
Abstract
Neuroendocrine neoplasms (NENs) of the lung encompass neuroendocrine tumors (NETs) composed of typical (TC) and atypical (AC) carcinoids and full-fledged carcinomas (NECs) inclusive of large cell neuroendocrine carcinoma (LCNEC) and small cell carcinoma (SCLC). NETs and NECs are thought to represent distinct and separate lesions with neither molecular overlap nor common developmental continuum. Two perspectives were addressed regarding the morphologic and molecular classification of lung NENs: (i) a supervised approach by browsing the traditional classification, the relevant gene alterations, and their clinical implications; and (ii) an unsupervised approach, by reappraising neoplasms according to risk factors and natural history of disease to construct an interpretation model relied on biological data. We herein emphasize lights and shadows of the current classification of lung NENs and provide an alternative outlook on these tumors focused on what we currently know about the biological determinants and the natural history of disease.
Similar content being viewed by others


Introduction
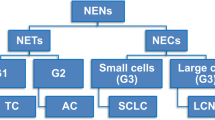
Lung neuroendocrine neoplasms (NENs) encompass four histologic subtypes whose terminology and defining criteria have been endorsed over the last three classifications by the World Health Organization (WHO). This classification will remain fundamentally unchanged in the forthcoming 5th edition of the WHO Blue Book on lung tumors. Accordingly, lung NENs include typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC), and small cell lung carcinoma (SCLC) (Fig. 1). Diagnostic criteria include mitotic count per 2 mm2 and necrosis alongside a wide constellation of cellular and architectural features fulfilling NE morphology, while immunohistochemical (IHC) markers are applied to reveal NE differentiation especially in high-grade SCLC and LCNEC (Table 1). The most important separation is between well differentiated tumors of low (i.e., TC as G1) to intermediate (i.e., AC as G2) grade (henceforth, NETs) and poorly differentiated full-fledged NE carcinomas (henceforth, NECs), which are all grouped together as high-grade neoplasms (tautologically G3) with small cell (SCLC) and/or large cell (LCNEC) appearance and no remarkable differences in survival between them [62] (Fig. 1). The morphologic approach to classification complies with a three-tier spectrum of clinical outcomes, survival rates, and therapy options, essentially because defining criteria are effective enough to highlight low-to-intermediate grade while failing to a large extent in the behavioral distinction of NECs. Such a dichotomy between NETs and NECs has strict biological grounds, inasmuch as NETs are not precursors to NECs and therapy options are closely dictated upon morphology-based tumor grading [75]. Molecular investigations have indeed supported the notion that NETs make up distinct and separate tumor entities as opposed to NECs, arguing against a causative relationship between NETs and NECs (Table 2).

Composition of lung neuroendocrine neoplasms. A binary subdivision into neuroendocrine tumors (NETs) and neuroendocrine carcinoma (NECs) is preliminary to the identification of typical carcinoid (TC) as G1 tumor, atypical carcinoid (AC) as G2 tumor, and NEC with small cells (namely small cell lung carcinoma, SCLC) and large cells (namely large cell neuroendocrine carcinoma, LCNEC), both of them making tautologically G3—high-grade neoplasms
Herein, we aimed to review lights and shadows of lung NEN classification by accounting for their clinicopathologic features, molecular traits, and natural history of disease.
Material and methods
An accurate survey of papers related to tumor classification and intratumor heterogeneity (not a systematic review nor meta-analysis) was conducted until the end of July 2020. A list of key questions regarding lung NENs was generated concerning diagnosis, prognosis, classification, tumor subtypes, carcinoid, small cell carcinoma, large cell neuroendocrine carcinoma, variant subtype, prediction, immunohistochemistry, clinical implications, genetic/epigenetic changes, and development of biomolecular models. Only articles dealing with the 2015 WHO classification were accounted for to assure comparability of both pathology criteria and clinical outcomes. The research was limited to available English literature in PubMed®, with only full-papers being considered along with some smaller studies or even case reports, if appropriate. Morphologic and molecular classification of lung NENs was addressed under two different perspectives: (i) a supervised one, by browsing the traditional classification through its own specific gene alterations and clinical implications, and (ii) an unsupervised one, by reappraising these neoplasms according to risk factors, molecular assembly, and developmental mechanisms.
A supervised approach: a classification based on morphology with clinical claims
Histologic classification
Lung NENs make up about 20% of all lung malignancies, with a large prevalence of NECs (LCNEC and SCLC) over NETs (TC and AC). These four histologic subtypes are distinguished upon mitotic count per 2 mm2, necrosis assessment, and cytologic detail appreciation (i.e., relative cytoplasm amount, nuclear chromatin pattern, nucleolar appearance, nucleus-cytoplasm ratio) (Table 1). NE morphology in lung NETs shows an extreme variety of features, with multiple patterning (e.g., insular, lobular, trabecular, follicular, and solid) and/or cellular variants (polygonal, spindle, oncocytic, mucinous, melanin-laden, etc.) within the same tumors (Table 1 and Fig. 2). Likewise, the same wide array of morphologic features can be documented in NECs, whether LCNEC or SCLC, where cellular details (cell size and nuclear chromatin patterning) serve as discriminants to distinguish LCNEC (large cells with coarser chromatin and prominent nuclei) from SCLC (small cells with finely dispersed chromatin and inconspicuous nucleoli) (Table 1 and Fig. 3). NECs have no upper limits in mitotic count, generally demonstrating around 50–60 mitoses in LCNEC and 70–80 mitoses in SCLC [75], with variable necrosis amount accounting for prognosis deterioration [55, 61].

Histomorphologic and immunohistochemical features of lung carcinoids. Typical carcinoid shows a variety of histological patterns, including organoid to trabecular (a), lobular (b), or spindle cells (c), but always with < 2 mitoses per 2 mm2 and absent necrosis. Immunohistochemistry for neuroendocrine markers documents diffuse and intense positivity for chromogranin A (d) and synaptophysin (d, inset). This atypical carcinoid featuring a trabecular architecture exhibits at least two mitoses (e, dotted white circle), which are also easily recognizable in another similarly patterned case (f, dotted white circle). Atypical carcinoid may exhibit punctate necrosis, in this case against a background of a solid-appearing tumor (g, dotted white circle). Immunohistochemistry for neuroendocrine markers in atypical carcinoid documents reactivity for synaptophysin (h), chromogranin A (h, inset) and INSM1 (i), all consistent with neuroendocrine morphology. Of note, INSM1 is also expressed in normal and hyperplastic neuroendocrine cells, as well as in tumorlets (not shown in the picture) (j)

Histomorphologic and immunohistochemical features of lung neuroendocrine carcinomas. LCNEC features organoid aggregates with peripheral palisading and plentiful mitoses (a), but even SCLC-like appearance (b). LCNEC is usually positive for INSM1 (c), while retinoblastoma nuclear decoration is often missing (d). SCLC is composed of small-sized tumor cells with prominent nuclear molding, inconspicuous nucleoli, and abundant necrosis (e), but spindling of neoplastic cells may be on record (f). Synaptophysin is largely retained in this case (g), along with INSM1 labeling (h), while chromogranin A is usually reduced to negative (g, inset), just like does retinoblastoma that is consistently unreactive (h, inset). Differential diagnosis between carcinoids (i) and SCLC (j) may be demanding upon morphology in cytology samples, but Ki-67 staining helps to operate such a separation confidently showing low expression in carcinoid (i, inset) and high expression in SCLC (j inset). Note that distinction of typical and atypical carcinoids rests on mitotic count and/or punctate necrosis, while neuroendocrine carcinomas are especially distinguished according to cell size appreciation.
The concurrence of combined variants is a nearly exclusive prerogative of approximately 25–30% NECs [75]. In SCLC, combined variants are defined by the association with any other subtype of non-small cell carcinoma (NSCC) (LCNEC, adenocarcinoma, squamous cell carcinoma, sarcomatoid carcinoma, or large cell carcinoma), while combined LCNEC shows adjuncts exclusively of other NSCC subtypes [42, 75]. Just the association of SCLC and LCNEC requires a threshold of 10% for either tumor component to be present because of the continuum for cell size and nuclear changes [75]. Combined variants of NETs (mostly AC) with adenocarcinoma or squamous carcinoma are de facto anecdotal findings [28, 36, 39] and those with NECs are undescribed [32, 75].
Classification of lung NENs is applicable as a whole to surgical specimens only, whereas cytology/small biopsy samples need more simplified terminology, especially in the event of crush artifacts [3, 11, 41]. TC and AC are grouped together as carcinoids, and LCNEC is only suggested as possible option of diagnosis.
The integration of immunohistochemistry
As a part of morphologic NE differentiation, lung NENs show consistent IHC expression of pan-NE markers, such as chromogranin A, synaptophysin, or insulinoma-associated protein 1 (INSM1), the latter being a newly released marker of NE differentiation in the lung [15, 16, 63], effective even in cytology/biopsy samples [1] (Table 1). In NSCC lacking NE morphology, the current guidelines discourage performing NE markers. A minority of SCLC (about 10–15%) shows low/absent levels of NE markers, realizing the so-called variant subtype, which molecularly straddle morphologically undifferentiated NSCC and LCNEC [65, 69, 83].
The IHC characterization is a defining criterion in LCNEC to rule out NSCC, but is recommended in SCLC to improve its differential diagnosis towards close histologic mimickers, such as poorly differentiated squamous cell carcinoma and basaloid carcinoma, small round cell tumors, NUT carcinoma, or hematologic malignancies [74]. No diagnostic role is currently supported in lung NENs for Ki-67 labeling index because of the significant overlap between tumor categories. However, it can be helpful in small biopsies with crush artifacts or cytology samples in order to avoid overdiagnosing carcinoids as SCLC [3, 41].
In a wider context of differential diagnosis, IHC markers may help to exclude histologic mimickers, especially when facing with NECs [47, 81]. Briefly, (i) positive expression of a couple of NE immunomarkers (expectedly chromogranin A and synaptophysin ± INSM1) in the lung serves to put NE differentiation into the proper context of morphologic features, sample types (cytology, biopsy, resection), and extent of positive reaction [81]. Of note, INSM1 reactivity is consistently found not only across the entire spectrum of lung NENs but also even in normal and reactive NE cells as documented in Fig. 2j at variance with Achaete-Scute family BHLH transcription factor 1 (ASCL1) which is mostly expressed by NECs. CD56 should be considered an indicator of SCLC [27] rather than a marker of NE differentiation due to lack of specificity; (ii) p40 [43, 44] and/or high molecular weight cytokeratins [70] exclude, if negative, poorly differentiated squamous cell carcinoma and basaloid carcinoma; (iii) TTF1 rules out, if positive, the NE-low variant subtype of SCLC [23, 83]; (iv) cytokeratins are useful to highlight either epithelial differentiation or staining pattern, i.e., dot-like vs. cytoplasmic diffuse (lung carcinoids may be proven cytokeratin-negative especially in peripheral tumors, but separation from paraganglioma is clinically negligible); (v) retinoblastoma identifies, if positive, carcinoids and some NECs straddling LCNEC and SCLC and facing with the variant subtypes of SCLC [69]; (vi) p53 abnormal expression, either diffuse staining or absence in all tumor cells, is commonly shared by subtypes of NECs but can also be found in carcinoids, albeit rarely [67]; (vii) lastly, demonstration of NUT protein identifies NUT carcinoma, regardless of the remaining IHC patterning [81].
Beyond diagnosis, several IHC biomarkers can be useful in the emerging clinical setting of targeted therapies and immuno-oncology setting of lung NENs. They include somatostatin receptors in NETs [60]; delta-like canonical Notch Ligand 3 (DLL3), an inhibitory NOTCH ligand, in SCLC [38]; mammalian target of rapamycin (mTOR) in NETs and LCNEC [59, 82]; and programmed death-ligand 1 (PD-L1) [76] or thymidylate synthase (TS) [24] in NECs.
Clinical correlations
A direct inference of the histological classification occurs in clinical behavior, according to which TC are low-grade (G1) malignant tumors with long survival and good prognosis even in the event of distant metastases, AC intermediate grade (G2) malignant tumors with significantly increased rates of distant metastases and considerably worse prognosis [6, 22, 62], and NEC full-fledged high-grade carcinomas (tautologically G3) with the highest likelihood of lymph node and visceral metastases and dismal prognosis in spite of therapy efforts [9, 25, 35] (Table 3).
Paraneoplastic syndromes can be observed in either NETs or NECs, whether endocrinological or non-endocrinological. SCLC account for the most frequent lung cancer associated with paraneoplastic syndromes (up to 20% of instances), with either ectopic hormone production (hyponatremia, Cushing syndrome) or autoimmune-mediated destruction upon onconeural neoantigen expression (paraneoplastic encephalomyelitis or dermatomyositis with poorer outcome and Lambert-Eaton myasthenic syndrome with more prolonged clinical course). In LCNEC, paraneoplastic syndromes are quite uncommon, with single records of ectopic adrenocorticotropic hormone syndrome (EACTH) and Lambert-Eaton syndrome or cancer-associated retinopathy. In NETs, TCs are the major reservoir of paraneoplastic syndromes (5%) as compared to AC (< 2%), mostly of endocrinological type (EACTH [29], GH-related acromegaly, histamine-related atypical carcinoid syndrome), but even of non-endocrinological type (giant cell arteritis, rheumatic polymyalgia, or polymyositis). Regarding demography, NECs by far prevail in elderly male smokers, while NETs, particularly TC, slightly exceed in younger female non-smokers (Table 3). Regarding treatment, NETs are primarily managed by surgery, deserving multimodal approaches to metastatic cases (especially AC) [4, 6, 21, 22], whereas NECs are mostly treated by (neo)adjuvant chemoradiotherapy [9, 21, 25, 26], a deserving surgery to very early-stage lesions (5% or less of patients) [31]. Options of targeted therapies and/or immunoncology scenarios, which are being increasingly explored in both NETs and NECs, also require accurate subtyping of lung NEN patients.
Molecular pathology
Pulmonary NENs encompass a substantially heterogeneous spectrum of molecular alterations, whose profiling could aid in the clinical management of patients by means of tailored treatments.
Regarding low-grade arm, Fernandez-Cuesta et al. performed whole-exome sequencing (WES) of 69 pulmonary carcinoids identifying alterations of chromatin remodeling genes, namely, menin 1 (MEN1), AT-rich interaction domain 1A (ARID1A), and eukaryotic translation initiation factor 1A X-linked (EIF1AX), as significantly mutated genes along with low-mutation burden (a mean somatic mutation rate of 0.4 mutations per megabase (Mb) of sequenced DNA) [12]. These molecular findings were confirmed by subsequent studies, which also argued against genetic separation of carcinoids likely due to the existence of common precursors [30, 67]. In a case series examined by Swarts et al. [73], 7/55 (13%) of pulmonary carcinoids harbored MEN1 mutations associated with reduced mRNA expression and poor prognosis. Also, in mutation-absent tumors, low MEN1 gene expression was correlated with an adverse disease outcome [73]. In keeping with the abovementioned data, WES analysis of 14 pulmonary carcinoids (10 TC and 4 AC) confirmed a low mean somatic mutation rate of 0.3 per Mb [34]. Moreover, the number of somatic alterations was associated with increasing proliferation activity with both Ki67 index (p < 0.01) and mitotic count (p < 0.01). On the other hand, Mucin 6 (MUC6) and spectrin alpha erythrocytic 1 (SPTA1) were recurrently mutated at a frequency of 21% (3/14) and 14% (2/14), respectively. Of note, pathway analysis of the mutated genes revealed enrichment of genes involved in mitogen-activated protein kinase (MAPK) signaling, regulation of the actin cytoskeleton and focal adhesion, and transforming growth factor (TGF)-β signaling [34]. Despite small sample size, 307 genes were found to be differentially expressed to a significant degree in 5 pulmonary carcinoids with favorable prognosis as compared with 5 cases with an adverse clinical course. Within all carcinoids as well as atypical carcinoids, RET proto-oncogene (RET) upregulation and orthopedia homeobox gene (OTP) and CD44 molecule (Indian blood group) downregulation were significantly associated with low 20-year survival of patients [71, 72]. Moreover, chromosomal instability, in terms of increased frequency and extent of chromosomal alterations, was found as a common event in atypical and metastasized carcinoids [79].
The literature data indicate that the genetic profile of pulmonary carcinoids is considerably different from that of high-grade NECs, which demonstrate an overall high-tumor mutation burden (> 7 per megabase pairs) [19, 64]. The mutation frequency increases with the tumor’s higher grade or biological malignancy as heralded in other lung cancer subtypes [46]. In this regard, in a paper by Vollbrecht et al., the SMAD family member 4 (SMAD4) mutation was found in TC subtype, while KIT proto-oncogene receptor tyrosine kinase (c-KIT), phosphatase and tensin homolog (PTEN), HNF1 homeobox A (HNF1A), and smoothened frizzled class receptor (SMO) were altered in AC [78]. On the other hand, Janus kinase 3 (JAK3), NRAS proto-oncogene (NRAS), RB transcriptional corepressor 1 (RB1), and Von Hippel-Lindau tumor suppressor (VHL1) were exclusively identified in SCLC, whereas the fibroblast growth factor receptor 2 (FGFR2) mutation was detected in LCNEC only [78]. Of note, JAK3 mutations were also found in LCNEC of the thymus, an organ where the classification criteria of NENs are the same as in the lung, which secondarily developed from preexisting AC [10]. In experimental models, inhibition of NOTCH gene signaling induces lung progenitor cells to form pulmonary neuroendocrine cells, from which tumors resembling early-stage SCLC grew in immunodeficient mice via RB and TP53 simultaneous blockage and transcriptome enrichment with cell cycle–related RNAs [7].
LCNEC make up the widest spectrum of molecular heterogeneity among lung NENs, with some of them being classified as SCLC-like LCNEC accounting for about 40%, NSCLC-like LCNEC accounting for about 50% and carcinoid-like LCNEC accounting for about 5% on the basis of different sets of altered genes [57, 58]. SCLC-like LCNEC share molecular alterations with SCLC and show RB1, tumor protein P53 (TP53), CREB binding protein (CREBBP), E1A binding protein P300 (EP300), and lysine methyltransferase 2A (KMT2A) gene mutations alongside V-myc myelocytomatosis viral oncogene homolog 1 (MYCL1) and FGFR1 amplifications. NSCLC-like LCNEC exhibit cyclin-dependent kinase inhibitor 2A (CDKN2A) deletion, transcription termination factor 1 (TTF1) amplifications, and Kelch-like ECH-associated protein 1 (KEAP1) and serine/threonine kinase 11 (STK11) mutations as observed in non-NE tumors. Finally, carcinoid-like LCNEC bear MEN1 mutations [8, 57]. Interestingly, there is also a molecular link between NE-low and RB1-wild type LCNEC and variant subtypes of SCLC [65] that exhibit either low levels of NE differentiation or larger cell morphology, thus challenging once again LCNEC as a unitary neoplastic entity [69].
In a more recent study by Simbolo et al. [68] that investigated a case series consisting of AC and LCNEC, three transcriptional clusters were identified. Cluster 1 comprised 20 LCNECs and one AC harboring concurrent inactivation of TP53 and RB1, in the absence of MEN1 mutations and RB1 nuclear decoration. Cluster 2 included 14 AC and eight LCNEC showing intermediate features: inactivation of TP53, 40.9%; MEN1, 22.7%; and RB1, 18.2%. Cluster 3 comprehended 20 AC and four LCNEC lacking RB1 alterations and having frequent MEN1 (37.5%) and TP53 mutations (16.7%). Moreover, menin nuclear immunostaining was lost in 75% of cases. Notably, patients in cluster C1 had a shorter cancer-specific survival than did patients in C2 or C3, thus reflecting the importance of gene signature for tumor behavior as already stated [2, 50, 51, 68].
Zhou et al. [84] performed capture-based ultra-deep targeted sequencing on tumor samples of LCNEC, large cell carcinoma (LCC), and SCLC, revealing a molecular signature consisting of RUNX family transcription factor 1 (RUNX1), Erb-B2 receptor tyrosine kinase 4 (ERBB4), breast cancer 1 (BRCA1), and EPH receptor A3 (EPHA3), distinctively mutated in LCNEC. A majority (60%) of LCNEC patients harbored copy number variations (CNVs). Notably, NFKB inhibitor alpha (NFKBIA) amplification was shared between LCNEC and LCC, while AKT serine/threonine kinase 2 (AKT2) amplification was private to LCNEC and SCLC. There were no common CNVs shared among the three investigated subtypes. Furthermore, genetic alterations in the PI3K/AKT/mTOR pathway were enriched in all three subtypes, thus indicating converging effector mechanisms on carcinogenesis.
Uccella et al., casting 6 TC, 4 AC, 11 LCNEC, and 8 SCLC, demonstrated a significantly higher expression of ribonuclease T2 (RNASET2), hypoxia-inducible factor 1 subunit alpha (HIF-1α), and its target carbonic anhydrase IX (CA IX) in NECs than NETs [77]. Moreover, in vitro data showed that an overexpression of RNASET2 is consequence of the activation of HIF-1α. The authors suggested that RNASET2 increased expression may contribute to phenotypic alterations of NECs, such as the high apoptotic rate and the extensive necrosis [77].
In a study by George et al., two molecular subgroups of LCNECs were identified by means of comprehensive genomic and transcriptomic analyses [20], namely, “type I LCNEC” with biallelic TP53 and serine/threonine kinase 11/Kelch-like ECH-associated protein 1 (STK11/KEAP1) alterations (37%), and “type II LCNECs” enriched for biallelic inactivation of TP53 and RB1 (42%). LCNECs form distinct transcriptional subgroups with closest similarity to SCLC. While type I LCNECs and SCLCs exhibited a neuroendocrine profile with ASCL1-high/delta-like canonical Notch Ligand 3 (DLL3) high/Notch Receptor 1 (NOTCH) low, type II LCNECs bore TP53 and RB1 alterations and differed from most SCLC tumors for reduced neuroendocrine markers, a pattern of ASCL1-low/DLL3-low/NOTCH-high and upregulation of immune-related pathways [20].
SCLC typically has one of the highest mutation rates in cancers, bearing biallelic inactivation of TP53 and RB1 as a hallmark [19, 40, 52], quite uncommon in TC and AC [5, 67]. Also, chromatin remodeling genes (CREBBP, EP300, KMT2A) are commonly found altered [12, 19, 40, 52]. Of note, in addition to mutual biallelic RB1 and TP53 alterations, NOTCH gene inactivation with simultaneous ASCL1 and canonical WNT signaling engagement is the basis of pulmonary and extra-pulmonary small cell carcinoma developing as secondary tumors from preexisting non-NE carcinomas, either spontaneous or induced by therapy [33, 49,50,51]. A novel molecular stratification of SCLC tumors has recently been proposed by Rudin et al. [65] by integrating experimental data with clinical material and based on the relative abundance of genes encoding four key transcriptional factors, namely, ASCL1, neuronal differentiation 1 (NEUROD1), POU class 2 homeobox 3 (POU2F3), and yes1-associated transcriptional regulator (YAP1), as ASCL1-high (SCLC-A), NEUROD1-high (SCLC-N), YAP1-high (SCLC-Y), and POU2F3-high (SCLC-P) subtypes. SCLC-Y and SCLC-P merged with NE-low variant subtypes of this tumor, which share similarities to either LCNEC or NSCC [69]. We have incorporated some molecular characteristics of both low- and high-grade lung NENs into Table 2.
An unsupervised approach: the issue of intratumor heterogeneity of classification
How to account for heterogeneity
The classification of lung NENs considers every different tumor category as a monolithic entity independent of each other rather than a combination of biologically diverse lesions, with major differences likely depending on natural history of disease (preclinical and clinical phases). It is tempting to hypothesize that the shorter the preclinical phase, the greater the clinical aggressiveness and the shorter the clinical course, with more severe gene alterations being responsible for undifferentiated tumors with cancer stem cell properties to arise. These tumors are likely to show resistance to therapy [51]. Conversely, the longer the preclinical phase, the lower the clinical aggressiveness and the more prolonged the clinical course, in the premise that over time accumulation of diverse gene alterations would cause variably differentiated tumors with progenitor cell properties to arise. These tumors are likely to be more amenable to therapy [50, 51]. In this scenario, tumor fate, clinical outcome, and susceptibility to therapies of lung NENs might be largely pre-determined at the onset and/or modeled over time by diverse molecular signatures, which in turn are the mirror of different risk factors acting persistently on lung tissue microenvironment [33, 50, 51]. Of note, while both the cells of origin and the driver genetic alterations are likely to play a role in shaping ultimate lung cancer phenotypes, the relationship between ancestors and cancer subtypes is not necessarily one-to-one, inasmuch as different sets of genetic/epigenetic alterations could result in variably differentiated and/or reprogrammed cancer cells [13, 14, 53, 54, 56, 66, 80]. This challenging interaction between ancestors and molecular alterations, in turn continuously under the influence of risk factors, gives rise to the important phenomena of intertumoral and intratumoral heterogeneity, subtype transdifferentiation, cell pliancy, and latency of lung tumors, which represent the pathologic and molecular bases of clinical outcome and potential targets of therapy [14, 50, 51, 56, 66]. All these issues, however, are only partially accounted for by the current classification, because defining criteria are not precise enough to intercept the great variety of neoplasms at the level of an individual patient’s cancer and thus to account for the natural history of tumors as the major determinant of their fate [51]. AC and LCNEC show the least reproducible diagnostic criteria and thus the broadest range of biological properties (prognosis, expression profiles, carcinogenesis paths, sensitivity to therapies), which is much less remarkable in either TC (well differentiated cells with negligible proliferation activity and no necrosis) or SCLC (undifferentiated small cells resembling stem cells with huge proliferation activity and geographic necrosis) (Table 1, 2, and 3). This intratumor heterogeneity of lung NEN classification can be unraveled by focusing on tumor grading, interobserver variability, expression profiles and single-gene alterations, survival analysis, omics studies, and pathogenesis models [45, 48, 50, 51].
Modeling the pathogenesis of lung neuroendocrine neoplasms
The homeostasis of epithelial cells is maintained by an asymmetrical self-renewal of stem cells putatively aligned along basal layer of stratified epithelia or lineage compartments (horizontal or de novo/basal-like propagation) and differentiation of progenitor elements committed to specific cell lineages (vertical or luminal-like evolution). In this regard, tumor population could variably comprise cancer stem cells (CSC), adult-differentiated elements genetically reprogrammed during tumorigenesis (cell pliancy), progenitor elements derived from CSCs, deprogrammed cells, and differentiating cells, thereby realizing a heterogeneous assembly whose relative composition might result from different gene alterations and hence developmental mechanisms [14, 51, 66]. Of note, retinoblastoma (RB1) and TP53 genes suppress self-renewal, whereas Notch marks stem cells and initiates deprogramming and transit amplification [37]. Accordingly, this reciprocal proportion of transformed cells could reflect different genetic/epigenetic mechanisms of tumorigenesis and/or microenvironment stimuli, which could be related to and/or maintained by risk factors, either exogenous or endogenous, acting during the natural history of disease. Therefore, we can schematically identify three different types of neoplastic lesions. Each one of them is likely to derive from different mechanisms [51]. This interpretation model is inspired by an unsupervised approach to genetic signatures, which are likely to shape the final morphologic appearance of tumor cells [50] (Fig. 4).
-
a)
An early maturation block caused by crucial genetic events (e.g., biallelic inactivation of RB1 and TP53, NOTCH silencing) would promote horizontal or de novo/basal-like expansion and/or re/deprogramming of differentiated and/or progenitor cells. These mechanisms result in tumors enriched in neoplastic stem cells which are undifferentiated, with high biological malignancy and short preclinical phase. While dysplastic/in situ lesions composed of undifferentiated tumor cells have been experimentally documented in mouse models [17, 18], in humans, the smoking-damaged normal epithelium associated with SCLC is genetically scrambled by similar molecular alterations. These tumors, which can be further modulated by additional genetic alterations giving rise to similar lesions still dominated by CSCs in pure or combined forms, might derive from primordial adult cells out of a neuroendocrine niche even capable of multidivergent evolution [33, 66]. Therefore, they would represent primary or early-aggressive high-grade NETs (PEA-HGNETs/NECs) as a function of their short natural history as reflected by very short clinical course [51]. Transversions C > A are usually present due to their close relationship with smoking. Classical SCLC usually feature diffuse growth of undifferentiated cells (sometimes mimicking hematologic malignancies) [18], with possible multidivergent differentiation but still in a background of diffuse pattern of growth. These tumors are usually diagnosed on biopsy/cytology samples because of advanced disease at presentation, show low mutational heterogeneity between primaries and metastases, and are resistant to therapy with dismal prognosis [51]. These tumors show very high Ki-67 labeling index (usually 60–100%), with homogeneous distribution of stained cells inside tumors. PEA-HGNETs/NECs could account for 70–75% of lung NETs and about 13% of all lung cancers [51].
-
b)
An intermediate maturation block caused by a variety of gene alterations, such as TP53 → RB1 mono/biallelic inactivation, NOTCH alteration, KRAS/LKB1/MEN1 mutation, MYC/MYCL/TERT/SDHA/RICTOR amplification, or epithelial-mesenchymal transition, would promote vertical or luminal-like mechanisms and/or re/deprogramming of differentiated cells giving rise to tumors enriched in progenitor cells. They show a greater spectrum of morphologic differentiation and less biological aggressiveness. Transversions C > A are still present due to their association with smoking. These tumors can be further modeled by additional genetic alterations and are enriched by progenitor and better differentiated cells. They would be likely to derive from neuroendocrine and non-neuroendocrine precursors such as carcinoids or their ancestors or NSCC. Therefore, they would represent differentiating or secondary high-grade NETs (DS-HGNETs/NECs) as a function of their longer natural history [33, 50, 51]. Carcinoids with elevated indices of proliferation, LCNEC resembling NSCC or carcinoid and SCLC are usually seen with organoid patterns of growth even in combined variants [33, 50, 51]. Other features of these DS-HGNETs/NECs are lower stage at presentation, diagnosis on resection specimens, better prognosis, higher mutational heterogeneity between primaries and related metastases [80], and greater curability rates. These tumors present with a variable Ki-67 labeling index (ranging from 10 to 60%), typically with a heterogeneous distribution inside tumors. DS-HGNETs/NECs could account for 20–25% of lung NENs and about 6% of all lung cancers [51].
-
c)
A late maturation block caused by genetic/epigenetic alterations without genetic segregation would promote vertical luminal-like mechanisms giving rise to tumors enriched in differentiated cells, with low-grade dysplastic lesions (e.g., diffuse idiopathic neuroendocrine cell hyperplasia, DIPNECH), long preclinical phase and best prognosis. Transversions C > A are quite uncommon, but these smoking-linked tumors could be at higher risk of progression to DS-HGNETs/NECs [50]. Tumors would derive from mature NE/preinvasive lesions (e.g., DIPNECH) for genetic re/deprogramming and represent indolent NETs (I-NETs) with long-term preclinical phase [51]. These tumors mostly feature TC or low proliferating AC (e.g., ≤ 3 mitoses per 2 mm2), with organoid pattern of growth, diagnosis performed on resection specimens, low stage at presentation with favorable clinical outcome, and best curability upon surgery. I-NETs exhibit a low Ki-67 labeling index (≤ 10%), typically featuring homogeneous distribution inside tumors. They could account for 5% of lung NETs and about 1% of all lung cancers [51].

Development model of lung neuroendocrine neoplasms. Mechanisms of horizontal or de novo/basal-like propagation from cancer stem cells early blocked-in differentiation give rise to primary early-aggressive high-grade tumors (PEA-HGNETs/NECs). They are characterized by undifferentiated cells resembling typical SCLC featuring diffuse pattern of growth. Mechanisms of vertical- or luminal-like propagation from progenitor cells with an intermediate block of differentiation cause more differentiated lesions to develop from pre-existing neuroendocrine tumors or non-small cell carcinoma (NET-like DS-HGNET/NEC and NSCC-like DS-HGNET/NEC, respectively). They are characterized by better differentiated cells featuring a variety of histologic appearances along with organoid patterns of growth. Lastly, there are indolent NETs (I-NETs) composed of fully differentiated cells (in red and green on the right of picture), which usually do not progress further over time but can contribute to DS-HGNETs/NECs by re/deprogramming events. PEA-HGNET/NEC, primary early-aggressive high-grade neuroendocrine tumor/carcinoma; DS-HGNET/NEC, differentiating secondary high-grade neuroendocrine tumor/carcinoma; I-NET, indolent NET
Of note, Ki-67 labeling index could be a major forerunner to this classification, with a homogeneous intratumor distribution in PEA-HGNETs/NECs and I-NETs, and an uneven distribution in DS-HGNETs/NECs due to coexistence of closely intermingling low and high proliferating tumor areas. Integration of the Ki-67 labeling index with the WHO classification could help to better stratify patients for different therapy options [48, 49].
Conclusions
The WHO classification of lung NENs is an indispensable tool in clinical practice. Separation between NETs and NECs is fundamental to provide accurate and adequate management of patients considering significant differences in molecular landscapes, survival expectancy, and therapy options. However, while privileging an unsupervised approach upon molecular signatures, it is possible to reappraise lung NENs according to differences in risk factors and pathogenesis models where intratumor heterogeneity of Ki-67 labeling index could play an important role.
References
Abe H, Takase Y, Sadashima E, Fukumitsu C, Murata K, Ito T, Kawahara A, Naito Y, Akiba J (2019) Insulinoma-associated protein 1 is a novel diagnostic marker of small cell lung cancer in bronchial brushing and cell block cytology from pleural effusions: validity and reliability with cutoff value. Cancer Cytopathol 127:598–605
Alcala N, Leblay N, Gabriel AAG, Mangiante L, Hervas D, Giffon T, Sertier AS, Ferrari A, Derks J, Ghantous A, Delhomme TM, Chabrier A, Cuenin C, Abedi-Ardekani B, Boland A, Olaso R, Meyer V, Altmuller J, Le Calvez-Kelm F, Durand G, Voegele C, Boyault S, Moonen L, Lemaitre N, Lorimier P, Toffart AC, Soltermann A, Clement JH, Saenger J, Field JK, Brevet M, Blanc-Fournier C, Galateau-Salle F, Le Stang N, Russell PA, Wright G, Sozzi G, Pastorino U, Lacomme S, Vignaud JM, Hofman V, Hofman P, Brustugun OT, Lund-Iversen M, Thomas de Montpreville V, Muscarella LA, Graziano P, Popper H, Stojsic J, Deleuze JF, Herceg Z, Viari A, Nuernberg P, Pelosi G, Dingemans AMC, Milione M, Roz L, Brcic L, Volante M, Papotti MG, Caux C, Sandoval J, Hernandez-Vargas H, Brambilla E, Speel EJM, Girard N, Lantuejoul S, McKay JD, Foll M, Fernandez-Cuesta L (2019) Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat Commun 10:3407
Aslan DL, Gulbahce HE, Pambuccian SE, Manivel JC, Jessurun J (2005) Ki-67 immunoreactivity in the differential diagnosis of pulmonary neuroendocrine neoplasms in specimens with extensive crush artifact. Am J Clin Pathol 123:874–878
Baudin E, Hayes AR, Scoazec JY, Filosso PL, Lim E, Kaltsas G, Frilling A, Chen J, Kos-Kudla B, Gorbunova V, Wiedenmann B, Nieveen van Dijkum E, Cwikla JB, Falkerby J, Valle JW, Kulke MH, Caplin ME, Participants EMAB, Participants EMAB (2019) Unmet medical needs in pulmonary neuroendocrine (carcinoid) neoplasms. Neuroendocrinology 108:7–17
Beasley MB, Lantuejoul S, Abbondanzo S, Chu WS, Hasleton PS, Travis WD, Brambilla E (2003) The P16/cyclin D1/Rb pathway in neuroendocrine tumors of the lung. Hum Pathol 34:136–142
Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E, Oberg K, Pelosi G, Perren A, Rossi RE, Travis WD, participants Ecc (2015) Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol 26:1604–1620
Chen HJ, Poran A, Unni AM, Huang SX, Elemento O, Snoeck HW, Varmus H (2019) Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J Exp Med 216:674–687
Clinical Lung Cancer Genome P, Network Genomic M (2013) A genomics-based classification of human lung tumors. Sci Transl Med 5:209ra153
Eldessouki I, Gaber O, Namad T, Wang J, Morris JC, Abdel Karim N (2018) Small or non-small cell lung cancer based therapy for treatment of large cell neuroendocrine cancer of the lung? University of Cincinnati Experience. J Oncol 2018:9761826
Fabbri A, Cossa M, Sonzogni A, Bidoli P, Canova S, Cortinovis D, Abbate MI, Calabrese F, Nannini N, Lunardi F, Rossi G, La Rosa S, Capella C, Tamborini E, Perrone F, Busico A, Capone I, Valeri B, Pastorino U, Albini A, Pelosi G (2017) Thymus neuroendocrine tumors with CTNNB1 gene mutations, disarrayed ss-catenin expression, and dual intra-tumor Ki-67 labeling index compartmentalization challenge the concept of secondary high-grade neuroendocrine tumor: a paradigm shift. Virchows Arch 471:31–47
Fabbri A, Cossa M, Sonzogni A, Papotti M, Righi L, Gatti G, Maisonneuve P, Valeri B, Pastorino U, Pelosi G (2017) Ki-67 labeling index of neuroendocrine tumors of the lung has a high level of correspondence between biopsy samples and surgical specimens when strict counting guidelines are applied. Virchows Arch 470:153–164
Fernandez-Cuesta L, Peifer M, Lu X, Sun R, Ozretic L, Seidal D, Zander T, Leenders F, George J, Muller C, Dahmen I, Pinther B, Bosco G, Konrad K, Altmuller J, Nurnberg P, Achter V, Lang U, Schneider PM, Bogus M, Soltermann A, Brustugun OT, Helland A, Solberg S, Lund-Iversen M, Ansen S, Stoelben E, Wright GM, Russell P, Wainer Z, Solomon B, Field JK, Hyde R, Davies MP, Heukamp LC, Petersen I, Perner S, Lovly C, Cappuzzo F, Travis WD, Wolf J, Vingron M, Brambilla E, Haas SA, Buettner R, Thomas RK (2014) Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun 5:3518
Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, Lambooij JP, Proost N, Gargiulo G, Berns A (2016) SOX2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell 30:519–532
Ferone G, Lee M, Sage J, Berns A (2020) Cells of origin of lung cancers: lessons from mouse studies. Genes Dev 34:1017–1032
Fujino K, Motooka Y, Hassan WA, Ali Abdalla MO, Sato Y, Kudoh S, Hasegawa K, Niimori-Kita K, Kobayashi H, Kubota I, Wakimoto J, Suzuki M, Ito T (2015) Insulinoma-associated protein 1 is a crucial regulator of neuroendocrine differentiation in lung cancer. Am J Pathol 185:3164–3177
Fujino K, Yasufuku K, Kudoh S, Motooka Y, Sato Y, Wakimoto J, Kubota I, Suzuki M, Ito K (2017) INSM1 is the best marker for the diagnosis of neuroendocrine tumors: comparison with CGA, SYP and CD56. Int J. Clin Exp Pathol 10:5393–5405
Gazdar AF (2018) Morphologic and other forms of heterogeneity in small Cell lung cancer: what can we learn from them? J Thorac Oncol 13:148–150
Gazdar AF, Bunn PA, Minna JD (2017) Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 17:725–737
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, Vaka D, Torres A, Wang MS, Korbel JO, Menon R, Chun SM, Kim D, Wilkerson M, Hayes N, Engelmann D, Putzer B, Bos M, Michels S, Vlasic I, Seidel D, Pinther B, Schaub P, Becker C, Altmuller J, Yokota J, Kohno T, Iwakawa R, Tsuta K, Noguchi M, Muley T, Hoffmann H, Schnabel PA, Petersen I, Chen Y, Soltermann A, Tischler V, Choi CM, Kim YH, Massion PP, Zou Y, Jovanovic D, Kontic M, Wright GM, Russell PA, Solomon B, Koch I, Lindner M, Muscarella LA, la Torre A, Field JK, Jakopovic M, Knezevic J, Castanos-Velez E, Roz L, Pastorino U, Brustugun OT, Lund-Iversen M, Thunnissen E, Kohler J, Schuler M, Botling J, Sandelin M, Sanchez-Cespedes M, Salvesen HB, Achter V, Lang U, Bogus M, Schneider PM, Zander T, Ansen S, Hallek M, Wolf J, Vingron M, Yatabe Y, Travis WD, Nurnberg P, Reinhardt C, Perner S, Heukamp L, Buttner R, Haas SA, Brambilla E, Peifer M, Sage J, Thomas RK (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524:47–53
George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F, Maas L, Muller C, Dahmen I, Delhomme TM, Ardin M, Leblay N, Byrnes G, Sun R, De Reynies A, McLeer-Florin A, Bosco G, Malchers F, Menon R, Altmuller J, Becker C, Nurnberg P, Achter V, Lang U, Schneider PM, Bogus M, Soloway MG, Wilkerson MD, Cun Y, McKay JD, Moro-Sibilot D, Brambilla CG, Lantuejoul S, Lemaitre N, Soltermann A, Weder W, Tischler V, Brustugun OT, Lund-Iversen M, Helland A, Solberg S, Ansen S, Wright G, Solomon B, Roz L, Pastorino U, Petersen I, Clement JH, Sanger J, Wolf J, Vingron M, Zander T, Perner S, Travis WD, Haas SA, Olivier M, Foll M, Buttner R, Hayes DN, Brambilla E, Fernandez-Cuesta L, Thomas RK (2018) Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun 9:1048
Gridelli C, Rossi A, Airoma G, Bianco R, Costanzo R, Daniele B, Chiara GD, Grimaldi G, Irtelli L, Maione P, Morabito A, Piantedosi FV, Riccardi F (2013) Treatment of pulmonary neuroendocrine tumours: state of the art and future developments. Cancer Treat Rev 39:466–472
Hendifar AE, Marchevsky AM, Tuli R (2017) Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol 12:425–436
Horie M, Miyashita N, Mattsson JSM, Mikami Y, Sandelin M, Brunnstrom H, Micke P, Nagase T, Saito A (2018) An integrative transcriptome analysis reveals a functional role for thyroid transcription factor-1 in small cell lung cancer. J Pathol
Ibe T, Shimizu K, Nakano T, Kakegawa S, Kamiyoshihara M, Nakajima T, Kaira K, Takeyoshi I (2010) High-grade neuroendocrine carcinoma of the lung shows increased thymidylate synthase expression compared to other histotypes. J Surg Oncol 102:11–17
Kalemkerian GP, Loo BW, Akerley W, Attia A, Bassetti M, Boumber Y, Decker R, Dobelbower MC, Dowlati A, Downey RJ, Florsheim C, Ganti AKP, Grecula JC, Gubens MA, Hann CL, Hayman JA, Heist RS, Koczywas M, Merritt RE, Mohindra N, Molina J, Moran CA, Morgensztern D, Pokharel S, Portnoy DC, Rhodes D, Rusthoven C, Sands J, Santana-Davila R, Williams CC, Hoffmann KG, Hughes M (2018) NCCN guidelines insights: small cell lung cancer, Version 2.2018. J Natl Compr Cancer Netw 16:1171–1182
Konala VM, Madhira BR, Ashraf S, Graziano S (2020) Use of immunotherapy in extensive-stage small cell lung cancer. Oncology:1–6
Kontogianni K, Nicholson AG, Butcher D, Sheppard MN (2005) CD56: a useful tool for the diagnosis of small cell lung carcinomas on biopsies with extensive crush artefact. J Clin Pathol 58:978–980
La Rosa S, Simbolo M, Franzi F, Uccella S, Imperatori A, Nardecchia E, Rotolo N, Dominioni L, Scarpa A, Sessa F (2018) Combined adenocarcinoma-atypical carcinoid of the lung. Targeted next-generation sequencing (NGS) suggests a monoclonal origin of the two components. Diagn Histopathol 24:120–123
La Rosa S, Volante M, Uccella S, Maragliano R, Rapa I, Rotolo N, Inzani F, Siciliani A, Granone P, Rindi G, Dominioni L, Capella C, Papotti M, Sessa F, Imperatori A (2019) ACTH-producing tumorlets and carcinoids of the lung: clinico-pathologic study of 63 cases and review of the literature. Virchows Arch 475:587–597
Laddha SV, da Silva EM, Robzyk K, Untch BR, Ke H, Rekhtman N, Poirier JT, Travis WD, Tang LH, Chan CS (2019) Integrative genomic characterization identifies molecular subtypes of lung carcinoids. Cancer Res 79:4339–4347
Lei Y, Feng H, Qiang H, Shang Z, Chang Q, Qian J, Zhang Y, Zhong R, Fan X, Chu T (2020) Clinical characteristics and prognostic factors of surgically resected combined small cell lung cancer: a retrospective study. Lung Cancer 146:244–251
Marchevsky AM, Wick MR (2015) Diagnostic difficulties with the diagnosis of small cell carcinoma of the lung. Semin Diagn Pathol 32:480–488
Meder L, Konig K, Ozretic L, Schultheis AM, Ueckeroth F, Ade CP, Albus K, Boehm D, Rommerscheidt-Fuss U, Florin A, Buhl T, Hartmann W, Wolf J, Merkelbach-Bruse S, Eilers M, Perner S, Heukamp LC, Buettner R (2016) NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer 138:927–938
Miyanaga A, Masuda M, Motoi N, Tsuta K, Nakamura Y, Nishijima N, Watanabe SI, Asamura H, Tsuchida A, Seike M, Gemma A, Yamada T (2020) Whole-exome and RNA sequencing of pulmonary carcinoid reveals chromosomal rearrangements associated with recurrence. Lung Cancer 145:85–94
Nicholson SA, Beasley MB, Brambilla E, Hasleton PS, Colby TV, Sheppard MN, Falk R, Travis WD (2002) Small cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol 26:1184–1197
Okazaki M, Sano Y, Soga Y, Katayama H, Sakao N, Shikatani Y, Yukumi S, Shigematsu H, Sugita A, Izutani H (2015) Combined atypical carcinoid tumour and squamous cell carcinoma of the lung. Intern Med 54:1385–1388
Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA (2019) Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell 179(403-416):e423
Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, He K (2019) DLL3: an emerging target in small cell lung cancer. J Hematol Oncol 12:61
Owens CL, Fraire AE (2011) Combined carcinoid tumor and squamous cell carcinoma of lung: expanding the spectrum of combined tumors of lung. Int J Surg Pathol 19:273–275
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, Altmuller J, Baessmann I, Becker C, de Wilde B, Vandesompele J, Bohm D, Ansen S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grutter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Lund-Iversen M, Sanger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Buttner R, Wolf J, Nurnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK (2012) Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44:1104–1110
Pelosi G, Rodriguez J, Viale G, Rosai J (2005) Typical and atypical pulmonary carcinoid tumor overdiagnosed as small-cell carcinoma on biopsy specimens: a major pitfall in the management of lung cancer patients. Am J Surg Pathol 29:179–187
Pelosi G, Sonzogni A, Galetta D, Perrone F, Braidotti P, Manzotti M, Fabbri A, Spaggiari L, Veronesi G, Viale G (2011) Combined small-cell carcinoma of the lung with quadripartite differentiation of epithelial, neuroendocrine, skeletal muscle, and myofibroblastic type. Virchows Arch 458:497–503
Pelosi G, Fabbri A, Bianchi F, Maisonneuve P, Rossi G, Barbareschi M, Graziano P, Cavazza A, Rekhtman N, Pastorino U, Scanagatta P, Papotti M (2012) DeltaNp63 (p40) and thyroid transcription factor-1 immunoreactivity on small biopsies or cellblocks for typing non-small cell lung cancer: a novel two-hit, sparing-material approach. J Thorac Oncol 7:281–290
Pelosi G, Rossi G, Cavazza A, Righi L, Maisonneuve P, Barbareschi M, Graziano P, Pastorino U, Garassino M, de Braud F, Papotti M (2013) DeltaNp63 (p40) distribution inside lung cancer: a driver biomarker approach to tumor characterization. Int J Surg Pathol 21:229–239
Pelosi G, Papotti M, Rindi G, Scarpa A (2014) Unraveling tumor grading and genomic landscape in lung neuroendocrine tumors. Endocr Pathol 25:151–164
Pelosi G, Fabbri A, Papotti M, Rossi G, Cavazza A, Righi L, Tamborini E, Perrone F, Settanni G, Busico A, Testi MA, Maisonneuve P, De Braud F, Garassino M, Valeri B, Sonzogni A, Pastorino U (2015) Dissecting pulmonary large-cell carcinoma by targeted next generation sequencing of several cancer genes pushes genotypic-phenotypic correlations to emerge. J Thorac Oncol 10:1560–1569
Pelosi G, Scarpa A, Forest F, Sonzogni A (2016) The impact of immunohistochemistry on the classification of lung tumors. Expert Rev Respir Med 10:1105–1121
Pelosi G, Pattini L, Morana G, Fabbri A, Faccinetto A, Fazio N, Valeri B, Sonzogni A (2017) Grading lung neuroendocrine tumors: controversies in search of a solution. Histol Histopathol 32:223–241
Pelosi G, Sonzogni A, Harari S, Albini A, Bresaola E, Marchio C, Massa F, Righi L, Gatti G, Papanikolaou N, Vijayvergia N, Calabrese F, Papotti M (2017) Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res 6:513–529
Pelosi G, Bianchi F, Dama E, Simbolo M, Mafficini A, Sonzogni A, Pilotto S, Harari S, Papotti M, Volante M, Fontanini G, Mastracci L, Albini A, Bria E, Calabrese F, Scarpa A (2018) Most high-grade neuroendocrine tumours of the lung are likely to secondarily develop from pre-existing carcinoids: innovative findings skipping the current pathogenesis paradigm. Virchows Arch 472:567–577
Pelosi G, Bianchi F, Hofman P, Pattini L, Strobel P, Calabrese F, Naheed S, Holden C, Cave J, Bohnenberger H, Dinter H, Harari S, Albini A, Sonzogni A, Papotti M, Volante M, Ottensmeier CH (2019) Recent advances in the molecular landscape of lung neuroendocrine tumors. Expert Rev Mol Diagn 19:281–297
Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordonez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ (2010) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463:184–190
Pozo K, Kelenis DP, Minna JD, Johnson JE (2018) Different originating cells underlie intertumoral heterogeneity in lung neuroendocrine tumors. Cancer Discov 8:1216–1218
Pozo K, Minna JD, Johnson JE (2018) Identifying a missing lineage driver in a subset of lung neuroendocrine tumors. Genes Dev 32:865–867
Proskuryakov SY, Gabai VL (2010) Mechanisms of tumor cell necrosis. Curr Pharm Des 16:56–68
Puisieux A, Pommier RM, Morel AP, Lavial F (2018) Cellular pliancy and the multistep process of tumorigenesis. Cancer Cell 33:164–172
Rekhtman N, Pietanza MC, Hellmann MD, Naidoo J, Arora A, Won H, Halpenny DF, Wang H, Tian SK, Litvak AM, Paik PK, Drilon A, Socci N, Poirier JT, Shen R, Berger MF, Moreira AL, Travis WD, Rudin CM, Ladanyi M (2016) Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin Cancer Res 22:3618–3629
Rekhtman N, Pietanza CM, Sabari J, Montecalvo J, Wang H, Habeeb O, Kadota K, Adusumilli P, Rudin CM, Ladanyi M, Travis WD, Joubert P (2018) Pulmonary large cell neuroendocrine carcinoma with adenocarcinoma-like features: napsin A expression and genomic alterations. Mod Pathol 31:111–121
Righi L, Volante M, Rapa I, Tavaglione V, Inzani F, Pelosi G, Papotti M (2010) Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr Relat Cancer 17:977–987
Righi L, Volante M, Tavaglione V, Bille A, Daniele L, Angusti T, Inzani F, Pelosi G, Rindi G, Papotti M (2010) Somatostatin receptor tissue distribution in lung neuroendocrine tumours: a clinicopathologic and immunohistochemical study of 218 ‘clinically aggressive’ cases. Ann Oncol 21:548–555
Rindi G, Klersy C, Inzani F, Fellegara G, Ampollini L, Ardizzoni A, Campanini N, Carbognani P, De Pas TM, Galetta D, Granone PL, Righi L, Rusca M, Spaggiari L, Tiseo M, Viale G, Volante M, Papotti M, Pelosi G (2014) Grading the neuroendocrine tumors of the lung: an evidence-based proposal. Endocr Relat Cancer 21:1–16
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, Busam KJ, de Krijger RR, Dietel M, El-Naggar AK, Fernandez-Cuesta L, Kloppel G, McCluggage WG, Moch H, Ohgaki H, Rakha EA, Reed NS, Rous BA, Sasano H, Scarpa A, Scoazec JY, Travis WD, Tallini G, Trouillas J, van Krieken JH, Cree IA (2018) A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol 31:1770–1786
Rooper LM, Sharma R, Li QK, Illei PB, Westra WH (2017) INSM1 demonstrates superior performance to the individual and combined use of synaptophysin, chromogranin and CD56 for diagnosing neuroendocrine tumors of the thoracic cavity. Am J Surg Pathol 41:1561–1569
Rossi G, Bertero L, Marchio C, Papotti M (2018) Molecular alterations of neuroendocrine tumours of the lung. Histopathology 72:142–152
Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, Massion PP, Minna JD, Oliver TG, Quaranta V, Sage J, Thomas RK, Vakoc CR, Gazdar AF (2019) Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 19:289–297
Semenova EA, Nagel R, Berns A (2015) Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev 29:1447–1462
Simbolo M, Mafficini A, Sikora KO, Fassan M, Barbi S, Corbo V, Mastracci L, Rusev B, Grillo F, Vicentini C, Ferrara R, Pilotto S, Davini F, Pelosi G, Lawlor RT, Chilosi M, Tortora G, Bria E, Fontanini G, Volante M, Scarpa A (2017) Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol 241:488–500
Simbolo M, Barbi S, Fassan M, Mafficini A, Ali G, Vicentini C, Sperandio N, Corbo V, Rusev B, Mastracci L, Grillo F, Pilotto S, Pelosi G, Pelliccioni S, Lawlor RT, Tortora G, Fontanini G, Volante M, Scarpa A, Bria E (2019) Gene expression profiling of lung atypical carcinoids and large cell neuroendocrine carcinomas identifies three transcriptomic subtypes with specific genomic alterations. J Thorac Oncol
Sonkin D, Thomas A, Teicher BA (2019) Are neuroendocrine negative small cell lung cancer and large cell neuroendocrine carcinoma with WT RB1 two faces of the same entity? Lung Cancer Manag 8:LMT13
Sturm N, Lantuejoul S, Laverriere MH, Papotti M, Brichon PY, Brambilla C, Brambilla E (2001) Thyroid transcription factor 1 and cytokeratins 1, 5, 10, 14 (34betaE12) expression in basaloid and large-cell neuroendocrine carcinomas of the lung. Hum Pathol 32:918–925
Swarts DR, Henfling ME, Van Neste L, van Suylen RJ, Dingemans AM, Dinjens WN, Haesevoets A, Rudelius M, Thunnissen E, Volante M, Van Criekinge W, van Engeland M, Ramaekers FC, Speel EJ (2013) CD44 and OTP are strong prognostic markers for pulmonary carcinoids. Clin Cancer Res 19:2197–2207
Swarts DR, Van Neste L, Henfling ME, Eijkenboom I, Eijk PP, van Velthuysen ML, Vink A, Volante M, Ylstra B, Van Criekinge W, van Engeland M, Ramaekers FC, Speel EJ (2013) An exploration of pathways involved in lung carcinoid progression using gene expression profiling. Carcinogenesis 34:2726–2737
Swarts DR, Scarpa A, Corbo V, Van Criekinge W, van Engeland M, Gatti G, Henfling ME, Papotti M, Perren A, Ramaekers FC, Speel EJ, Volante M (2014) MEN1 gene mutation and reduced expression are associated with poor prognosis in pulmonary carcinoids. J Clin Endocrinol Metab 99:E374–E378
Thunnissen E, Borczuk AC, Flieder DB, Witte B, Beasley MB, Chung JH, Dacic S, Lantuejoul S, Russell PA, den Bakker M, Botling J, Brambilla E, de Cuba E, Geisinger KR, Hiroshima K, Marchevsky AM, Minami Y, Moreira A, Nicholson AG, Yoshida A, Tsao MS, Warth A, Duhig E, Chen G, Matsuno Y, Travis WD, Butnor K, Cooper W, Mino-Kenudson M, Motoi N, Poleri C, Pelosi G, Kerr K, Aisner SC, Ishikawa Y, Buettner RH, Keino N, Yatabe Y, Noguchi M (2017) The use of immunohistochemistry improves the diagnosis of small cell lung cancer and its differential diagnosis. An International reproducibility study in a demanding set of cases. J Thorac Oncol 12:334–346
Travis W, Brambilla E, Burke A, Marx A, Nicholson A (2015) WHO classification of tumours of the lung, pleura, thymus and heart. IARC Press, Lyon
Tsuruoka K, Horinouchi H, Goto Y, Kanda S, Fujiwara Y, Nokihara H, Yamamoto N, Asakura K, Nakagawa K, Sakurai H, Watanabe SI, Tsuta K, Ohe Y (2017) PD-L1 expression in neuroendocrine tumors of the lung. Lung Cancer 108:115–120
Uccella S, La Rosa S, Scaldaferri D, Monti L, Maragliano R, Sorrenti E, Gariboldi M, Taramelli R, Sessa F, Acquati F (2018) New insights into hypoxia-related mechanisms involved in different microvascular patterns of bronchopulmonary carcinoids and poorly differentiated neuroendocrine carcinomas. Role of ribonuclease T2 (RNASET2) and HIF-1alpha. Hum Pathol 79:66–76
Vollbrecht C, Werner R, Walter RF, Christoph DC, Heukamp LC, Peifer M, Hirsch B, Burbat L, Mairinger T, Schmid KW, Wohlschlaeger J, Mairinger FD (2015) Mutational analysis of pulmonary tumours with neuroendocrine features using targeted massive parallel sequencing: a comparison of a neglected tumour group. Br J Cancer 113:1704–1711
Warth A, Herpel E, Krysa S, Hoffmann H, Schnabel PA, Schirmacher P, Mechtersheimer G, Blaker H (2009) Chromosomal instability is more frequent in metastasized than in non-metastasized pulmonary carcinoids but is not a reliable predictor of metastatic potential. Exp Mol Med 41:349–353
Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, Kong CS, Berghoff AS, Schmitt A, Reinhardt HC, Park KS, Preusser M, Kundaje A, Greenleaf WJ, Sage J, Winslow MM (2018) Intertumoral heterogeneity in SCLC is influenced by the cell type of origin. Cancer Discov 8:1316–1331
Yatabe Y, Dacic S, Borczuk AC, Warth A, Russell PA, Lantuejoul S, Beasley MB, Thunnissen E, Pelosi G, Rekhtman N, Bubendorf L, Mino-Kenudson M, Yoshida A, Geisinger KR, Noguchi M, Chirieac LR, Bolting J, Chung JH, Chou TY, Chen G, Poleri C, Lopez-Rios F, Papotti M, Sholl LM, Roden AC, Travis WD, Hirsch FR, Kerr KM, Tsao MS, Nicholson AG, Wistuba I, Moreira AL (2019) Best practices recommendations for diagnostic immunohistochemistry in lung cancer. J Thorac Oncol 14:377–407
Zhang Z, Wang M (2017) PI3K/AKT/mTOR pathway in pulmonary carcinoid tumours. Oncol Lett 14:1373–1378
Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, Gazdar AF (2018) Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res 7:32–49
Zhou Z, Zhu L, Niu X, Shen S, Zhao Y, Zhang J, Ye J, Han-Zhang H, Liu J, Liu C, Lu S (2019) Comparison of genomic landscapes of large cell neuroendocrine carcinoma, small cell lung carcinoma, and large cell carcinoma. Thorac Cancer 10:839–847
Acknowledgments
This work is dedicated to the memory of Carlotta, an extraordinarily lively girl who died an untimely death due to cancer in the prime of her life. The authors are indebted with Ms. Cecilia Pelosi for accurate proofreading of the manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest.
Funding
Open Access funding provided by Università degli Studi di Milano.
Author information
Authors and Affiliations
Contributions
GP conceived and designed the study, drafted, and finalized the manuscript; JM designed the study, drafted, and finalized the manuscript; MB, FB, PH, VH, MR, IK, LC, LP, AS, GV, SH, FF, and MP critically revised and finalized the manuscript. All authors approved the submitted version.
Corresponding author
Ethics declarations
This is a review article on already existing literature data; thus, no approval by Internal Review Board was required. The study does not involve humans and/or animals for which specific approval by the ethics committee and/or informed consent are required. Moreover, the study does not have implications for public health or general welfare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Metovic, J., Barella, M., Bianchi, F. et al. Morphologic and molecular classification of lung neuroendocrine neoplasms. Virchows Arch 478, 5–19 (2021). https://doi.org/10.1007/s00428-020-03015-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-020-03015-z