharide-induced innate immune responses in primary hepatocytes downregulates woodchuck hepatitis virus replication via interferon-independent pathways. Cell Microbiol 11:1624–1637
Thompson AJ, Colledge D, Rodgers S, Wilson R, Revill P, Desmond P, Mansell A, Visvanathan K, Locarnini S (2009) Stimulation of the interleukin-1 receptor and Toll-like receptor 2 inhibits hepatitis B virus replication in hepatoma cell lines in vitro. Antivir Ther 14:797–808
Zhang X, Ma Z, Liu H, Liu J, Meng Z, Broering R, Yang D, Schlaak JF, Roggendorf M, Lu M (2012) Role of Toll-like receptor 2 in the immune response against hepadnaviral infection. J Hepatol 57:522–528
Preiss S, Thompson A, Chen X, Rodgers S, Markovska V, Desmond P, Visvanathan K, Li K, Locarnini S, Revill P (2008) Characterization of the innate immune signalling pathways in hepatocyte cell lines. J Viral Hepat 15:888–900
Xu Y, Kock J, Lu Y, Yang D, Lu M, Zhao X (2011) Suppression of hepatitis B virus replication in Tupaia hepatocytes by tumor necrosis factor alpha of Tupaia belangeri. Comp Immunol Microbiol Infect Dis 34:361–368
Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, Desmond P, Locarnini S (2007) Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 45:102–110
Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A (2011) The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol 55:762–769
Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y, Zhang Q, Wang J, Zhang Z, Shen F, Yuan Z (2008) Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol 128:400–408
Böttcher JP, Schanz O, Wohlleber D, Abdullah Z, Debey-Pascher S, Staratschek-Jox A, Höchst B, Hegenbarth S, Grell J, Limmer A, Atreya I, Neurath MF, Busch DH, Schmitt E, van Endert P, Kolanus W, Kurts C, Schultze JL, Diehl L, Knolle PA (2013) Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep 3:779–795
Kern M, Popov A, Scholz K, Schumak B, Djandji D, Limmer A, Eggle D, Sacher T, Zawatzky R, Holtappels R, Reddehase MJ, Hartmann G, Debey-Pascher S, Diehl L, Kalinke U, Koszinowski U, Schultze J, Knolle PA (2010) Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 138:336–346
Liu J, Jiang M, Ma Z, Dietze KK, Zelinskyy G, Yang D, Dittmer U, Schlaak JF, Roggendorf M, Lu M (2013) TLR1/2 ligand-stimulated mouse liver endothelial cells secrete IL-12 and trigger CD8+ T cell immunity in vitro. J Immunol 191:6178–6190
Wang S, Chen Z, Hu C, Qian F, Cheng Y, Wu M, Shi B, Chen J, Hu Y, Yuan Z (2013) Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol 190:5142–5151
Zhang X, Kraft A, Boering R, Schlaak JF, Dittmer U, Lu M (2012) Preclinical development of TLR ligands as drugs for the treatment of chronic viral infection. Expert Opin Drug Discov 7:561–597
Krieg AM (2002) CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20:709–760
Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, Wader T, Tluk S, Liu M, Davis HL, Krieg AM (2004) Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol 34:251–262
Menne S, Tennant BC, Liu KH, Ascenzi MA, Baldwin BH, Bellezza CA, Cote PJ, Zheng X, Wolfgang G, Turnas D (2011) Anti-viral efficacy and induction of an antibody response against surface antigen with the TLR7 Agonist GS-9620 in the Woodchuck Model of Chronic HBV Infection. J Hepatol 54:S441
Lanford RE, Guerra B, Chavez DC, Hodara VL, Zheng X, Wolfgang G, Tumas D (2011) Therapeutic efficacy of a TLR7 agonist for HBV chronic infection in chimpanzees. J Hepatol 54:S45
Romagne F (2007) Current and future drugs targeting one class of innate immunity receptors: the Toll-like receptors. Drug Discov Today 12:80–87
Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR (2003) Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi. 529. Expert Rev Vaccines 2:219–229
Dupont J, Altclas J, Lepetic A, Lombardo M, Vázquez V, Salgueira C, Seigelchifer M, Arndtz N, Antunez E, von Eschen K, Janowicz Z (2006) A controlled clinical trial comparing the safety and immunogenicity of a new adjuvanted hepatitis B vaccine with a standard hepatitis B vaccine. Vaccine 24:7167–7174
Kanzler H, Barrat FJ, Hessel EM, Coffman RL (2007) Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 13:552–559
Sablan BP, Kim DJ, Barzaga NG, Chow WC, Cho M, Ahn SH, Hwang SG, Lee JH, Namini H, Heyward WL (2012) Demonstration of safety and enhanced seroprotection against hepatitis B with investigational HBsAg-1018 ISS vaccine compared to a licensed hepatitis B vaccine. Vaccine 30:2689–2696
Cooper C, Mackie D (2011) Hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine: a review of HEPLISAV safety and efficacy. Expert Rev Vaccines 10:417–427
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Wu J, Chen M, Lin Y, Xia Y, Sun C, Wang J, Guo Y, Song J, Zhang E, Wang B, Zhen X, Schlaak JF, Lu M, Yang D (2014) Polyinosinic-polycytidylic acid (polyIC) treatment leads to the interferon-dependent clearance of hepatitis B virus in hydrodynamic injection mouse mode. J Virol 88:10421–10431
Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, Akira S, Marvel J, Bonnefoy-Berard N (2006) TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol 36:1684–1693
Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E (2010) When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood 116:3494–3504
Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY (2004) TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A 101:3029–3034
Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N (2009) TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol 182:1860–1867
Asprodites N, Zheng L, Geng D, Velasco-Gonzalez C, Sanchez-Perez L, Davila E (2008) Engagement of Toll-like receptor-2 on cytotoxic T-lymphocytes occurs in vivo and augments antitumor activity. FASEB J 22:3628–3637
Geng D, Zheng L, Srivastava R, Velasco-Gonzalez C, Riker A, Markovic SN, Davila E (2010) Amplifying TLR-MyD88 signals within tumor-specific T cells enhances antitumor activity to suboptimal levels of weakly immunogenic tumor antigens. Cancer Res 70:7442–7454
Deres K, Schild H, Wiesmüller KH, Jung G, Rammensee HG (1989) In vivo priming of virus-specific cytotoxic T lymphocytes with synthetic lipopeptide vaccine. Nature 342:561–564
Heathcote J, McHutchison J, Lee S, Tong M, Benner K, Minuk G, Wright T, Fikes J, Livingston B, Sette A, Chestnut R (1999) A pilot study of the CY-1899 T-cell vaccine in subjects chronically infected with hepatitis B virus. The CY1899 T Cell Vaccine Study Group. Hepatology 30:531–536
Jackson DC, Lau YF, Le T, Suhrbier A, Deliyannis G, Cheers C, Smith C, Zeng W, Brown LE (2004) A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc Natl Acad Sci U S A 101:15440–15445
Vitiello A, Ishioka G, Grey HM, Rose R, Farness P, LaFond R, Yuan L, Chisari FV, Furze J, Bartholomeuz R et al (1995) Development of a lipopeptide-based therapeutic vaccine to treat chronic HBV infection. I. Induction of a primary cytotoxic T lymphocyte response in humans. J Clin Invest 95:341–349
Acknowledgments
This work was supported by Grants of Deutsche Forschungsgemeinschaft (TRR60 and GK1949) and National Science Foundation, China (81202312).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue “Therapeutic vaccination in chronic hepatitis B—approaches, problems, and new perspectives”.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, E., Lu, M. Toll-like receptor (TLR)-mediated innate immune responses in the control of hepatitis B virus (HBV) infection. Med Microbiol Immunol 204, 11–20 (2015). https://doi.org/10.1007/s00430-014-0370-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-014-0370-1