
Abstract
SMARCB1-deficient sinonasal adenocarcinoma is a rare variant of SWI/SNF-deficient malignancies with SMARCB1 loss and adenocarcinoma features. More than 200 high-grade epithelial sinonasal malignancies were retrieved. A total of 14 cases exhibited complete SMARCB1 (INI1) loss and glandular differentiation. SMARCA2 and SMARCA4 were normal, except for one case with a loss of SMARCA2. Next-generation sequencing (NGS) and/or fluorescence in situ hybridization (FISH) revealed an alteration in the SMARCB1 gene in 9/13 cases, while 2/13 were negative. Two tumors harbored SMARCB1 mutations in c.157C > T p.(Arg53Ter) and c.842G > A p.(Trp281Ter). One harbored ARID1B mutations in c.1469G > A p.(Trp490Ter) and MGA c.3724C > T p.(Arg1242Ter). Seven tumors had a SMARCB1 deletion. One carried an ESR1 mutation in c.644-2A > T, and another carried a POLE mutation in c.352_374del p.(Ser118GlyfsTer78). One case had a PAX3 mutation in c.44del p.(Gly15AlafsTer95). Histomorphology of SMARCB1-deficient adenocarcinoma was oncocytoid/rhabdoid and glandular, solid, or trabecular in 9/14 cases. Two had basaloid/blue cytoplasm and one showed focal signet ring cells. Yolk sac tumor-like differentiation with Schiller-Duval-like bodies was seen in 6/14 cases, with 2 cases showing exclusively reticular-microcystic yolk sac pattern. Follow-up of a maximum of 26 months (median 10 months) was available for 8/14 patients. Distant metastasis to the lung, liver, mediastinum, bone, and/or retroperitoneum was seen in 4/8 cases. Locoregional failure was seen in 75% of patients, with 6/8 local recurrences and 3 cervical lymph node metastases. At the last follow-up, 5 of 8 (62%) patients had died of their disease 2 to 20 months after diagnosis (median 8.2 months), and 3 were alive with the disease. The original diagnosis was usually high-grade non-intestinal-type adenocarcinoma or high-grade myoepithelial carcinoma. A correct diagnosis of these aggressive tumors could lead to improved targeted therapies with potentially better overall disease-specific survival.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Poorly differentiated or undifferentiated sinonasal carcinomas represent a challenging area in head and neck pathology. Over the recent years, advances in molecular techniques have led to significant progress in understanding the molecular underpinnings of sinonasal malignancies, with significant developments in the histological and pathogenetic classification of entities included in the historical spectrum of “sinonasal undifferentiated carcinoma (SNUC)” and poorly differentiated unclassified carcinoma [1]. SNUC, a diagnosis of exclusion, should be made only in cases of relatively monomorphic, sometimes basaloid-looking, high-grade/poorly differentiated tumor cells with evidence of epithelial origin and the absence of features pointing toward other entities. Recently, up to 80% of SNUC cases were reported to harbor hotspot mutations in the IDH2 gene [2, 3], while mutations in the IDH1 gene have been reported only rarely [3]. Another novel entity with a very poor prognosis is NUT carcinoma, composed of undifferentiated primitive cells with irregular overlapping nuclei with prominent nucleoli and defined by pathogenic fusions of the NUTM1 gene, most commonly NUTM1::BRD4 [4].
Recent molecular-genetic findings have aided in subclassifying primary sinonasal carcinomas, prompting the inclusion of several new entities in the 5th edition of the WHO Classification of Head and Neck Tumors [5]. A major change has been the recognition of subtypes of carcinomas defined by genetic defects leading to the inactivation of different protein subunits in the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex [6]. This led to the identification of 4 sinonasal entities driven by SWI/SNF deficiency complexes: SMARCB1 (INI1)-deficient sinonasal carcinoma (lacking gland formation and frequently displaying a non-descript basaloid and less frequently eosinophilic/oncocytoid morphology), SMARCB1-deficient sinonasal adenocarcinoma with unequivocal glands or yolk sac-like pattern, SMARCA4-deficient carcinoma lacking glandular or squamous immunophenotypes, and SMARCA4-deficient subset (~ 80%) of sinonasal teratocarcinosarcoma [6]. The most common of these four, the SMARCB1 (INI-1)-deficient sinonasal carcinoma, is an aggressive tumor that was first reported in 2014 independently by two research groups [7, 8]. It is a rare form of cancer arising in the nasal cavity and paranasal sinuses, and it is characterized by the loss or inactivation of the SWI/SNF-complex genes that play crucial roles in the regulation of cell growth and division [9].
Primary sinonasal adenocarcinomas (SNAC) are rare tumors encompassing a wide morphological spectrum. They are divided into three groups: intestinal-type sinonasal adenocarcinoma (ITAC), non-intestinal-type sinonasal adenocarcinoma (non-ITAC), and adenocarcinomas of salivary gland subtypes [10]. The classification of SNAC has developed in the last two decades [11]. High-grade non-intestinal SNACs are particularly heterogeneous, with highly variable morphology [12] including several molecularly defined new entities [13, 14].
SMARCB1-deficient sinonasal adenocarcinoma is a rare SWI/SNF-deficient malignancy defined by the presence of unequivocal glandular differentiation and/or by the presence of other features of adenocarcinoma [15]. Glandular differentiation has been very rarely reported in SMARCB1 (INI-1)-deficient sinonasal carcinomas, although it is possible that this feature has been underrecognized [16,17,18,19]. The tumors are usually composed of oncocytoid/plasmacytoid tumor cells in rounded nests, trabeculae, reticular, microcystic, and glandular patterns and could be misdiagnosed as high-grade non-intestinal adenocarcinoma, myoepithelial carcinoma, or even metastatic yolk sac tumor [20].
The use of next-generation sequencing platforms in clinical practice has allowed for further subclassification of tumors previously grouped together. The discovery of a subset of tumors with several unique molecular features has opened up the door to potential therapeutic targets [21]. These targetable subsets include not only NUT carcinoma [22] and SNUC with a novel isocitrate dehydrogenase (IDH) mutation [23] but also SWI/SNF-deficient sinonasal malignancies [24]. We aim to summarize our experience with a subset of SWI/SNF-deficient sinonasal adenocarcinomas diagnosed in three major consult centers in head and neck pathology (AS, AA, and AF).
Materials and methods
Case selection
In their consultation practice, two of the authors (AA and AS) encountered cases of sinonasal undifferentiated malignancies reminiscent of salivary myoepithelial carcinoma or high-grade sinonasal non-ITAC that showed a complete loss of SMARCB1 immunoexpression. Indeed, some of these cases were submitted for consultation with a diagnosis of sinonasal myoepithelial carcinoma or high-grade sinonasal non-ITAC, and a diagnosis of SMARCB1-deficient adenocarcinoma was not raised by the primary pathologist. These consult cases prompted us to perform a computer search of our routine and consult files for high-grade sinonasal non-ITAC carcinomas and SNUCs. Seventy-five and one hundred thirty high-grade malignant tumors of sinonasal localization with epithelial features and available tissue material were retrieved from the authors’ files, respectively (AS and AA). All cases were evaluated morphologically and examined immunohistochemically (IHC) by cytokeratin cocktail AE1-AE3 antibodies and antibodies to CK7, CK5/6, p63/p40, SOX10, S100, SALL4, glypican-3, and SWI/SNF proteins (Table 1). Next-generation sequencing (NGS) using the Illumina Oncology TS500 and TST170 DNA panels was performed in 44 cases, including 9 cases that demonstrated loss of SWI/SNF proteins by immunohistochemistry (AS) and 4 cases (AA). The latter cases were subjected to selected immunohistochemical and molecular studies (Table 2).
Histological and immunohistochemical studies
For conventional microscopy, the excised tissues were fixed in formalin, processed routinely, embedded in paraffin (FFPE), cut, and stained with hematoxylin and eosin.
For immunohistochemistry, 4-μm-thick sections were cut from paraffin blocks and mounted on positively charged slides (TOMO, Matsunami Glass IND, Osaka, Japan). Sections were processed on a BenchMark ULTRA (Ventana Medical Systems, Tucson, AZ), deparaffinized, and subjected to heat-induced epitope retrieval by immersion in a CC1 solution (pH 8.6) at 95 °C. All primary antibodies used in this study are summarized in Table 1.
Visualization was performed using the ultraView Universal DAB Detection Kit (Roche, Tucson, AZ) and the ultraView Universal Alkaline Phosphatase Red Detection Kit (Roche, Tucson, AZ). The slides were counterstained with Mayer’s hematoxylin. Appropriate positive and negative controls were employed.
Molecular studies
TruSight Oncology 500 Kit (TS500)
Mutation analysis and fusion transcript detection were performed using the TruSight Oncology 500 Kit (Illumina, San Diego, CA). RNA was extracted using the Maxwell RSC DNA FFPE Kit and the Maxwell RSC Instrument (Promega, Madison, WI) according to the manufacturer’s instructions and quantified using the Qubit HS RNA Assay Kit (Thermo Fisher Scientific, Waltham, MA). DNA was extracted using the QIAsymphony DSP DNA mini (Qiagen, Hilden, Germany) and quantified using the Qubit BR DNA Assay Kit (Thermo Fisher Scientific, Waltham, MA). The quality of DNA was assessed using the FFPE QC kit (Illumina) and the quality of RNA using the Agilent RNA ScreenTape Assay (Agilent, Santa Clara, CA). DNA samples with Cq < 5 and RNA samples with DV200 ≥ 20 were used for further analysis. After DNA enzymatic fragmentation with the KAPAFrag Kit (KAPA Biosystems, Wilmington, MA), DNA and RNA libraries were prepared with the TruSight Oncology 500 Kit (Illumina) according to the manufacturer’s protocol. Sequencing was performed on the NovaSeq 6000 sequencer (Illumina) following the manufacturer’s recommendations. Data analysis was performed using the TruSight Oncology 500 v2.2 Local App (Illumina). Variant annotation and filtering were performed using the Omnomics NGS analysis software (Euformatics, Espoo, Finland). A custom variant filter was set up including only non-synonymous variants with coding consequences, read depth greater than 50, and benign variants according to the ClinVar database [25] were also excluded. The remaining subset of variants was checked visually, and suspected artefactual variants were excluded.
VariantPlex Kit (VST)
DNA was extracted using the Qiasymphony DSP DNA mini (Qiagen, Hilden, Germany), and input DNA clean-up was performed using the Archer PreSeq™ DNA QC Assay Protocol. DNA was quantified using the Qubit HS DNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The Archer VariantPlex Solid Tumor Kit was used (VST, ArcherDX Inc., Boulder, CO). Library preparation was performed following the Archer VariantPlex™ Protocol for Illumina (ArcherDx) and the product Insert VariantPlex™ Solid Tumor Panel. Final libraries were diluted 1:100,000 and quantified following the Library Quantification for Illumina (KAPA Biosystems, Wilmington, MA), normalized, and pooled. The libraries were sequenced on NovaSeq 6000 (Illumina) following the manufacturer’s recommendations. Variant annotation and filtering were performed using Archer Analysis software v7 (ArcherDx) with the parameters mentioned above.
Fluorescent in situ hybridization (FISH)
For the detection of SMARCB1 deletion, ZytoLight® SPEC SMARCB1/22q12 Dual Color Probe was used (ZytoVision GmbH, Bremerhaven, Germany). The FISH procedure was performed as described elsewhere [26]. The heterozygous deletion was defined as one green signal (SMARCB1) compared to two orange control signals (KREMEN1), and the homozygous deletion as zero green signals (SMARCB1) compared to at least one orange control signal (KREMEN1).
Results
Demographic and clinical features
A total of 14 cases demonstrated clear morphologic evidence of glandular differentiation and loss of SMARCB1 immunoexpression, and these were included in the study (Table 2). They occurred in 11 men and 2 women with ages ranging from 36 to 92 years (mean 59 years). Clinical data were not available for one patient. The tumors arose in the nasal cavity (7), maxillary sinus (1), ethmoid sinus (1), paranasal sinus NOS (4), and sinonasal tract NOS (1). Imaging revealed extensive involvement of the paranasal sinuses with or without the involvement of the nasal cavity and frequent involvement of the skull base (Fig. 1a, b).

SMARCB1-deficient sinonasal adenocarcinoma T3N0M0: coronal noncontrast CT scan (A) and axial noncontrast CT scan (B)
Of eight patients with detailed tumor staging information, six (75%) presented with stage T4 disease with extensive involvement of the bony confinements of the sinonasal cavities and variable infiltration into periorbital or skull base tissues. Synchronous regional lymph node involvement and distant metastases were detected in one patient (Table 3).
Treatment consisted of radical surgical resection combined with chemotherapy and/or radiation in eight patients. One patient received only supportive (palliative) care after a diagnostic biopsy. For the remaining six patients, detailed information regarding therapy was not available (Table 3).
Follow-up data were available for 8 patients, and the follow-up period ranged from shortly after diagnosis to 26 months (median, 10). Distant metastases were recorded in 4 of 8 cases (50%). The sites of distant metastases included the lungs (n = 2), the liver (n = 1), the mediastinum (n = 2), the bones (n = 1), and the retroperitoneum (n = 1). They occurred from presentation to 24 months after diagnosis (median, 10 months). Regional failure was seen in 75% of patients, with 6/8 local recurrences and 3 regional recurrences to cervical lymph nodes. At the last follow-up, 5 of 8 (62%) patients had died of their disease 2 to 20 months after diagnosis (median, 8.2 months), and 3 were alive with the disease (Table 3).
Histopathological and immunohistochemical finding
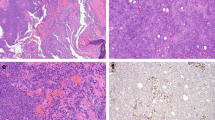
In most of the cases, tumor histomorphology was predominantly solid, with trabecular and alveolar growth patterns. The tumor cells were large with eosinophilic, oncocytoid, plasmacytoid, and/or rhabdoid appearance. In nine cases (9/14, 64%), the dominant cell morphology was oncocytoid/rhabdoid (Fig. 2a–c). However, in two cases a basaloid/blue cytoplasm was observed (Table 2 and Fig. 2d). All cases demonstrated varying proportions of glandular changes, including alveolar/acinar with abortive microglandular differentiation, trabecular, and solid/cribriform/insular patterns. A focal signet-ring cell pattern was noted in one case (Table 2 and Fig. 3a, b). Variable luminal and stromal mucin-like secretion was noted in some cases, one case with prominent myxoid stroma. Areas with yolk sac tumor-like differentiation with Schiller-Duval body-like structures were found in six cases (6/14; 43%) with two cases showing exclusively reticular-microcystic yolk sac pattern (Table 2 and Fig. 3c–e). Seven cases with high-grade malignant histomorphology and two cases with intermediate-grade malignant histomorphology were observed (Table 2). In all cases, the Ki-67 index was high (> 40%).

SMARCB1-deficient sinonasal adenocarcinoma is characterized by the presence of oncocytoid/plasmacytoid cell morphology, with variable but unequivocal gland formation (A–C) defined by open glandular structures (A), abortive microglandular differentiation (B), and small nests (C). Less common pattern is basaloid/blue phenotype with uniformly high-grade cytomorphology (D)

Cribriform structures may be present in solid areas (A), and focal signet-ring cell pattern was noted in one case (B). Fibrous stroma with intermingled glands with a sieve-like morphology or floating tumor cell strips embedded within mucoid stroma was present in two cases (C) along with structures resembling the Schiller-Duval bodies. (D) Two cases showing exclusively reticular-microcystic yolk sac pattern (E) positive for SALL4 (inset)
In immunohistochemical stainings, all cases were completely negative for SMARCB1 (INI-1) proteins (Fig. 4a). SMARCA2 and SMARCA4 had normal patterns of expression (retained) except for case 14, which has shown SMARCA2 loss. In all cases, the neoplastic cells showed strong staining for cytokeratins AE1/AE3 and/or OSCAR (9/9), CK7 (9/9), and EMA (4/4), while S100, SOX10, CK20, CDX2, p63, p40, GATA3, NUT, and AR were mostly negative. Proliferative activity was high, with the Ki-67 index reaching 40–80% (mean 56%) (Fig. 4b). Focal p63 staining was seen in 5 cases, CDX2 staining in 2 cases, and weak focal synaptophysin in 3 cases. Immunohistochemical markers for yolk sac tumor (SALL4 or glypican-3) decorated 6 cases (Fig. 4c–d), corresponding to their yolk sac tumor-like histologies.

Representative example of complete loss of SMARCB1 immunohistochemistry in tumor cells with positive internal control in lymphocytes and fibroblasts (A). Proliferative activity was high with Ki-67 > 40% (B). Germ cell markers of yolk sac-like differentiation, SALL4 (C) and glypican-3 (D) highlighted 6 cases
Molecular findings
Next-generation sequencing (NGS) and/or fluorescence in situ hybridization (FISH) revealed alteration in the SMARCB1 gene in nine cases (9/13, 69%), while two cases were negative and two cases were not analyzable. In one case, molecular analysis was not done. (Table 2). Among the positive cases, two had nonsense mutations truncating the SMARCB1 gene, namely SMARCB1 c.157C > T p.(Arg53Ter) and c.842G > A, p.(Trp281Ter). Seven cases had deletions in the SMARCB1 gene detected by NGS or FISH. One case was negative for both SNV and CNV analysis by NGS and also negative for SMARCB1 deletion by FISH. Another one was negative for SNV and CNV by NGS only. In addition, one of these tumors harbored mutations in genes ARID1B c.1469G > A p.(Trp490Ter) and MGA c.3724C > T p.(Arg1242Ter); another case had a splicing mutation in the ESR1 gene c.644-2A > T and the other had a mutation in gene POLE c.352_374del p.(Ser118GlyfsTer78). In addition, in one case, there was a mutation in the gene PAX3 c.44del p.(Gly15AlafsTer95). The complete results of molecular testing are illustrated in Table 2.
Discussion
SWI/SNF (switching/sucrose non-fermentable) genes were first described in Saccharomyces yeast in 1984 as genes required to enable mating-type switching and sucrose metabolism [27]. In the process of evolution from yeast to mammals, the SWI/SNF complex (aka BAF complex) has evolved into a large ATP-dependent chromatin remodeling complex with many subunits encoded by a variety of genes with a markedly heterogeneous structure and function. Due to the tumor suppressor role of this complex, somatic mutations in SWI/SNF genes are involved in the tumorigenesis of multiple human neoplasms [28, 29]. Other tumor suppressor genes, such as RB1, TP53, MYC, and BRCA1, are known to interact with the SWI/SNF complex, and therefore mutations in these partner genes can affect its function and increase cell proliferation [28,29,30,31]. Up to 25% of human cancers carry mutations in at least one of nine SWI/SNF subunit genes including SMARCA1 and 2, SMARCB1, ARID1A/B, PBRM1, and ARID2 [32].
SMARCB1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily b, member 1) is also known as INI1 (for integrase interactor 1) and SNF5, the latter of which was initially named based on a conserved region of this protein in the SWI/SNF complex in yeast [33]. SMARCB1, located on chromosome 22q11.2, functions as a tumor suppressor gene. Mutations in the SMARCB1 gene were first described in rhabdoid tumors in 1998, evidencing for the first time the link between the SWI/SNF complex and human cancer [33]. SMARCB1 mutations have been implicated in the pathogenesis of several malignancies, including atypical teratoid rhabdoid tumor, malignant rhabdoid tumor, epithelioid sarcoma, renal medullary carcinoma, myoepithelial carcinoma, epithelioid malignant peripheral nerve sheath tumor, and extraskeletal myxoid chondrosarcoma [8]. Although these diverse neoplasms exhibit many distinct clinicopathologic features, they all tend to share the presence of “rhabdoid” cells, defined as large cells with abundant eosinophilic cytoplasm and eccentrically placed nuclei with open chromatin and prominent nucleoli. A meta-analysis of 10,849 patients from 15 studies found that 5% of human cancers had alterations in SMARCB1 [34, 35, 36]. Such tumors mostly but not exclusively comprise rare, high-grade lethal cancers [32, 34].
Since the first description of malignant rhabdoid tumors in pediatric patients, various histological patterns in SWI/SNF-mutated tumors have been reported. Such tumors comprise, but are not limited to, rhabdoid eosinophilic cells, blue-basaloid cells, small cells, and clear cells, as well as glandular, sarcomatoid, yolk sac-like, and mixed histological patterns [15, 37]. A rare histologic subgroup of SWI/SNF-deficient neoplasms and the subject of our investigation is SMARCB1-deficient sinonasal adenocarcinoma defined by glandular features. In our limited series, we found that a majority of SMARCB1-deficient sinonasal adenocarcinomas exhibited a dominant eosinophilic pattern with an oncocytoid/rhabdoid appearance, followed by a solid blue-basophilic cell pattern. Glandular structures including tubular, microglandular, cribriform, solid, and signet-ring patterns were identified in all our cases, comparable to other published series also showing intracytoplasmic and intraluminal mucin confirmed by mucin staining [15]. Of interest is the finding of yolk sac tumor-like differentiation with Schiller-Duval body-like structures in 34% of our cases, with both basophilic and non-pink cell appearances.
Three potential pitfalls can be expected in the diagnosis of SMARCB1-deficient sinonasal adenocarcinoma. First, the common eosinophilic-glandular pattern of this tumor could be misclassified as a high-grade non-ITAC. Loss of immunohistochemical SMARCB1 staining, which was uniformly observed in all our cases, is an essential criterion to avoid this diagnostic error. A second differential diagnostic error could be due to the basophilic/blue glands with endometrioid/yolk sac-like structures that might be interpreted as features of ITAC. Although this misinterpretation is more likely due to the focal CK20 and CDX2 positivity seen in these cases, there was a lack of diffuse CK20/CDX2 staining, which, along with the loss of SMARCB1 staining and focal positivity for glypican-3 and/or SALL4, helped in differentiating these entities. Shah et al. also highlighted other differential diagnoses in this setting, such as extragonadal or metastatic yolk sac tumor and metastatic hepatocellular carcinoma [15]. These diagnoses could be inferred from positive staining for glypican-3 and SALL4; however, these entities are extremely rare in the sinonasal tract. Moreover, negative SMARCB1 and other germ cell markers would rule out these diagnoses. Third, the microglandular/cribriform and eosinophilic appearance of cells with focal p63 staining can be interpreted as myoepithelial differentiation. Our cases were entirely negative for S100 and SOX10, which, in combination with SMARCB1, negativity excludes myoepithelial carcinoma.
SWI/SNF-deficient malignancies pursue a highly aggressive clinical course, resulting in widespread disease dissemination either at the time of diagnosis or soon afterwards and causing the death of the patient soon after diagnosis, despite an apparently curative therapeutic intent. Systemic chemotherapy has shown no success so far [23]. To date, satisfactory systemic chemotherapy has not been established for these patients. This disappointing finding underlines the urgent need for effective systemic therapy to allow sufficient intermediate to long-term disease control. However, there are a number of scientific investigations aimed at deciphering the vulnerable molecular sites secondary to SWI/SNF mutation in these tumors [38]. Gene sequencing and molecular subgrouping of each SWI/SNF-mutated tumor will help identify a target suitable for tailored therapy [35]. Recently, SWI/SNF-deficiency has increasingly emerged as pivotal in cancer immunogenicity and hence a promising biomarker when predicting response to immune-checkpoint inhibition therapy utilizing several recently established drugs [23]. Immunotherapy targeting PDL1 [35, 39], employing the PRC2 (EZH2 subunit) inhibitor tazemetostat [15], as well as inhibitors acting against protein kinases, MYC, MDM2/4, and the proteasome are major examples of these efforts [32, 35].
Summary
In this limited series, it appears that SMARCB1-deficient SNACs show a predilection for male patients, and in contrast to non-glandular tumors, may occur with greater frequency in the nasal cavity. The original diagnosis in most cases of SMARCB1-deficient SNAC was HG non-ITAC and less frequently HG myoepithelial carcinoma or HG ITAC. Differential diagnosis is challenging, but the availability of immunohistochemical antibodies against SWI/SNF proteins represents an emerging effective tool for screening these neoplasms. For the first time, we have shown an NGS-detectable mutation in the SMARCB1 gene in a subset of cases. Recent advances in molecular profiling have led to major updates and revisions in the classification of high-grade sinonasal carcinomas. Although the majority of these tumors are characterized by aggressive biologic behavior, the identification of these mutations could potentially lead to improved targeted therapeutic options and improved overall disease-specific survival.
Data availability
Data supporting the findings of this study are available within the article. The complete datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Agaimy A, Franchi A, Lund VJ, Skálová A, Bishop JA, Triantafyllou A, Andreasen S, Gnepp DR, Hellquist H, Thompson LDR, Rinaldo A, Ferlito A (2020) Sinonasal undifferentiated carcinoma (SNUC): from an entity to morphologic pattern and back again-a historical perspective. Adv Anat Pathol 27(2):51–60
Dogan S, Chute DJ, Xu B et al (2017) Frequent IDH2 R172 mutations in undifferentiated and poorly-differentiated sinonasal carcinomas. J Pathol 242(4):400–408
Mito JK, Bishop JA, Sadow PM et al (2018) Immunohistochemical detection and molecular characterization of IDH-mutant sinonasal undifferentiated carcinomas. Am J Surg Pathol 42(8):1067–1075
French CA (2010) NUT midline carcinoma. Cancer Genet Cytogenet 203(1):16–20
Bishop JA, Thompson LDR, Loney EL (eds) (2022) Chapter 2: nasal, paranasal, and skull base tumours. In: WHO classification of tumours editorial board. Head and neck tumours. [Internet; beta version ahead of print]. Lyon (France): International Agency for Research on Cancer; [cited 2023 May 8]. (WHO classification of tumours series, 5th ed.; vol. 9). Available from: https://tumourclassification.iarc.who.int/chapters/52/1
Agaimy A (2023) SWI/SNF-deficient sinonasal carcinomas. Adv Anat Pathol 30(2):95–103
Agaimy A, Koch M, Lell M et al (2014) SMARCB1(INI1)-deficient sinonasal basaloid carcinoma: a novel member of the expanding family of SMARCB1-deficient neoplasms. Am J Surg Pathol 38:1274–1281
Bishop JA, Antonescu CR, Westra WH (2014) SMARCB1 (INI-1)-deficient carcinomas of the sinonasal tract. Am J Surg Pathol 38:1282–1289
Agaimy A, Hartmann A, Antonescu CR et al (2017) SMARCB1 (INI-1)-deficient sinonasal carcinoma: a series of 39 cases expanding the morphologic and clinicopathologic spectrum of a recently described entity. Am J Surg Pathol 41:458–471
Leivo I (2016) Sinonasal adenocarcinoma: update on classifcation, immunophenotype and molecular features. Head Neck Pathol 10:68–74
Leivo I (2007) Update on sinonasal adenocarcinoma: classification and advances in immunophenotype and molecular genetic make-up. Head Neck Pathol 1(1):38–43
Stelow EB, Jo VY, Mills SE et al (2011) A histologic and immunohistochemical study describing the diversity of tumors classifed as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol 35:971–980
Baneckova M, Agaimy A, Andreasen S et al (2018) Mammary analog secretory carcinoma of the nasal cavity: characterization of 2 cases and their distinction from other low-grade sinonasal adenocarcinomas. Am J Surg Pathol 42(6):735–743
Klubíčková N, Mosaieby E, Ptáková N et al (n.d.) High-grade non-intestinal type sinonasal adenocarcinoma with ETV6::NTRK3 fusion, distinct from secretory carcinoma by immunoprofile and morphology. Virchows Arch 2023 in press
Shah AA, Jain D, Ababneh E, Agaimy A, Hoschar AP, Griffith CC, Magliocca KR, Wenig BM, Rooper LM, Bishop JA (2020) SMARCB1 (INI-1)-deficient adenocarcinoma of the sinonasal tract: a potentially under-recognized form of sinonasal adenocarcinoma with occasional yolk sac tumor-like features. Head Neck Pathol 14(2):465–472
Zamecnik M, Rychnovsky J, Syrovatka J (2018) Sinonasal SMARCB1 (INI1) deficient carcinoma with yolk sac tumor diferentiation: report of a case and comparison with INI1 expression in gonadal germ cell tumors. Int J Surg Pathol 26:245–249
Li CY, Han YM, Xu K et al (2021) Case report: SMARCB1 (INI-1)-deficient carcinoma of the nasal cavity with pure yolk sac tumor differentiation and elevated serum AFP levels. Onco Targets Ther 14:2227–2233
Hazir B, Şímsek B, Erdemír A et al (2022) Sinonasal SMARCB1 (INI1) deficient carcinoma with yolk sac tumor differentiation: a case report and treatment options. Head Neck Pathol 16:596–601
Ng JKM, Chan JYK, Li JJX et al (2022) SMARCB1 (INI1)-deficient sinonasal carcinoma with yolk sac differentiation showing co-loss of SMARCA4 immunostaining- a case report and literature review. Head Neck Pathol 16:934–941
Skalova A, Agaimy, A, Vaněček T, Klubíčková N, Kiss, K, Baneckova M, Michal M (2023) SMARCB1-deficient sinonasal adenocarcinoma: rare variant of SWI/SNF-deficient malignancy often misclassified as high-grade non-intestinal adenocarcinoma or myoepithelial carcinoma. Laboratory Investigation, (USCAP 2022 Abstracts: Head and Neck Pathology (1014–1081)), 1072–1073. https://doi.org/10.1016/j.labinv.2023.100092
Reiersen DA, Pahilan ME, Devaiah AK (2012) Meta-analysis of treatment outcomes for sinonasal undifferentiated carcinoma. Otolaryngol Head Neck Surg 147:7–14
French CA (2013) The importance of diagnosing NUT midline carcinoma. Head Neck Pathol 7(1):11–16
Alzumaili B, Sadow PM (2023) IDH2 -Mutated sinonasal tumors: a review. Adv Anat Pathol 30(2):104–111
Agaimy A (2023) SWI/SNF-deficient Malignancies: optimal candidates for immune-oncological therapy? Adv Anat Pathol 30(3):211–217
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D1067. https://doi.org/10.1093/nar/gkx1153
Šteiner P, Andreasen S, Grossmann P et al (2018) Prognostic significance of 1p36 locus deletion in adenoid cystic carcinoma of the salivary glands. Virchows Arch 473(4):471–480. https://doi.org/10.1007/s00428-018-2349-6
Stern M, Jensen R, Herskowitz I (1984) Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol 178(4):853–68
Reddy D, Bhattacharya S, Workman JL (2023) (mis)-Targeting of SWI/SNF complex(es) in cancer. Cancer Metastasis Rev. https://doi.org/10.1007/s10555-023-10102-5
Li Z, Zhao J, Tang Y (2023) Advances in the role of SWI/SNF complexes in tumours. J Cell Mol Med 27:1023–1031
Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R (2000) BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102(2):257–65
Lee D, Kim JW, Seo T, Hwang SG, Choi EJ, Choe J (2002) SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem 277(25):22330–22337
Mittal P, Roberts CWM (2020) The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol 17:435–448
Brzeski J, Podstolski W, Olczak K, Jerzmanowski A (1999) Identification and analysis of the Arabidopsis thaliana BSH gene, a member of the SNF5 gene family. Nucleic Acids Res 27(11):2393–2399
Versteege I, Sévenet N, Lange J et al (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Cooper GW, Hong AL (2022) SMARCB1-Deficient cancers: novel molecular insights and therapeutic vulnerabilities. Cancers (Basel) 14(15):3645
Wang N, Qin Y, Du F, Wang X, Song C (2022) Prevalence of SWI/SNF genomic alterations in cancer and association with the response to immune checkpoint inhibitors: a systematic review and meta-analysis. Gene 5(834):146638
Agaimy A (2022) Proceedings of the North American Society of Head and Neck Pathology, Los Angeles, CA, March 20, 2022: SWI/SNF-deficient Sinonasal Neoplasms: An Overview. Head Neck Pathol 16(1):168–178
Carugo A, Minelli R, Sapio L, Soeung M, Carbone F, Robinson FS, Tepper J, Chen Z, Lovisa S, Svelto M, Amin S, Srinivasan S, Del Poggetto E, Loponte S, Puca F, Dey P, Malouf GG, Su X, Li L, Lopez-Terrada D, Rakheja D, Lazar AJ, Netto GJ, Rao P, Sgambato A, Maitra A, Tripathi DN, Walker CL, Karam JA, Heffernan TP, Viale A, Roberts CWM, Msaouel P, Tannir NM, Draetta GF, Genovese G (2019) p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell 35(2):204-220.e9
Ngo C, Postel-Vinay S (2022) Immunotherapy for SMARCB1-deficient sarcomas: current evidence and future developments. Biomedicines 10(3):650
Funding
This study was supported by study grant SVV 260652 from the Ministry of Education, Czech Republic (Natálie Klubíčková) and the Cooperatio Program, research area SURG (Natálie Klubíčková), and the project National Institute for Cancer Research—NICR (Programme EXCELES, ID Project No. LX22NPO5102)—Funded by the European Union—Next Generation EU (Alena Skálová, Martina Bradová).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Alena Skálová and Abbas Agaimy. The first draft of the manuscript was written by Touraj Taheri, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the institutional review board of the Faculty of Medicine in Pilsen, Charles University. The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Informed consent
No patient consent was required for this study.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skálová, A., Taheri, T., Bradová, M. et al. SMARCB1-deficient sinonasal adenocarcinoma: a rare variant of SWI/SNF-deficient malignancy often misclassified as high-grade non-intestinal-type sinonasal adenocarcinoma or myoepithelial carcinoma. Virchows Arch (2023). https://doi.org/10.1007/s00428-023-03650-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00428-023-03650-2