
Abstract
For many cancer types, the immune system plays an essential role in their development and growth. Based on these rather novel insights, immunotherapeutic strategies have been developed. In the past decade, immune checkpoint blockade has demonstrated a major breakthrough in cancer treatment and has currently been approved for the treatment of multiple tumor types. Adoptive cell therapy (ACT) with tumor-infiltrating lymphocytes (TIL) or gene-modified T cells expressing novel T cell receptors (TCR) or chimeric antigen receptors (CAR) is another strategy to modify the immune system to recognize tumor cells and thus carry out an anti-tumor effector function. These treatments have shown promising results in various tumor types, and multiple clinical trials are being conducted worldwide to further optimize this treatment modality. Most successful results were obtained in hematological malignancies with the use of CD19-directed CAR T cell therapy and already led to the commercial approval by the FDA. This review provides an overview of the developments in ACT, the associated toxicity, and the future potential of ACT in cancer treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past few decades, the potency of the immune system in the development and treatment of cancer has been a major focal point of research [1]. Although targeted therapy and immunotherapy with immune checkpoint blockade have greatly improved the survival of, amongst others, melanoma and non-small cell lung cancer patients, a large proportion of patients still develop disease progression upon these therapies [2, 3]. Adoptive cell therapy (ACT) may provide an additional treatment option for these patients and comprises the intravenous transfer of either tumor-resident or peripheral blood modified immune cells into cancer patients to mediate an anti-tumor function. Currently, ACT can be classified into three different types with each their own mechanism of action, namely ACT with tumor-infiltrating lymphocytes (TIL), ACT using T cell receptor (TCR) gene therapy, and ACT with chimeric antigen receptor (CAR) modified T cells [4]. The use of other immune cell types such as natural killer cells as a basis for cell therapy is also an area of current research. However, many hurdles have to be overcome in order for this to be an effective anti-cancer treatment [5] and lie beyond the scope of this review.
The first studies with TIL were performed by Rosenberg and coworkers at the Surgery Branch in the National Institutes of Health (SB, NIH, Bethesda, Maryland, US), where TIL were grown from different murine tumors and showed anti-tumor activity in vivo [6]. The current TIL therapy consists of ex vivo expansion of TIL from resected tumor material and adoptive transfer into the patient following a lymphodepleting preparative regimen and subsequent support of interleukin-2 (IL-2). With this regimen, remarkable objective tumor responses of around 50% have been achieved in patients with metastatic melanoma in several phase I/II clinical trials [7,8,9]. After the successes seen with TIL in melanoma patients, the production of TIL from other solid tumor types has also been studied. Up until now, it has been possible to grow out TILs from non-melanoma tumor types such cervical cancer [10], renal cell cancer [11], breast cancer [12], and non-small cell lung cancer [13] with varying rates of tumor reactivity.
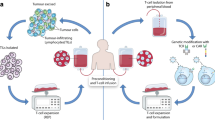
Next to the naturally occurring TILs in tumors and the thereupon-based treatment options, peripheral blood T cells can be isolated and genetically modified in vitro to express TCRs that target specific tumor antigens for the use of ACT. With the use of this method, large pools of tumor specific T cells can be generated [14, 15], with potent anti-tumor activity and objective clinical responses observed in up to 30% of treated patients [16]. For the recognition by the modified TCR, antigen presentation via the major histocompatibility complex (MHC) is required. However, it is well known that many cancer types can escape T cell-mediated immune responses by downregulation or loss of their MHC expression [17]. To circumvent the need for the presence of MHC on tumor cells for the recognition by tumor-specific T cells, artificial receptors such as CAR molecules have been developed. ACT with CAR-modified T cells holds the capacity of the same effector function as TCR-modified T cells, but independently of MHC expression [18]. Besides the use of protein antigens, other antigens such as carbohydrates [19] or glycolipid antigens [20, 21] have also been explored. Impressive clinical responses have already been seen in hematological malignancies with CD19-specific CAR T-cells [22], which led to the exploration of using CAR therapy in solid tumors as well [23]. Table 1 summarizes these three different treatment modalities of ACT and Fig. 1 shows an overview of the process for adoptive cell therapy with TIL, TCR gene therapy and CAR-modified T cells.

Schematic overview of the processes for adoptive cell therapy (ACT) of tumor-infiltrating lymphocytes (TIL), ACT with T cell receptor (TCR) gene therapy and ACT with chimeric antigen receptor (CAR)-modified T cells. In ACT with TIL, tumor-resident T cells are isolated and expanded ex vivo after surgical resection of the tumor. Thereafter, the TILs are further expanded in a rapid expansion protocol (REP). Before intravenous adoptive transfer into the patient, the patient is treated with a lymphodepleting conditioning regimen. In ACT with genetically modified peripheral blood T cells, TCR gene therapy and CAR gene therapy can be distinguished. For both treatment modalities, peripheral blood T cells are isolated via leukapheresis. These T cells are then transduced by viral vectors to either express a specific TCR or CAR, respectively
Although most studies with ACT in solid tumors have been performed in melanoma, the role of ACT in the treatment of other tumor types is growing. Recently, an overview of initiated trials conducted with ACT since May 2015 was published by Fournier et al. [27], where an impressive 121 new clinical trials were described (including ACT in non-solid tumors). This illustrates the need for up-to-date knowledge on ACT in this quickly developing field. The aim of this review is to give a comprehensive overview of the previous developments and the current status of ACT, as the potential of ACT as treatment modality in cancer continues to rise.
Adoptive cell therapy with tumor-resident T cells
The presence of TIL in neoplastic tissue is thought to indicate an anti-tumor immune response by the host and correlates with clinical outcome in several tumor types, especially in melanoma [28, 29]. Dr. S. Rosenberg (SB, NIH, Bethesda, Maryland, US) was the first to demonstrate the anti-tumor activity of TIL in vivo in murine models in the 1980s of the past century [6]. Combining T cell growth factor IL-2 with the TIL infusion product resulted in a greater therapeutic potency of TIL compared to lymphokine-activated killer (LAK) cells produced from peripheral blood lymphocytes in the presence of IL-2 in mice with metastases from various tumor types. Addition of cyclophosphamide to TIL and IL-2 further potentiated the anti-tumor effect of TIL [30]. These early murine studies formed the basis for the original and still most commonly used TIL treatment protocol.
In the original treatment protocol of TIL in metastatic melanoma, patients underwent resection of one or more metastases with a total diameter of at least 2–3 cm. The resected tumor was fragmented or enzymatically digested and subsequently cultured in the presence of IL-2, which resulted in proliferation of TIL. This initial outgrowth phase took approximately 14 days. Once culture consisted mostly of CD3+ T cells, their specificity was tested during a short culture in the presence of an autologous or HLA-matched tumor cell line by quantification of interferon-γ (IFN-γ) [7]. This selection step, however, was time-consuming and complex. Follow-up studies showed that TIL production without this pre-selection for tumor reactivity, so-called “young TIL,” resulted in comparable clinical responses [31, 32] and became the current standard treatment protocol. At least 50 × 106 TILs from this initial outgrowth phase are required to be further expanded in a rapid expansion protocol (REP) in the presence of a soluble anti-CD3 antibody, IL-2 and irradiated allogeneic or autologous feeder cells. During this 14 days lasting expansion phase, up to approximately 1 × 1011 cells are obtained. These TILs are harvested and prepared for infusion into the patient [33]. Prior to infusion, patients will undergo lymphodepleting, but non-myeloablative (NMA) chemotherapy consisting of 2 days intravenous (i.v.) cyclophosphamide (60 mg/kg) followed by 5 days fludarabine (25 mg/m2). Shortly after infusion of TIL, patients receive i.v. high-dose (HD) bolus IL-2 (720,000 IU/kg every 8 h until maximal tolerance) [7, 24, 34]. Subsequent support with IL-2 is thought to further enhance the survival and clinical efficacy of TIL [35].
Multiple studies have been performed evaluating the role of preconditioning lymphodepleting regimens. Lymphodepleting regimens cause a short, but deep lymphopenia and neutropenia, with full bone marrow recovery within 7–10 days, not requiring hematopoietic stem cell support [36]. Murine models had shown that response rates upon TIL improved after prior lymphodepletion by total body irradiation (TBI) or chemotherapy [30, 37]. These models showed that depletion of endogenous lymphocytes created “physical space,” resulted in less competition for homeostatic cytokines IL-7 and IL-15 [38] and removed immunosuppressive lymphoid and myeloid populations [39]. Incorporating an intensified lymphodepleting regimen by combining cyclophosphamide/fludarabine with either 2 Gy or 12 Gy TBI, improved the overall response rate from 49% to respectively 52% and 72% in small cohorts of 25 metastatic melanoma patients. Significantly higher levels of IL-7 and IL-15 were measured in patients treated with the intensified lymphodepleting regimen (p = 0.02 and p = 0.005 respectively). However, this intensified regimen did result in more acute toxicity and prolonged organ dysfunction and required hematopoietic stem cell support for bone marrow recovery [34]. In a randomized controlled phase II trial by the NIH in 2016, the clinical benefit of addition of 12 Gy TBI to NMA chemotherapy could not be reproduced [40]. A currently recruiting phase II trial conducted by the Sheba Medical Center, Israel, evaluating the effect of a reduced intensity lymphodepleting regimen prior to TIL is expected to give further insight into the optimal preparative regimen (NCT03166397).
Most studies conducted with TIL have been performed in patients with advanced (cutaneous) melanoma, but the place of TIL in the current standard treatment protocol is still being investigated. Promising clinical responses have been observed in patients with metastatic uveal melanoma [41] and may provide a novel treatment modality for this group of patients, as no standard treatment is available yet. The currently recruiting phase III randomized clinical trial comparing ipilimumab to TIL treatment (NCT02278887) should provide more evidence for the role of TIL as anti-tumor treatment in metastatic melanoma. Successful production of TIL was also achieved in renal cell carcinoma [11], breast cancer [12], cervical cancer [10], gastrointestinal cancers [42, 43], cholangiocarcinoma [44], pancreatic cancer [45], head and neck cancer [46], ovarian cancer [47], and non-small cell lung cancer [13]. However, the antitumor reactivity of TIL obtained from these other solid tumors still remains a challenge. It has been demonstrated that TILs isolated from melanoma show consistent antitumor reactivity [48] and that high mutational load and high neo-antigen rates are significantly associated with clinical benefit upon TIL therapy in patients with metastatic melanoma [49]. The production and reactivity of TIL products from other solid tumor types is highly variable, most likely as a result of heterogeneity in mutational and neo-antigen burden, in lymphocytic infiltration with variations of CD4+ and CD8+ T cells, in myeloid infiltrate composition and other as yet unknown factors [50]. Further research is needed in this field in order to potentiate TIL treatment for other solid tumors.
Adoptive cell therapy with genetically modified peripheral blood T cells
Antigen receptor gene engineering with TCRs or CARs is a developing and promising new anti-tumor modality [15]. In contrast to TIL, peripheral blood lymphocytes are enhanced to create anti-tumor reactivity by transduction with tumor-specific receptors. This modification can be performed with relevant tumor-reactive TCR for TCR gene therapy or synthetic antibody-based receptors for CAR T cell therapy [36].
TCR gene therapy
In TCR gene therapy, tumor antigen recognition is achieved by the introduction of a novel T cell receptor into T cells. Autologous T cells are redirected to recognize tumor antigens by engraftment of genes encoding TCR-α and β chains. TCR-modified T cells exert antigen recognition in an MHC-dependent manner [23, 51]. Examples of targetable antigens are tissue-specific antigens like melanoma differentiation antigens, cancer/testis (C/T) antigens, overexpressed antigens, and viral antigens. In most clinical trials, peripheral blood T cells for genetic modification are obtained via leukapheresis and are transduced by gamma-retroviral or lentiviral vectors that incorporate the TCR genes into the host genome, which results in high-level expression of the introduced TCR [36, 52, 53]. Other means of genetic engineering that are currently in development include the transposon/transposase system, such as Sleeping Beauty [54], or Crispr/Cas9 based technology [55]. These technologies do not require the production of lenti- or gamma-retroviral vectors, and may therefore provide a more flexible and cheap platform. As in ACT with TIL, most clinical protocols with TCR gene therapy have incorporated preconditioning of the patient with a lymphodepleting regimen prior to T cell infusion, to facilitate engraftment and expand the lifespan of the modified T cells. In addition, IL-2 administration following T cell infusion has been used [56].
The first evidence of the feasibility and clinical potency of TCR gene therapy targeting the melanoma differentiation antigen MART-1, present in approximately 80–95% of melanomas [57, 58], was demonstrated in 17 patients with progressive metastatic melanoma. Gene transfer efficiencies of 17–62% were achieved and an objective partial tumor response was seen in two (13%) of the treated patients [25]. In a subsequent clinical trial, 36 patients with metastatic melanoma were treated with high-avidity TCRs targeting the melanoma differentiation antigens gp100 or MART-1 and objective tumor responses were observed in 19% and 30% respectively [16].
Next to melanoma differentiation antigens as a target for TCR gene therapy, notable clinical responses have also been achieved when targeting C/T antigens. The genes encoding these antigens are normally expressed during embryogenesis, but are epigenetically silenced later in life, except in spermatocytes. Cancers oftentimes aberrantly re-express these genes, hence the name C/T antigens. These antigens include NY-ESO-1, MAGE-family, and SSX [59]. NY-ESO-1 is expressed in up to 52% of melanomas [60], 82% of neuroblastomas [61], 80–100% of synovial sarcomas and mixoid and round cell liposarcomas [62], 43% of ovarian cancer [63] and to a lesser extent in multiple other tumor types. Because of its restricted expression in normal tissues in combination with a widespread expression in cancer, NY-ESO-1 has been frequently used as a target in TCR gene therapy. In a phase II clinical trial, 5/11 (45%) patients with progressive advanced melanoma and 4/6 (67%) patients with synovial sarcoma had an objective tumor response after infusion of 1.6–130 × 109 retrovirally transduced autologous T cells with a NY-ESO-1 targeting high affinity TCR [64]. Similar response rates have been observed when targeting MAGE-A3, which is expressed in 62% of melanomas [65]. In a clinical phase I/II trial, nine pretreated patients with either advanced melanoma (n = 7), synovial sarcoma (n = 1), or esophageal cancer (n = 1) were treated with anti-MAGE-A3 TCR gene-modified T cells in a dose-escalating manner. The first three patients were treated with 3 × 1010 transduced cells and the remaining patients were treated with 1 × 1011 transduced cells, with a transduction efficiency of 70% (CD8+ T cells). Of these patients, five (56%), including four melanoma patients and the single synovial sarcoma patient, had an objective tumor response [66].
Tumor regression has also been seen in patients with T cells targeting carcinoembryonic antigen (CEA), which is overexpressed in colorectal adenocarcinoma but is also present in normal epithelial cells [67]. Three patients were treated with 2 × 108–4 × 108 CEA reactive-TCR transduced T cells, and one of these patients achieved a partial response. However, all 3 patients developed a severe transient colitis [68].
Viral antigens such as human papillomavirus (HPV) as possible targets for TCR therapy have also been explored for HPV-associated epithelial cancers including cervical, oropharyngeal, vulvar, vaginal, anal, and penile cancer. T cells engineered with the TCR recognizing the HPV-16 E6 epitope from metastatic anal cancer also showed recognition of HPV-16 positive cervical and head and neck cancer cell lines and this epitope may thus be a potential target for TCR therapy [69].
Transduction of T cells in an early differentiation stage (central memory CD8+ T cells) seems to result in greater anti-tumor responses when combined with tumor-antigen vaccination and exogenous IL-2 in preclinical murine models [70]. It has also been shown that the cytokines IL-7 and IL-15, which play a role in the development of central memory T cells in vivo, favor the generation of this T cell subset in culture systems [70, 71]. In most cancer immunotherapy studies, CD8+ T cells have been the main focus because of their known strong cytolytic capacity [72]. However, recent evidence shows that CD4+ T cells also exert anti-tumor efficacy [44] and CD4+ T cells have already been part of the TCR infusion product used in early clinical trials [16, 64, 66]. In a validated good manufacturing practice (GMP) production process, CD4+ as well as CD8+ T cells are retrovirally transduced and expanded in the presence of IL-7 and IL-15 in combination with anti-CD3/CD28 bead selection and activation [73]. This protocol is being evaluated in a phase I/IIa trial in HLA-A:*02-01 MART-1 positive patients with advanced melanoma, including patients with uveal melanoma (NCT02654821), and should provide more insight into the feasibility and safety of TCR therapy in melanoma.
In summary, these results demonstrate that TCR therapy can be a potent anti-tumor treatment in various cancer types. However, as the antigens explored up until now are not solely expressed by the tumor, the identification of antigens restricted to tumors is essential to further increase the efficacy and safety of TCR therapy.
CAR T cell therapy
CARs are hybrid receptors and are currently genetically constructed to contain a scFv of a monoclonal antibody as the antigen-binding extracellular domain, an intracellular CD3ζ chain as the TCR signaling domain and an additional co-signaling domain, mainly CD28 and 4-1BB (CD137) or others, to deliver co-stimulation [23, 74]. Multiple methods to transfer CARs to T cells have been developed, but most commonly used is transfer by retroviral infection, which has proven to be efficacious and safe [75]. Induction of cytotoxic activity of the manufactured T cell is a result of antigen-binding to the scFv, leading to downstream signaling through phosphorylation of CD3ζ and additional signaling cascades via the co-stimulating domains [76], similar to signaling following T cell activation through the TCR complex. Unlike TCR gene therapy, CAR T cells show target recognition in an MHC-independent manner, as was first demonstrated by the groups of Kuwana and Eshhar in the late 1980s in the first generation CARs [18, 77]. Since this first discovery, CAR therapy has undergone major improvements and thus far most research has been performed in hematological malignancies such as B cell lymphoma and leukemia. The co-receptor CD19 showed to be an optimal target [78,79,80] as it is expressed early during B cell development and expression is maintained until plasma cell differentiation. B cell malignancies originating from these B cell differentiation states also express CD19. As CD19 is also expressed on normal B cells, treatment with CD19 CAR T cells will result in a transient or lasting B cell aplasia and hypogammaglobulinemia [81]. In 2003, the group of Sadelain at the Memorial Sloan-Kettering Cancer Center (New York, US) were the first to show successful transduction of peripheral blood lymphocytes with CD19 CARs in immunodeficient mice with various B cell malignancies resulting in tumor reduction and even long-term eradication [82].
The engineering of CARs has evolved over time and resulted in four generations of CAR molecules. In 1993, first-generation CAR consisted of a scFv and intracellular CD3ζ domain which mediated the production of IL-2 and non-MHC-restricted cell lysis upon activation in murine models [83]. However, the presence of costimulatory signals lead to better T cell activation (by providing signal two) and resulted in better T cell proliferation [84]. These second and third generation CARs additionally contained costimulatory domains to enhance T cell survival, activation and expansion [20, 85, 86]. Second generation CARs carry the costimulatory domains CD28 [87] or 4-1BB [85]. These showed enhanced TCR signaling, production of cytotoxic cytokines such as IL-2, proliferation and survival [20, 85, 87, 88]. Third generation CARs aim to encompass the signaling capacity of two costimulatory domains, mostly CD28 in combination with 4-1BB or OX-40. Addition of proliferative cytokines such as IL-12 or costimulatory ligands such as 4-1BBL have proven to further potentiate the anti-tumor capacity of second generation CAR T cells in preclinical studies and are currently known as the fourth generation CAR T cells [89, 90]. These CAR T cells can also be referred to as T cells redirected for universal cytokine killing (TRUCKs), which can deliver a transgenic product to the targeted tissue. By using nuclear factor of activated T cells (NFAT) to induce cytokines such as IL-12, the area around the CAR-targeted tissue is made more favorable for an immune response [91].
In xenograft models comparing the efficacy of different CAR constructs, CARs consisting of two signaling domains (CD3ζ plus CD28) and 4-1BB ligand showed the greatest anti-tumor efficacy and also increased persistence in the peripheral blood compared to first generation CAR constructs [92]. With second generation CARs, complete response rates of around 40% have been demonstrated in acute lymphoblastic leukemia (ALL) murine models treated with 5–10 × 106 CD19 CD28 or 4-1BB CAR T cells [82, 93, 94]. The first clinical trial to show clinically significant responses in patients with ALL was performed by Sadelain and co-workers in 2013. In this trial, five patients with relapsed B-ALL not previously treated with allogenic hematopoietic stem cell transplantation (allo-HSCT) were treated with 1.5–3 × 106/kg CD19 CD28 CAR T cells after prior conditioning treatment with cyclophosphamide (1.5–3.0 g/m2) and subsequent allo-HSCT as per protocol and complete remissions were seen in all treated patients (n = 4) [26]. Finalization of this clinical trial in 2014 with a total of 16 patients resulted in a complete response rate of 88% [95]. In a case series by Grupp et al., two children with relapsed and refractory pre-B cell ALL received 1.4 × 106–1.2 × 107/kg CD19 4-1BB CAR T cells and both patients showed a complete remission, one of which was ongoing 11 months post-treatment (current status unknown) [80]. In a following phase I dose-escalation trial, 21 patients with relapsed or refractory ALL or non-Hodgkin lymphoma (NHL) were either treated with 1 × 106/kg/dose, 3 × 106/kg/dose or the entire CD19 CAR T cell product if the product did not meet the required dosage amounts. The maximum tolerated dose was 1 × 106/kg/dose, all toxicities were temporary and a complete response rate of 67% (14/21 patients) was reached [96]. More recently in 2017, patients with refractory diffuse B cell lymphoma, primary mediastinal B cell lymphoma, or transformed follicular lymphoma were treated in a multicenter phase II trial with 2 × 106/kg CD19 CD28 CAR T cells following low dose preconditioning regimens with cyclophosphamide (500 mg/m2/day) and fludarabine (30 mg/m2/day) for 3 days. Of 101 treated patients, an objective response was seen in 82% and 54% of patients showed a complete response, of which 40% were durable complete responses [97]. These response rates were reproducible, as in another study complete response rates of 57% were seen in 28 patients with refractory B cell lymphoma treated with a median of 5.79 × 106/kg/dose CD19 4-1BB CAR T cells [98].
As in ACT with TIL, preconditioning lymphodepletion is commonly used in the clinical treatment protocol with CAR therapy. When patients with chemotherapy-refractory chronic lymphocytic leukemia were treated solely with CAR T cells (without lymphodepleting regimen), no clinical benefit and less persistence of the CAR T cells was observed. However, it is important to note that these patients in this small study also received a lower dose of CAR T cells [78]. The clinical successes with CAR T cell therapy has recently led to the FDA approval of two CD19 CAR therapies for ALL and NHL in 2017, namely axicabtagene ciloleucel (Yescarta) with costimulatory molecule CD28 and tisagenlecleucel (Kymriah) with costimulatory molecule 4-1BB [99]. Although high complete remission rates have been demonstrated with the use of CD19 CAR T cells, resistance via the loss of CD19 has been observed in 28% of young adult and pediatric patients with acute leukemia in an international trial [100].
As stated above, most of the research with CAR T cell therapy has been performed in hematological malignancies, but also other B cell lineage-restricted targets like CD22 and B cell maturation antigen (BCMA) are currently under investigation. Moreover, CAR T cell technology is being explored in solid tumors, however achieving limited clinical activity thus far. For example in sarcomas targeting ERBB2/HER2 [101], renal cell cancer targeting carbonic anhydrase IX (CAIX) [102], non-small cell lung cancer and cholangiocarcinoma targeting epidermal growth factor (EGFR) [103], and neuroblastoma targeting GD2 [104] and other solid tumors (targeting shared antigens including mesothelin and CEA) [105]. Recently, CAR T cells directed against the colorectal cancer antigen GUCY2C were investigated in murine models showing increased antigen-dependent T cell activation, cytokine production and killing of GUCY2C-expressing tumor cells [106].
Toxicity
The toxicity observed with ACT can grossly be divided in three groups: toxicity due to the lymphodepleting preparative regimen, immune-related toxicity and cytokine-related toxicity. During treatment with TIL, toxicities are predominantly caused by the lymphodepleting preparative regimens, resulting in pancytopenia and febrile neutropenia, and the supportive IL-2 infusions [8, 107, 108]. These toxicities are also seen in TCR gene therapy [64] and CAR therapy [97]. The most prevalent side effects seen in TIL treatment due to IL-2 include chills, high fever, hypotension, oliguria, and edema due to the systemic inflammatory and capillary leak syndrome effects and can usually be treated with supportive measures [109]. Reports have been published of autoimmune phenomena such as vitiligo or uveitis, however, these side effects are not frequently seen and uveitis usually responds well to local corticoid treatment [108].
The potency of TCR therapy lies with the recognition of antigens on tumor cells, often not tumor-specific and thus also expressed on healthy tissues. This can lead to the occurrence of “on-target, off-tumor” toxicity [36]. Targeting melanoma differentiation antigens such as MART-1 and gp100 can result in severe skin rash, uveitis, and ototoxicity due to expression of these antigens in these organs [16] and these effects seem to be dose-dependent [110]. Some of the used TCR, such as NY-ESO-1 and MAGE-A3 have undergone affinity maturation in conducted clinical trials to increase the affinity of the TCR for the target MHC-peptide complex [64, 66]. This process may increase the chance of cross-reactivity to other targets as well, and may have caused some of the severe toxicities that were seen thus far with TCRs directed at C/T antigens. The C/T antigen MAGE-A3 is not known to be present in normal tissue, but two separate TCR-based therapies directed to MAGE-A3 did result in fatal neurotoxicity and cardiotoxicity. Neurological complications may be due to cross-reactivity to MAGE-A12 which is expressed in a subset of neurons in the brain and the cardiac events may be due to cross-recognition of the muscle protein Titin [66, 111], perhaps as a result of TCR affinity enhancement. On the other hand, treatment with the C/T antigen NY-ESO-1 specific TCR transduced T cells so far seem to be safe [64]. B cell aplasia is an expected “on target off tumor” side effect of CD19 CAR T cell therapy and can be managed with replacement therapy with intravenous immunoglobulin [79].
Another common toxicity seen in ACT is cytokine release syndrome (CRS). CRS is a non-antigen-specific adverse event and is a result of activation most probably of the infused T cells through antigen recognition, leading to massive cytokine release, including IFN-γ, IL-1, and IL-6 [112]. Toxicity from CRS has a very heterogeneous presentation, but usually involves fever, hypotension, tachycardia, and respiratory insufficiency and it is potentially fatal, as described by van den Berg et al. in a case report of a patient with metastatic melanoma treated with a TCR recognizing MART-1 [110]. The severity of CRS is correlated with tumor burden [26, 113]. Severe and life-threatening CRS can effectively be treated with tocilizumab (human monoclonal antibody against the IL-6 receptor) or siltuximab (monoclonal antibody against IL-6) and sometimes corticosteroids [112, 114, 115]. Additional targeted immunosuppressive agents like infliximab (anti-TNF-α antibody) and anakinra (anti-IL-1R antibody) have been used in some very severe cases [112], the latter of which has recently also been shown to be effective against neurotoxicity in CAR T cell therapy in a murine model [116].
Neurological complications, including confusion, delirium and occasional seizures and cerebral edema, occasionally resulting in death, have been observed with the use of CD19-specific CAR T cells [117]. The underlying pathogenesis of this neurotoxicity remains currently unknown. Recently, endothelial cell activation and increased blood-brain barrier permeability were found to play a role at the initiation of this toxicity [118]. Intensive research is currently ongoing to further elucidate the underlying pathogenesis of both CRS and the neurotoxicity, identify useful biomarkers and optimize current treatment algorithms [119].
Lastly, an IgE-mediated anaphylactic reaction has been observed in a patient treated with autologous T cells electroporated with mRNA encoding for a CAR derived from a murine antibody to mesothelin [120]. This reaction may have been induced by “foreign” CAR moieties and current strategies for CAR therapy involve the use of humanized or even fully human scFv to circumvent these IgE-mediated responses.
Future prospective of adoptive cell therapy and conclusions
The ultimate goal of adoptive cell therapy for malignancies is to create an optimized personalized cellular product solely reactive to the tumor. In the past decades, the production of TIL, TCRs, and CARs have all undergone developments to improve its efficacy as anti-cancer treatment. TIL are a heterogeneous cell product and the presence of antigen-reactive TIL is a key determinant of anti-tumor reactivity [121]. Enrichment for tumor-reactive TIL through CD137 [122] or PD-1 [123] selection is currently being investigated as a method to increase the anti-tumor reactivity.
Genetic editing of TIL may also improve its functionality, as has already been demonstrated with Zinc finger nuclease engineering which decreases the PD-1 expression in TIL, improving the efficacy [124].
In TCR therapy, it is of major importance that targetable antigens are identified which are expressed on the tumor, but not on healthy tissue to decrease the “on-target, off-tumor” toxicity and to further potentiate its effector function. Currently, oncogenes such as BRAFV600E driver mutations are also being explored as possible therapeutic targets for ACT and have already shown clinical activity in a patient with advanced melanoma [125]. HPV antigens E6 and E7 are being explored as targets for ACT as treatment of HPV-associated cancers [10, 69]. Neo-antigens that arise as a result of tumor-specific mutations appear exquisite targets for TCR gene therapy as these antigens are, like viral antigens, fully foreign to the immune system. Gene therapy targeting these antigens are also being developed.
As in ACT with TCR gene therapy, the main challenges in CAR therapy comprise new target discovery, reduction of toxicity, and improvement of cell trafficking. Because of the correlation between tumor burden and toxicity from CAR T cell therapy [26, 113], tumor reduction before therapy or dose adaptation strategies could possibly be applied to reduce side effects. More promising are current investigations with CAR T cells engineered to contain suicide genes or switches (for example iCasp9) which are evaluated in preclinical and clinical studies [126]. CAR T cells seem to express inhibitory receptors such as PD-1 in an exhausted state, which upregulates PD-L1 on tumor cells. Combinatorial approaches of CAR T cell therapy and PD-1 blockade have resulted in improved CAR T cell activity and tumor eradication in preclinical experiments and clinical studies are currently ongoing [127, 128]. Furthermore, the combination of CAR T cell therapy with oncolytic virus-driven production of a bispecific T cell engager showed enhanced efficacy in a mouse model compared to both monotherapies [129].
In conclusion, ACT represents a personalized immunotherapeutic approach that has developed rapidly in recent years. Great successes have already been seen with the use of TIL treatment in melanoma and CAR therapy in hematologic malignancies. However, further optimization of this promising treatment modality is warranted to enhance the anti-tumor effect and reduce the associated toxicity. More than 100 clinical trials have been initiated since 2015 and are currently being conducted with ACT [27] and should provide new insights in the efficacy and further developments of this treatment modality.
Availability of data and material
Not applicable. All cited published original research in this manuscript are publically available.
Abbreviations
- ACT:
-
Adoptive cell therapy
- ALL:
-
Acute lymphoblastic leukemia
- BCMA:
-
B cell maturation antigen
- BRAF:
-
B-Raf
- C/T:
-
Cancer/testis
- CAIX:
-
Carbonic anhydrase IX
- CAR:
-
Chimeric antigen receptor
- CD:
-
Cluster of differentiation
- CEA:
-
Carcinoembryonic antigen
- CRS:
-
Cytokine release syndrome
- EGFR:
-
Epidermal growth factor receptor
- ERBB2:
-
erb-b2 receptor tyrosine kinase 2
- FDA:
-
Food and Drug Administration
- GMP:
-
Good manufacturing practice
- gp100:
-
Glycoprotein 100
- GUCY2C:
-
Guanylate cyclase 2C
- Gy:
-
Gray
- HD:
-
High dose
- HER2:
-
Human epidermal growth factor receptor 2
- HLA:
-
Human leukocyte antigen
- HPV:
-
Human papillomavirus
- HSCT:
-
Hematopoietic stem cell transplantation
- i.v.:
-
Intravenous
- iCasp9:
-
Inducible caspase-9
- IFN:
-
Interferon
- IL:
-
Interleukin
- IMC:
-
Immunocore
- IU:
-
International unit
- kg:
-
Kilogram
- LAK:
-
Lymphokine-activated killer
- m:
-
Meter
- MAGE:
-
Melanoma antigen gene
- MART-1:
-
Melanoma antigen recognized by T cells 1
- mg:
-
Milligram
- MHC:
-
Major histocompatibility complex
- NFAT:
-
Nuclear factor of activated T cells
- NHL:
-
Non-Hodgkin lymphoma
- NIH:
-
National Institutes of Health
- NMA:
-
Non-myeloablative
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed death ligand-1
- REP:
-
Rapid expansion protocol
- SB:
-
Surgery branch
- scFv:
-
Single-chain variable fragment
- SSX:
-
Synovial sarcoma X breakpoint
- TBI:
-
Total body irradiation
- TCR:
-
T cell receptor
- TIL:
-
Tumor-infiltrating lymphocytes
- TNF:
-
Tumor necrosis factor
- TRUCK:
-
T cells redirected for universal cytokine killing
- US:
-
United States
References
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10
Luke JJ, Flaherty KT, Ribas A, Long GV (2017) Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 14:463–482
Mayor M, Yang N, Sterman D, Jones DR, Adusumilli PS (2016) Immunotherapy for non-small cell lung cancer: current concepts and clinical trials. Eur J Cardiothorac Surg 49:1324–1333
June CH, Riddell SR, Schumacher TN (2015) Adoptive cellular therapy: a race to the finish line. Sci Transl Med 7:280ps287
Morvan MG, Lanier LL (2016) NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 16:7–19
Spiess PJ, Yang JC, Rosenberg SA (1987) In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J Natl Cancer Inst 79:1067–1075
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17:4550–4557
Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, Hölmich LR, Hendel HW, Met Ö, Andersen MH, thor Straten P, Svane IM (2016) Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res 22:3734–3745
Besser MJ, Shapira-Frommer R, Itzhaki O et al (2013) Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 19:4792–4800
Stevanovic S, Draper LM, Langhan MM et al (2015) Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol 33:1543–1550
Andersen R, Westergaard MCW, Kjeldsen JW, Müller A, Pedersen NW, Hadrup SR, Met Ö, Seliger B, Kromann-Andersen B, Hasselager T, Donia M, Svane IM (2018) T-cell responses in the microenvironment of primary renal cell carcinoma-implications for adoptive cell therapy. Cancer Immunol Res 6:222–235
Lee HJ, Kim YA, Sim CK, Heo SH, Song IH, Park HS, Park SY, Bang WS, Park IA, Lee M, Lee JH, Cho YS, Chang S, Jung J, Kim J, Lee SB, Kim SY, Lee MS, Gong G (2017) Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget 8:113345–113359
Ben-Avi R, Farhi R, Ben-Nun A, Gorodner M, Greenberg E, Markel G, Schachter J, Itzhaki O, Besser MJ (2018) Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol Immunother 67:1221–1230
Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD (2002) Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A 99:16168–16173
Schumacher TN (2002) T-cell-receptor gene therapy. Nat Rev Immunol 2:512–519
Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CCR, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–546
Garrido F, Ruiz-Cabello F, Aptsiauri N (2017) Rejection versus escape: the tumor MHC dilemma. Cancer Immunol Immunother 66:259–271
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 86:10024–10028
Mezzanzanica D, Canevari S, Mazzoni A, Figini M, Colnaghi MI, Waks T, Schindler DG, Eshhar Z (1998) Transfer of chimeric receptor gene made of variable regions of tumor-specific antibody confers anticarbohydrate specificity on T cells. Cancer Gene Ther 5:401–407
Krause A, Guo HF, Latouche JB, Tan C, Cheung NKV, Sadelain M (1998) Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med 188:619–626
Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans P (2000) Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia 2:449–459
Maude SL, Shpall EJ, Grupp SA (2014) Chimeric antigen receptor T-cell therapy for ALL. Hematology Am Soc Hematol Educ Program 2014:559–564
June CH, O’Connor RS, Kawalekar OU et al (2018) CAR T cell immunotherapy for human cancer. Science 359:1361–1365
Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE (1994) Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 86:1159–1166
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–129
Brentjens RJ, Davila ML, Riviere I et al (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra138
Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L (2017) Trial watch: adoptively transferred cells for anticancer immunotherapy. Oncoimmunology 6:e1363139
Clemente CG, Mihm MC Jr, Bufalino R et al (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310
Burton AL, Roach BA, Mays MP, Chen AF, Ginter BA, Vierling AM, Scoggins CR, Martin RC, Stromberg AJ, Hagendoorn L, McMasters K (2011) Prognostic significance of tumor infiltrating lymphocytes in melanoma. Am Surg 77:188–192
Rosenberg SA, Spiess P, Lafreniere R (1986) A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233:1318–1321
Itzhaki O, Hovav E, Ziporen Y, Levy D, Kubi A, Zikich D, Hershkovitz L, Treves AJ, Shalmon B, Zippel D, Markel G, Shapira-Frommer R, Schachter J, Besser MJ (2011) Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J Immunother 34:212–220
Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, Phan GQ, Kammula US, Hughes MS, Citrin DE, Restifo NP, Wunderlich JR, Prieto PA, Hong JJ, Langan RC, Zlott DA, Morton KE, White DE, Laurencot CM, Rosenberg SA (2010) CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res 16:6122–6131
Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA (2003) Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–342
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang JP, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA (2008) Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 26:5233–5239
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17:2105–2116
Haanen JB (2013) Immunotherapy of melanoma. EJC Suppl 11:97–105
Mills CD, North RJ (1983) Expression of passively transferred immunity against an established tumor depends on generation of cytolytic T cells in recipient. Inhibition by suppressor T cells. J Exp Med 157:1448–1460
Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP (2005) Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202:907–912
Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, Rosenberg SA, Restifo NP (2005) CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 174:2591–2601
Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, Klebanoff CA, Hughes MS, Restifo NP, Langhan MM, Shelton TE, Lu L, Kwong MLM, Ilyas S, Klemen ND, Payabyab EC, Morton KE, Toomey MA, Steinberg SM, White DE, Rosenberg SA (2016) Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol 34:2389–2397
Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL, Wunderlich JR, Danforth DN, Zlott D, Paria BC, Sabesan AC, Srivastava AK, Xi L, Pham TH, Raffeld M, White DE, Toomey MA, Rosenberg SA, Kammula US (2017) Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol 18:792–802
Turcotte S, Gros A, Hogan K, Tran E, Hinrichs CS, Wunderlich JR, Dudley ME, Rosenberg SA (2013) Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol 191:2217–2225
Turcotte S, Gros A, Tran E, Lee CCR, Wunderlich JR, Robbins PF, Rosenberg SA (2014) Tumor-reactive CD8+ T cells in metastatic gastrointestinal cancer refractory to chemotherapy. Clin Cancer Res 20:331–343
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–645
Hall M, Liu H, Malafa M, Centeno B, Hodul PJ, Pimiento J, Pilon-Thomas S, Sarnaik AA (2016) Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer 4:61
Junker N, Andersen MH, Wenandy L, Dombernowsky SL, Kiss K, Sørensen CH, Therkildsen MH, von Buchwald C, Andersen E, Straten PT, Svane IM (2011) Bimodal ex vivo expansion of T cells from patients with head and neck squamous cell carcinoma: a prerequisite for adoptive cell transfer. Cytotherapy 13:822–834
Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, Tanaka K (1995) Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res 1:501–507
Yannelli JR, Hyatt C, McConnell S, Hines K, Jacknin L, Parker L, Sanders M, Rosenberg SA (1996) Growth of tumor-infiltrating lymphocytes from human solid cancers: summary of a 5-year experience. Int J Cancer 65:413–421
Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, Salim M, Vallon-Christersson J, Törngren T, Kvist A, Ringnér M, Svane IM, Jönsson G (2017) Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun 8:1738
Radvanyi LG (2015) Tumor-infiltrating lymphocyte therapy: addressing prevailing questions. Cancer J 21:450–464
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68
Houot R, Schultz LM, Marabelle A, Kohrt H (2015) T-cell-based immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res 3:1115–1122
Sinn PL, Sauter SL, McCray PB Jr (2005) Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors–design, biosafety, and production. Gene Ther 12:1089–1098
Peng PD, Cohen CJ, Yang S, Hsu C, Jones S, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA (2009) Efficient nonviral sleeping beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther 16:1042–1049
Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK (2018) CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 131:311–322
Linnemann C, Schumacher TN, Bendle GM (2011) T-cell receptor gene therapy: critical parameters for clinical success. J Invest Dermatol 131:1806–1816
Chen YT, Stockert E, Jungbluth A, Tsang S, Coplan KA, Scanlan MJ, Old LJ (1996) Serological analysis of Melan-A(MART-1), a melanocyte-specific protein homogeneously expressed in human melanomas. Proc Natl Acad Sci U S A 93:5915–5919
Barrow C, Browning J, MacGregor D et al (2006) Tumor antigen expression in melanoma varies according to antigen and stage. Clin Cancer Res 12:764–771
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT (2002) Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 188:22–32
Goydos JS, Patel M, Shih W (2001) NY-ESO-1 and CTp11 expression may correlate with stage of progression in melanoma. J Surg Res 98:76–80
Oba-Shinjo SM, Caballero OL, Jungbluth AA et al (2008) Cancer-testis (CT) antigen expression in medulloblastoma. Cancer Immun 8:7
Jungbluth AA, Antonescu CR, Busam KJ, Iversen K, Kolb D, Coplan K, Chen YT, Stockert E, Ladanyi M, Old LJ (2001) Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer 94:252–256
Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, Intengan M, Beck A, Keitz B, Santiago D, Williamson B, Scanlan MJ, Ritter G, Chen YT, Driscoll D, Sood A, Lele S, Old LJ (2003) NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res 63:6076–6083
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CCR, Levy CL, Li YF, el-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29:917–924
Roeder C, Schuler-Thurner B, Berchtold S et al (2005) MAGE-A3 is a frequent tumor antigen of metastasized melanoma. Arch Dermatol Res 296:314–319
Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36:133–151
Hammarstrom S (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9:67–81
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA (2011) T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19:620–626
Draper LM, Kwong ML, Gros A et al (2015) Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res 21:4431–4439
Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP (2005) Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102:9571–9576
Cha E, Graham L, Manjili MH, Bear HD (2010) IL-7 + IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res Treat 122:359–369
Berg RE, Forman J (2006) The role of CD8 T cells in innate immunity and in antigen non-specific protection. Curr Opin Immunol 18:338–343
Gomez-Eerland R, Nuijen B, Heemskerk B, van Rooij N, van den Berg JH, Beijnen JH, Uckert W, Kvistborg P, Schumacher TN, Haanen JBAG, Jorritsma A (2014) Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Hum Gene Ther Methods 25:277–287
Sadelain M, Riviere I, Riddell S (2017) Therapeutic T cell engineering. Nature 545:423–431
Scholler J, Brady TL, Binder-Scholl G et al (2012) Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4:132ra153
Chu F, Cao J, Neelalpu SS (2018) Versatile CAR T-cells for cancer immunotherapy. Contemp Oncol (Pozn) 22:73–80
Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y (1987) Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun 149:960–968
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118:4817–4828
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365:725–733
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368:1509–1518
Ghorashian S, Pule M, Amrolia P (2015) CD19 chimeric antigen receptor T cell therapy for haematological malignancies. Br J Haematol 169:463–478
Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Rivière I, Sadelain M (2003) Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 9:279–286
Eshhar Z, Waks T, Gross G, Schindler DG (1993) Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 90:720–724
Jenkins MK, Burrell E, Ashwell JD (1990) Antigen presentation by resting B cells. Effectiveness at inducing T cell proliferation is determined by costimulatory signals, not T cell receptor occupancy. J Immunol 144:1585–1590
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D (2004) Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18:676–684
Sadelain M (2017) CD19 CAR T cells. Cell 171:1471
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol 20:70–75
Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H (2001) Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol 167:6123–6131
Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M (2007) T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med 13:1440–1449
Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ (2012) Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119:4133–4141
Chmielewski M, Abken H (2015) TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther 15:1145–1154
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M (2015) Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28:415–428
Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, la Perle K, Quintas-Cardama A, Larson SM, Sadelain M (2007) Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res 13:5426–5435
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH (2009) Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17:1453–1464
Davila ML, Riviere I, Wang X et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra225
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL (2015) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385:517–528
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY (2017) Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 377:2531–2544
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, Wasik M, Levine BL, Lacey SF, Melenhorst JJ, Porter DL, June CH (2017) Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med 377:2545–2554
By the Numbers (2018) Novel Drugs Approved by the FDA, 2011–2017. Cancer Discov 8:380
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, de Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA (2018) Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 378:439–448
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu MF, Liu H, Hicks J, Rainusso N, Dotti G, Mei Z, Grilley B, Gee A, Rooney CM, Brenner MK, Heslop HE, Wels WS, Wang LL, Anderson P, Gottschalk S (2015) Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 33:1688–1696
Lamers CH, Sleijfer S, van Steenbergen S et al (2013) Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 21:904–912
Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, Han W (2016) Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 59:468–479
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118:6050–6056
Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q (2018) Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol 11:22
Magee MS, Abraham TS, Baybutt TR, Flickinger JC Jr, Ridge NA, Marszalowicz GP, Prajapati P, Hersperger AR, Waldman SA, Snook AE (2018) Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res 6:509–516
Ellebaek E, Iversen TZ, Junker N, Donia M, Engell-Noerregaard L, Met Ö, Hölmich L, Andersen R, Hadrup S, Andersen M, thor Straten P, Svane I (2012) Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J Transl Med 10:169
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–2357
Marabondo S, Kaufman HL (2017) High-dose interleukin-2 (IL-2) for the treatment of melanoma: safety considerations and future directions. Expert Opin Drug Saf 16:1347–1357
van den Berg JH, Gomez-Eerland R, van de Wiel B, Hulshoff L, van den Broek D, Bins A, Tan HL, Harper JV, Hassan NJ, Jakobsen BK, Jorritsma A, Blank CU, Schumacher TNM, Haanen JBAG (2015) Case report of a fatal serious adverse event upon Administration of T Cells Transduced with a MART-1-specific T-cell receptor. Mol Ther 23:1541–1550
Cameron BJ, Gerry AB, Dukes J et al (2013) Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5:197ra103
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124:188–195
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371:1507–1517
Le RQ, Li L, Yuan W et al (2018) FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist 23:943–947
Shimabukuro-Vornhagen A, Godel P, Subklewe M et al (2018) Cytokine release syndrome. J Immunother Cancer 6:56
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, Traversari C, Bordignon C, Ciceri F, Ostuni R, Bonini C, Casucci M, Bondanza A (2018) Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24:739–748
Turtle CJ, Hanafi LA, Berger C et al (2016) Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8:355ra116
Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Harju-Baker S, Özpolat T, Fink KR, Riddell SR, Maloney DG, Turtle CJ (2017) Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 7:1404–1419
Wang Z, Han W (2018) Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res 6:4
Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH (2013) T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 1:26–31
Weiss SA, Han SW, Lui K, Tchack J, Shapiro R, Berman R, Zhong J, Krogsgaard M, Osman I, Darvishian F (2016) Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma. Hum Pathol 57:116–125
Seliktar-Ofir S, Merhavi-Shoham E, Itzhaki O, Yunger S, Markel G, Schachter J, Besser MJ (2017) Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front Immunol 8:1211
Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, Yang JC (2010) Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother 33:956–964
Beane JD, Lee G, Zheng Z, Mendel M, Abate-Daga D, Bharathan M, Black M, Gandhi N, Yu Z, Chandran S, Giedlin M, Ando D, Miller J, Paschon D, Guschin D, Rebar EJ, Reik A, Holmes MC, Gregory PD, Restifo NP, Rosenberg SA, Morgan RA, Feldman SA (2015) Clinical scale zinc finger nuclease-mediated gene editing of PD-1 in tumor infiltrating lymphocytes for the treatment of metastatic melanoma. Mol Ther 23:1380–1390
Veatch JR, Lee SM, Fitzgibbon M, Chow IT, Jesernig B, Schmitt T, Kong YY, Kargl J, Houghton AMG, Thompson JA, McIntosh M, Kwok WW, Riddell SR (2018) Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest 128:1563–1568
Zhang E, Xu H (2017) A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J Hematol Oncol 10:1
Chen N, Morello A, Tano Z, Adusumilli PS (2017) CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology 6:e1273302
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS (2016) Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126:3130–3144
Wing A, Fajardo CA, Posey AD Jr, Shaw C, da T, Young RM, Alemany R, June CH, Guedan S (2018) Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res 6:605–616
Author information
Authors and Affiliations
Contributions
All authors contributed equally to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Disclosures of conflicts of interest
MR declares to have no conflicts of interests. SW declares to have no conflicts of interests. Through JH, NKI has received compensation for advisory roles from BMS, Merck, Roche, NEON therapeutics, Pfizer, and Ipsen; and NKI has received grants from BMS, Merck, Novartis and NEON therapeutics.
Author’s information
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rohaan, M.W., Wilgenhof, S. & Haanen, J.B.A.G. Adoptive cellular therapies: the current landscape. Virchows Arch 474, 449–461 (2019). https://doi.org/10.1007/s00428-018-2484-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-018-2484-0