
Abstract
We evaluated six cases of diffuse large B-cell lymphoma (DLBCL) involving the red pulp of the spleen. All had B symptoms and an aggressive clinical course. The lymphoma cells proliferated diffusely and non-cohesively in the cords of the red pulp. The lymphoma involved the bone marrow in four of the five patients and the liver in all four of the patients examined. However, lymph node (LN) involvement was rare at presentation, and systemic LN involvement was not observed even in the terminal phase. The lymphoma cells infiltrated the intrasinusoidal/intravascular and interstitial spaces of the involved tissues and were detected in the peripheral blood in two of the six patients. CD5-expressing lymphoma cells were detected in four of the five patients examined. Because these cases had some unique clinical features and occurred in distinct splenic sites, we proposed that primary splenic DLBCL manifesting in red pulp is a distinct clinicopathological entity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extranodal diffuse large B-cell lymphomas (DLBCLs) have specific clinicopathologic features that are dependent on the organ of origin [1]. Primary splenic DLBCL is a rare type of lymphoma. The majority of the DLBCLs are derived from the white pulp of the spleen and form one large, or multiple nodules [2]. Before the advent of immunohistological examination, the absence of mass formation and the diffuse infiltration of large neoplastic cells into the red pulp might have been diagnosed as malignant histiocytosis [3]. Kuratsune et al. [4] reported the first case of DLBCL that non-cohesively proliferated into the splenic red pulp and demonstrated clinicopathological features of malignant histiocytosis. Since their initial report, only 18 cases of DLBCLs non-cohesively infiltrating the splenic red pulp have been reported [4–13]. These reports support the existence of DLBCL manifesting in the splenic red pulp (DLBCLRP). Kroft et al. [8] identified two cases of DLBCLRP involving the bone marrow (BM) and the liver, and Morice et al. [10] reported two cases of DLBCLRP with prominent BM intravascular/intrasinusoidal lymphomatous infiltration.
Because there have been few case reports of DLBCLRP, there is no comprehensive list of its clinicopathological features. To clarify the features of this rare lymphoma, we describe six new cases of DLBCLRP and present an analysis of the clinicopathological features of an additional 18 previously reported cases [4–13]. After reviewing the data, we developed seven characteristic features of DLBCLRP. Based on these features, we propose that DLBCLRP is a distinct clinicopathologic entity. Moreover, we discuss the relationship between DLBCLRP and an Asian variant of intravascular lymphomatosis (IVL) with splenomegaly [14].
Materials and methods
Retrospective analyses were conducted for six patients clinicopathologically diagnosed with primary splenic large B-cell lymphoma manifesting in red pulp in our hospital from 2000 to 2007. Two hundred and ninety two patients were diagnosed with malignant lymphoma, including 228 with B-cell lymphomas (DLBCL 129, follicular lymphoma 58, marginal zone lymphoma 19, mantle cell lymphoma 6, etc), 45 with T-cell lymphomas, and 12 with Hodgkin’s lymphomas during this 8-year period. Thirteen patients were diagnosed with primary splenic lymphoma, two with splenic marginal zone lymphoma, one with follicular lymphoma, and ten with DLBCL. Two patients were diagnosed as having DLBCL with a micronodular pattern. Eight patients were clinically diagnosed with DLBCLRP; however, two were excluded based on the unavailability of splenic tissues. All procedures were performed with informed consent of the patients.
Tissue specimens obtained from surgery, biopsy, or necropsy were fixed in formalin and embedded in paraffin. The paraffin-embedded sections were dewaxed with xylene. The sections were stained with hematoxylin–eosin (HE) for light microscopic examination, and by the streptavidin–biotin–peroxidase method (Nichirei Co., Tokyo, Japan) for immunohistochemical analysis. The panel of primary antibodies included L26 (CD20), JCB117 (CD79a), F7.2.38 (CD3), UCHL-1 (CD45RO), 1F8 (CD21), 124 (Bcl-2), PG-B6p (Bcl-6), MIB-1 (Ki-67), MUM1p (MUM1), IgM polyclonal (DAKO A/S, Glostrup, Denmark), MT1 (CD43), 4C7 (CD5), 56C6 (CD10), 1B12 (CD23), 1G12 (CD30), IgD polyclonal (Novocastra, Newcastle upon Tyne, UK), and SP4 (Cyclin D1, Lab Vision, Fremont, CA, USA). The primary antibodies were replaced with mouse or rabbit serum as a negative control.
Epstein–Barr virus (EBV)-encoded RNA (EBER) was detected by in situ hybridization (ISH). ISH was performed using the BioGenex Automated Staining System (i6000; BioGenex; San Ramon, CA, USA). The paraffin-embedded sections were dewaxed with xylene, treated with proteinase K, and hybridized with a fluorescein-conjugated oligonucleotide EBER probe (PR005-10X, BioGenex). To visualize the bound probe, a Super Sensitive polymer-HRP ISH Detection System (DF300-YCX, BioGenex) was used according to the manufacturer’s instructions.
Karyotypes were obtained at the time of diagnosis from all six patients from spleen (n = 5), and lymph node (LN; n = 1) samples, as previously described [15]. Chromosomal abnormalities were described according to ISCN [16].
Serum samples were analyzed with an immunofluorescence kit using anti-EBV capsid antigen (VCA), early antigen, and EBV-encoded nuclear antigen (TFB Inc., Tokyo, Japan). Antibodies against the human immunodeficiency virus (HIV) and HTLV-1 were examined using chemiluminescent enzyme immunoassay kits (Abbot Japan and Fuji Rebio Inc., Tokyo, Japan).
We characterized the degree of extramedullary hematopoiesis in the splenic samples as erythropoietic (grade 1), granulopoietic (grade 2), and trilineage hematopoietic (grade 3). Hemophagocytosis of normal splenic histiocytes was characterized as grade 1 (<10) and grade 2 (>10) erythrophagocytes in 10 high power fields. In bone marrow samples, grade 1 indicated thrombophagocytosis and grade 2 indicated leukoerythrophagocytosis.
Results
Clinical features
In this series, five of the six patients were men (age, 64–81 years; median, 69 years). At presentation, all patients were febrile. Four patients (nos. 1, 3–5) had splenomegaly without lymphadenopathy. One patient (no. 2) had localized LN swelling at presentation and a second patient (no. 6) had localized LN swelling at the time of surgery, which suggested the presence of a primary splenic lymphoma. All patients had high levels of lactate dehydrogenase (LDH), soluble interleukin-2 receptor, hypoalbuminemia, and moderately elevated C-reactive protein (Table 1). Differential blood counts revealed the following: Marked lymphocytopenia was detected in all patients except 1 (no. 5), thrombocytopenia was detected in five patients (nos. 1–5), and anemia was detected in four patients (nos. 2, 4–6). In two patients (nos. 1 and 4), lymphoma cells were detected in the peripheral blood (Table 2). Cholestasis and hepatocellular damage with hepatomegaly was observed in four patients (nos. 1, 2, 5, and 6), suggesting hepatic involvement.
Characteristic clinical features of DLBCLRP (Table 2) included BM involvement in four of the five patients (nos. 1, 2, 5, and 6; determined at admission), and hepatic infiltration in four patients (nos. 1, 5, and 6, and no. 2 determined at biopsy and necropsy, respectively). Enlarged LN were rare and limited to localized areas. Systemic lymphadenopathy was not observed even during the terminal stage of the disease. Hemophagocytosis by normal histiocytes was observed in the spleen and BM of all six patients.
We assessed two treatment groups: (a) three patients (nos. 2–4) were treated with six courses of CHOP (combination of doxorubicin, vincristine, cyclophosphamide, and prednisolone), and (b) the remaining three patients (nos. 1, 5, and 6) were treated with six courses of rituximab and CHOP followed by two courses of rituximab, etoposide, and nimustine hydrochloride with intrathecal administration of methotrexate, Ara-C, and prednisolone for prophylaxis of central nervous system lymphoma.
Follow-up (1–75 months; median, 38 months) information was obtained for all six patients. Two patients (nos. 4 and 2) of the former group died of lymphoma, within 1 and 5 months of the onset of the disease, respectively. All three patients of the latter group survived following first complete remission.
All patients were negative for HIV and HTLV-1. Antibodies to EBV indicated an old infection. M-protein and autoantibodies, including Coombs’ test, were not detected in any of the patients.
Pathologic features
Spleen size and weight were abnormally high in all informative cases (nos. 1, 3, 4, 5, and 6). In one patient (no. 2), spleen weight was not available (Table 2). The weight of the spleen, which is 80 to 120 g in healthy Japanese adults, was 220 to 900 g (median, 546 g) in the patients. Cut surfaces of the spleens showed beef red color and a lack of mass formation. In all six patients, the splenic red pulp was expanded and diffusely infiltrated by non-cohesive large lymphoma cells (nos. 1, 3, 4, 5, and 6 before therapy, and no. 2 at necropsy). Lymphoma cells were present in the cord and had infiltrated the sinuses to various degrees (Fig. 1a–c). In one patient (no. 5), a small number of large lymphoma cells were scattered among numerous T cells/histiocytes, and the patient was diagnosed with T-cell/histiocyte-rich B-cell lymphoma. All patients had extramedullary hematopoiesis of erythroblasts. Trilineage extramedullary hematopoiesis was observed in two patients (nos. 1 and 5). In all six patients, there were an abnormally high number of histiocytes, which were engaged in hemophagocytosis. In two (nos. 1 and 3) of these patients, there was intensive hemophagocytosis.

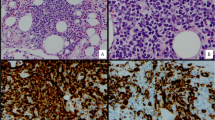
Lymphoma cells infiltrate the Billroth’s cords and sinuses of the splenic red pulp and the vascular wall. a Medium power field of the affected spleen. The tumor cells have diffusely infiltrated the Billroth’s cords of red pulp, case 3 HE staining. Original magnification, ×100. b Medium power field of the affected spleen. Tumor cells have diffusely infiltrated the Billroth’s cords as well as the sinusoid, case 4 HE staining. Original magnification, ×100. c High power magnification of PAS-stained sections showing the basement membrane of the sinusoid. Note the intrasinusoidal and extrasinusoidal lymphoma cell infiltrations, case 1. Original magnification, ×350. d Monomorphous lymphoma cell infiltration in the vascular wall. Short arrows indicate the vascular endothelial cells and long arrows indicate the arterial adventitia. Case 3 HE staining. Original magnification, ×40
In one patient (no. 1), CD20 and CD34 immunohistochemistry revealed large lymphoma cells that were visible in the sinusoids and a few were present in the cords (Fig. 2a,b). In the remaining patients, the lymphoma cells had diffusely infiltrated the interstitial spaces (Fig. 2e). It is possible that the intravascular lymphomatous infiltration of this lymphoma disappeared due to the destruction of endothelial cells by lymphoma cells in the significantly infiltrated lesions. Thrombophagocytosis in the BM was observed in all patients, and leukoerythrophagocytosis was detected in one of the five patients examined (no. 6).

Intrasinusoidal/intravascular lymphoma cell infiltration in the bone marrow, liver, lymph node, and adipose tissue. a CD20 immunostaining showing the predominantly intrasinusoidal pattern of marrow infiltration by large B-cell lymphoma, case 1. Original magnification, ×450. b CD34 immunoperoxidase staining of bone marrow sinusoidal cell lining (arrow). Large lymphoma cells are present in a sinus, case 1. Original magnification, ×350. c High power field of affected liver. Note an intrasinusoidal infiltration of large lymphoma cells, case 1 HE staining. Original magnification, ×350. d CD34 immunoperoxidase staining highlights the intravascular lymphoma cells from diffuse lymphomatous infiltration of lymph node parenchyma. Arrows show sinusoidal cell lining, case 2. Original magnification, ×400. e Medium power field of the affected BM. The tumor cells diffusely infiltrated in the cords of BM, case 6. Original magnification, ×100. f Perisplenic adipose tissue. Note the intravascular infiltrate of large lymphoma cells, case 4 HE staining. Original magnification, ×150
In four patients examined (nos. 1, 5, and 6 at biopsy, and no. 2 at necropsy), lymphoma cells had infiltrated into the sinusoidal and portal areas of the liver (Fig. 2c).
In one patient (no. 2), the lymph nodes were replaced completely by lymphoma cells. Intravascular infiltration of lymphoma cells was not visible in the HE-stained samples. However, diffuse infiltration into the vascular spaces was clearly detected by immunostaining for CD34 (Fig. 2d). Furthermore, in one patient (no. 4), intravascular infiltration of lymphoma cells was prominent in the adipose tissue of the splenic hilus (Fig. 2f), and in another patient (no. 1) infiltration into the subcutaneous vessels was observed. In three patients (nos. 2–4), lymphoma cells had infiltrated below the endothelial cells or into the vascular wall (Fig. 1d).
Immunohistochemistry of lymphoma cells
All patients had lymphoma cells positive for CD20 and Bcl-2 and negative for EBER, Cyclin D1, and CD23 (Table 3). The cells in two patients (nos. 3 and 6) were positive for CD10. Of the remaining four patients with CD10-negative lymphoma cells, the cells were negative for bcl-6 in three patients (nos.1, 4, and 5), and positive for bcl-6 and MUM1 in one patient (no. 2). CD5-positive cells were found in four of five patients by flow cytometry and three of five patients by immunohistochemistry. Cells of five patients examined were positive for surface membrane (sm) IgM and negative for smIgD (Table 3). Positive rate of Ki67 varied from 10% to 80%.
Chromosomal analysis
Chromosomes were analyzed in all six patients. Two patients (nos. 5 and 6) had normal karyotypes. The remaining four patients (nos. 1–4) were CD5-positive and carried complex chromosomal aberrations. One of these patients (no. 4) showed the 11q13 aberrations, whereas another (no. 2) showed the 3q27 abnormality (Table 4).
Discussion
Here, we have reported six cases of DLBCLRP. To further clarify the clinicopathological characteristics of DLBCLRP, we analyzed a total of 24 cases (Tables 5 and 6) of DLBCLRP, including six cases encountered by one of the authors (M. Kashimura). In the present series, 18 of the 24 patients were men and the age range of all the patients was 40–81 years (median, 64.5 years).
The size and weight of the spleens that were examined had increased in all informative cases. Spleen weight data were not available for five of the 24 patients. The weight of the spleen, which is 80–120 g and 110–170 g in healthy adults of Japan and western countries, respectively, was 220 to 2,600 g (median, 1,300 g) in the present cases.
We found that DLBCLRP has seven characteristic clinicopathological features as shown in Table 7 and a pattern of infiltration of the spleen. Lymphoma cells were present in the cord in all but one case of the 24 cases of splenic infiltration. In the remaining patient, the lymphoma cells were restricted to the sinusoids [7].
At presentation, lymphoma cells were frequently found in the BM (13/20, 65%) and the liver (11/17, 65%). However, LN involvement was rare and limited to a localized area (10/21, 48%), and LN swelling was not significant. Systemic LN swelling was not observed, even during the terminal phase of the disease.
Another characteristic of DLBCLRP is an intrasinusoidal infiltration of lymphoma cells in the BM (3/7, 43%) and the liver (8/8, 100%) as well as interstitial infiltration of lymphoma cells in the BM (10/11, 91%) and the liver (7/8, 88%). Furthermore, we observed intravascular infiltration of lymphoma cells in the LN (no. 2) and the adipose tissue (nos. 1 and 4), and the presence of lymphoma cells in the peripheral blood (10/23, 43%). This rate appears higher than that observed for nodal DLBCL.
Moreover, we observed lymphoma cell surface expression of the CD5 antigen in eight of the ten patients (80%) [8, 10, 12], including four of our original six patients. Low intensity of the lymphoma cell CD5 antigen might not be detectable by immunohistochemistry (Tables 3 and 6).
None of the patients had a history of chronic lymphocytic leukemia. Transformation of splenic marginal zone lymphoma (SMZL) into DLBCL [17, 18], which might be associated with CD5-positive transformation, must be ruled out in order to diagnose DLBCLRP as the de novo DLBCL. Despite a careful study, we were unable to find proliferative lesions of medium-sized lymphoma cells in any of our cases, which is usually observed when DLBCL transforms from SMZL. Lymphoma cells of all five patients examined were positive for smIgM; however, they were negative for smIgD, which is unusual for SMZL cells [19, 20]. Although four of the six patients (nos. 1–4) showed complex chromosomal aberrations, none of them had trisomy 3 or allelic loss of 7q21-32, which is usually observed with SMZL [21, 22]. Clinically, autoimmune phenomena and the presence of M-protein have been frequently observed in SMZL patients [17, 20, 23–25], but these were not observed in our cases. Therefore, our cases are not likely to be associated with SMZL transformation.
Therefore, these four patients (nos. 1–4) were diagnosed with de novo CD5-positive DLBCL [26]. Two of these patients with complex chromosomal aberration had chromosome abnormalities involving 3q27 (no. 2) and 11q13 (no. 4), which have been previously reported in CD5-positive DLBCL [27]. Yamagichi et al. [28] recently reported four morphological variants of de novo CD5-positive DLBCL. Using the molecular classification system by Hans et al. [29], lymphoma cells of 82% of the patients were of the non-germinal center B-cell type and the others were germinal center B-cell type. Interestingly, intravascular and/or intrasinusoidal lymphomatous infiltration was observed in 38% of the cells. Although data were not shown, the bone marrow, liver, and spleen were reported to be the most frequently involved anatomical sites. De novo CD5-positive DLBCL is heterogeneous in morphological, molecular, and clinical aspects. A potion of the cases with de novo CD5-positive DLBCL with splenomegaly may be DLBCLRP. While lymphoma cells of two other patients (nos. 5 and 6) in this study were CD5 negative, clinicopathological features other than CD5 were identical in these six patients (Table 7).
This high rate of CD5-positive lymphoma cells may be an aberrant expression of the aggressive lymphoma cells [30–32]. In previous genetic studies, it was suggested that de novo CD5-positive DLBCL originates from somatically mutated CD5 progenitor B cells [33, 34]. The mechanism of expression of CD5 in this lymphoma remains unsolved.
The lymphoma cells positive for CD10 in two patients (nos. 3 and 6) were considered germinal center B-cell type. Of the remaining four patients that were negative for CD10, three were negative for bcl-6 (nos. 1, 4, and 5) and one was positive for bcl-6 and MUM1 (no. 2). Therefore, later these were considered non-germinal center B-cell type (Table 3) [29]. Clinicopathological features were not clearly distinguishable among patients with germinal center B-cell-like DLBCL and those with non-germinal center B-cell-type DLBCL, except for the appearance of the lymphoma cells in the peripheral blood. However, our limited sample size makes it difficult to regard this feature as significant.
DLBCLRP is a clinically highly aggressive lymphoma compared with conventional CD5-negative DLBCL and de novo CD5-positive DLBCL. The patients with de novo CD5-positive DLBCL showed more aggressive clinical features and parameters (LDH level, clinical stage, and international prognostic index (IPI) score) than those with CD5-negative DLBCL [26]. All six patients in this study had fever, high LDH levels, clinical stage 4B, and a high-risk IPI score.
Another characteristic feature of DLBCLRP is hemophagocytosis of normal histiocytes observed in all the BMs (intensive cases; 1/5, 20%) and spleens (intensive cases; 2/6, 33%). This might be due to the high cytokine concentrations produced by these aggressive lymphoma cells [35]. But none fulfilled the criteria for adult hemophagocytic syndrome [36–38]. Moreover, the frequent presence of B symptoms (14/15) was a characteristic of DLBCLRP. On the basis of these clinicopathological features, we propose that DLBCLRP is a distinct clinicopathological entity.
The clinicopathological features of the cases of the Asian variant of IVL (AIVL) with splenomegaly are similar to those of DLBCLRP. Splenectomy samples from two patients with AIVL had cut surfaces that were beef red in color and there was a diffuse, large, B-cell lymphoma cell infiltrate in the red pulp [14]. However, the international consensus meeting of intravascular large B-cell lymphoma (IVLBCL) proposed a new definition of IVLBCL, which included cases from both western and Asian countries [39]. They added the criterion of a concomitant minimal extravascular location of the neoplastic cells [40] to the WHO criteria, which states that the neoplastic lymphocytes might only be present in the lumina of the small vessels, such as capillaries [41]. DLBCLRP produced interstitial infiltration as well as intrasinusoidal lymphomatous infiltration in the BM, liver, and LN (Fig. 2d and Table 5). The infiltration of DLBCLRP lymphoma cells into the liver and BM was a more characteristic feature of the SMZL than AIVL. However, further studies are required to clarify these relationships.
Briefly, DLBCLRP is an aggressive lymphoma with B symptoms. During the early phase, it spreads to the liver and/or BM via intrasinusoidal and/or interstitial infiltration. Because our characterization of DLBCLRP was based on the observations of a limited number of patients, further studies are required to confirm that DLBCLRP is a distinct clinicopathologic entity.
References
Mann RB (1999) Are there site-specific differences among extranodal aggressive B-cell neoplasms? Am J Clin Pathol 111:S144–S150
Harris NL, Aisenberg AC, Meyer JE et al (1984) Diffuse large cell (histiocytic) lymphoma of the spleen. Clinical and pathologic characteristics of ten cases. Cancer 54:2460–2467
Burke JS (1981) Surgical pathology of the spleen: an approach to the differential diagnosis of splenic lymphomas and leukemias. Part II. Diseases of the red pulp. Am J Surg Pathol 5:681–694
Kuratsune H, Machii T, Aozasa K et al (1988) B cell lymphoma showing clinicopathological features of malignant histiocytosis. Acta Haematol 79:94–98
Betman HF, Vardiman JW, Lau J (1994) T-cell-rich B-cell lymphoma of the spleen. Am J Surg Pathol 18:323–324
Faravelli A, Gambini S, Perego D et al (1995) Splenic lymphoma: unusual case with exclusive red pulp involvement. Pathologica 87:692–695
Kobrich U, Falk S, Karhoff M et al (1992) Primary large cell lymphoma of the splenic sinuses: a variant of angiotrophic B-cell lymphoma (neoplatic angioendotheliomatosis)? Hum Pathol 2:1184–1187
Kroft SH, Howard MS, Picker LJ et al (2000) De novo CD5+ diffuse large B-cell lymphomas. A heterogeneous group containing an unusual form of splenic lymphoma. Am J Clin Pathol 114:523–533
Mollejo M, Algara P, Mateo MS et al (2003) Large B-cell lymphoma presenting in the spleen: identification of different clinicopathologic conditions. Am J Surg Pathol 27:895–902
Morice WG, Rodriguez FJ, Hoyer JD et al (2005) Diffuse large B-cell lymphoma with distinctive patterns of splenic and bone marrow involvement: clinicopathologic features of two cases. Mod Pathol 18:495–502
Palutke M, Eisenberg L, Narang S et al (1988) B lymphocytic lymphoma (large cell) of possible splenic marginal zone origin presenting with prominent splenomegaly and unusual cordal red pulp distribution. Cancer 62:593–600
Salgado C, Feliu E, Montserrat E et al (1993) B-type large-cell primary splenic lymphoma with massive involvement of the red pulp. Acta Haematol 89:46–49
Stroup RM, Burke JS, Sheibani K et al (1992) Splenic involvement by aggressive malignant lymphomas of B-cell and T-cell types. A morphologic and immunophenotypic study. Cancer 69:413–420
Murase T, Nakamura S, Tashiro K et al (1997) Malignant histiocytosis-like B-cell lymphoma, a distinct pathologic variant of intravascular lymphomatosis: a report of five cases and review of the literature. Br J Haematol 99:656–664
Hashimoto K, Miura I, Chyubachi A et al (1995) Correlations of chromosome abnormalities with histologic and immunologic characteristics in 49 patients from Akita, Japan with non-Hodgkin lymphoma. Cancer Genet Cytogenet 81:56–65
Mitelman F (ed) ISCN (1995). An international system for human cytogenetic nomenclature. Karger, Basel
Berger F, Felman P, Thieblemont C et al (2000) Non-MALT marginal zone B-cell lymphomas: a description of clinical presentation and outcome in 124 patients. Blood 95:1950–1956
Camacho FI, Mollejo M, Mateo MS et al (2001) Progression to large B-cell lymphoma in splenic marginal zone lymphoma: a description of a series of 12 cases. Am J Surg Pathol 25:1268–1276
Mollejo M, Menárguez J, Lloret E, Sánchez A, Campo E, Algara P, Cristóbal E, Sánchez E, Piris MA (1995) Splenic marginal zone lymphoma: a distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am J Surg Pathol 19:1146–1157
Van Huyen JP, Molina T, Delmer A et al (2000) Splenic marginal zone lymphoma with or without plasmacytic differentiation. Am J Surg Pathol 24:1581–1592
Dierlamm J, Michaux L, Wlodarska I et al (1996) Trisomy 3 in marginal zone B-cell lymphoma: a study based on cytogenetic analysis and fluorescence in situ hybridization. Br J Haematol 93:242–249
Mateo M, Mollejo M, Villuendas R et al (1999) 7q31-32 allelic loss is a frequent finding in splenic marginal zone lymphoma. Am J Pathol 154:1583–1589
Chacón JI, Mollejo M, Muñoz E et al (2002) Splenic marginal zone lymphoma: clinical characteristics and prognostic factors in a series of 60 patients. Blood 100:1648–1654
Franco V, Florena AM, Iannitto E (2003) Splenic marginal zone lymphoma. Blood 101:2464–2472
Iannitto E, Ambrosetti A, Ammatuna E et al (2004) Splenic marginal zone lymphoma with or without villous lymphocytes. Hematologic findings and outcomes in a series of 57 patients. Cancer 101:2050–2057
Yamaguchi M, Seto M, Okamoto M et al (2002) De novo CD5+ diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients. Blood 99:815–821
Yoshioka T, Miura I, Kume M et al (2005) Cytogenetic features of de novo CD5-positive diffuse large B-cell lymphoma: chromosome aberrations affecting 8p21 and 11q13 constitute major subgroups with different overall survival. Genes Chromosomes Cancer 42:149–157
Yamaguchi M, Nakamura N, Suzuki R et al (2008) De novo CD5+ diffuse large B-cell lymphoma: results of a detailed clinicopathological review in 120 patients. Haematologica 93(8):1195–1202
Hans CP, Weisenburger DD, Greiner TC et al (2004) Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103:275–282
Ballesteros E, Osborne BM, Matsushima AY (1998) CD5+ low-grade marginal zone B-cell lymphomas with localized presentation. Am J Surg Pathol 22:201–207
Ferry JA, Yang WI, Zukerberg LR et al (1996) CD5+ extranodal marginal zone B-cell (MALT) lymphoma. A low grade neoplasm with a propensity for bone marrow involvement and relapse.. Am J Clin Pathol 105:31–37
Tiesinga JJ, Wu CD, Inghirami C (2000) CD5+ follicle center lymphoma. immunophenotyping detects a unique subset of “floral” follicular lymphoma. Am J Clin Pathol 114:912–921
Katzenberger T, Lohr A, Schwarz S et al (2003) Genetic analysis of de novo CD5+ diffuse large B-cell lymphomas suggests an origin from a somatically mutated CD5+ progenitor B cell. Blood 101:699–702
Nakamura N, Hashimoto Y, Kuze T et al (1999) Analysis of the immunoglobulin heavy chain gene variable region of CD5-positive diffuse large B-cell lymphoma. Lab Invest 79:925–933
Imashuku S (1997) Differential diagnosis of hemophagocytic syndrome: underlying disorders and selection of the most effective treatment. Int J Hematol 66:135–151
Tsuda H (1997) Hemophagocytic syndrome in children and adults. Int J Hematol 65:215–226
Wong KF, Chan JK (1992) Reactive hemophagocytic syndrome—a clinicopathologic study of 40 patients in an Oriental population. Am J Med 93:177–180
Yao M, Cheng AL, Su IJ et al (1994) Clinicopathological spectrum of hemophagocytic syndrome in Epstein–Barr virus associated peripheral T-cell lymphoma. Br J Haematol 87:535–543
Ponzoni M, Ferreri AJ, Campo E et al (2007) Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol 25:3168–3173
Ponzoni M, Arrigoni G, Gould VE et al (2000) Lack of CD 29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol 31:220–226
Gatter KC, Warnke RA (2001) Intravascular large B-cell lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW (eds) Tumors of haematopoietic and lymphoid tissues. IARC, Lyon
Conflict of interest statement
The authors declare no competing financial interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kashimura, M., Noro, M., Akikusa, B. et al. Primary splenic diffuse large B-cell lymphoma manifesting in red pulp. Virchows Arch 453, 501–509 (2008). https://doi.org/10.1007/s00428-008-0673-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-008-0673-y