
Abstract
Fulminant Epstein–Barr virus (EBV+) T-cell lymphoma in immunocompetent elderly patients is rare and its character has not been well defined. This study analyzed the clinicopathological features of five elderly patients (group A: 50–84 years) and compared them with those of eight children and young adult patients with systemic T-cell lymphomas (group B: 10–34 years). Group A more commonly presented with generalized lymphadenopathy (n = 3) than did group B (n = 1). Chronic active EBV infection (n = 3) and hydroa vacciniforme-like eruptions (n = 1) were seen in group B, while group A showed no evidence of chronic EBV infection, but did show chronic hepatitis B or C virus infections (n = 3). The histological and immunophenotypical findings were similar. All patients died within 1 to 14 months of diagnosis. These findings suggest that EBV+ T-cell lymphoma in elderly patients is a unique disease with an underlying derangement of T-cell immunity and failure to eradicate infected virus. Additional factors related to senility may play a role in the disruption of homeostasis between the virus and the host’s immune system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary infection with Epstein–Barr virus (EBV) is usually asymptomatic, and EBV has been shown to be latent in B cells for life in normal hosts after primary infection; however, some children and more often, adolescents and young adults, develop infectious mononucleosis (IM) [26]. IM is characterized by fever, hepatosplenomegaly, lymphadenopathy, and an increase in activated CD8+ T lymphocytes in the peripheral blood [1, 26]. Although IM usually follows a self-limited course without severe complications, uncommonly, acute EBV infections with fulminant manifestations such as persistent fever, severe hepatosplenomegaly, severe cytopenia, coagulopathy, central nervous system abnormalities, and vascular dysfunction have been demonstrated, and histiocytic erythrophagocytosis has been found in the bone marrow and secondary lymphoid organs of most of these patients [15, 28]. Those fulminant cases have been reported mainly in Asian countries, including Taiwan and Japan, and have been described under a variety of terms including fulminant EBV+ T-cell lymphoproliferative disease(LPD) of childhood, and sporadic fatal infectious mononucleosis. These diseases occur shortly after primary EBV infection or are associated with a long prodromal syndrome referred to as chronic active EBV (CAEBV) infection.
CAEBV infection is characterized by chronic or recurrent infectious mononucleosis-like symptoms persisting for at least 6 months and by an unusual pattern of anti-EBV antibodies [24]. Severe CAEBV infection is a more severe form of the disease, with a high mortality and high morbidity and life-threatening complications such as virus-associated hemophagocytic syndrome, interstitial pneumonia, lymphoma, coronary aneurysms, and central nervous system involvement [10, 16, 21, 22, 25]. Many studies have reported that clonal expansion of EBV-infected T or natural killer (NK) cells might be associated with CAEBV infection [7, 8, 10, 11, 14, 16, 17, 23, 31].
A new World Health Organization classification for fulminant EBV+ T-cell lymphoproliferative disorders, which will appear in 2008, establishes a new disease category of systemic EBV+ T-cell lymphoma. The disease is defined as a life-threatening illness of children and young adults characterized by a clonal proliferation of EBV-infected T cells with an activated cytotoxic phenotype.
Whereas systemic EBV+ T-cell lymphomas most commonly occur in children and young adults, the similar entity of fulminant EBV+ T-cell lymphoma can occur in immunocompetent adult patients. In our institution, we have noted several cases of fulminant EBV+ systemic T-cell lymphomas in elderly patients with no known immunodeficiency. Those cases have clinicopathological features similar to those of the systemic T-cell lymphoma of children and young adults, but some differences were also observed. Herein, we describe the clinicopathological features and molecular and immunohistochemical findings of 14 cases of fulminant EBV+ T-cell lymphoma to define the characteristics of systemic EBV+ T-cell lymphoma in elderly patients and to elucidate the relationship of these tumors to those in children and young adult patients.
Materials and methods
Patients
The criteria for the inclusion of case were as follows: (1) an aggressive clinical presentation with or without hemophagocytic syndrome; (2) EBER positivity in a majority of infiltrated lymphocytes; (3) clonal T-cell proliferation, as supported by either T-cell receptor gene rearrangement studies, or marked cytological atypia of CD3-positive cells; and (4) available paraffin block(s) for additional studies. This study was approved by the Institutional Review Board in accordance with the Declaration of Helsinki.
The surgical pathology files of the Department of Pathology, Samsung Medical Center from 1995 to 2007 were searched. Of the 20 cases that showed EBV+ T-cell lymphoproliferation, 13 cases were selected. Of the seven cases excluded, the clinical history was unavailable for two, one case had a history of rheumatoid arthritis, two cases were of indolent cutaneous lymphoproliferative disorders, one case was a typical hemophagocytic lymphohistiocytosis (HLH) according to the definition of Henter et al. [6], and one case showed polyclonal T-cell receptor gene rearrangements.
All patients were previously healthy, with no opportunistic infections or other indications of congenital immunodeficiency, nor had they received immunosuppressive medications. We segregated our 13 cases into two groups: elderly patients more than 50 years old, and children and young adults.
Formalin-fixed, paraffin-embedded tissue blocks of liver, bone marrow, lymph node, pharynx, tonsil, larynx, stomach, duodenum, colon, retroperitoneum, and skin biopsies were used for evaluation. All slides were reviewed by two pathologists.
Immunophenotype studies
Immunohistochemical analysis was performed on paraffin sections using monoclonal and polyclonal antibodies for the detection of lineage-specific or lineage characteristic antigens including CD3 (Dakopatts, Copenhagen, Denmark), CD20 (Dakopatts), CD56 (Monosan, Uden, The Netherlands), CD4 (Novocastra, Newcastle upon Tyne, UK), CD8 (Novocastra), βF1 (Endogen, Rockford, IL, USA), and granzyme B (Zymed, South San Francisco, CA, USA).
EBV studies
Serological studies of EBV infection were performed, including the detection of antiviral capsid antigen IgG and IgM (EBV-VCA IgG and IgM) and antiearly antigen IgG (EBV-EA IgG) using indirect immunofluorescence methods, and antinuclear antigen (EBNA) using an anticomplement immunofluorescence method.
EBV RNA was detected by an in situ hybridization technique. Paraffin-embedded sections (5 μm) were dewaxed with xylene, followed by treatment with proteinase K, and hybridized with FITC-conjugated EBER-1 and EBER-2 oligonucleotide probes (Novocastra). Following incubation with anti-FITC antibody tagged with alkaline phosphatase, slides were covered with nitrobluetetrazolium, 5-bromo-4-chloro-3-indolyl phosphate, and 1 M levamisole. We used EBV-negative lymphoid tissues and the hybridization mixture without EBV oligonucleotides as negative controls.
Real-time quantitative polymerase chain reaction (PCR) assays for EBV DNA were performed using instruments from Roche Molecular Diagnostics (Mannheim, Germany) and the LightCycler EBV quantification kit as described [5]. The amount of EBV DNA was calculated as the number of virus copies per 5 μl whole blood. The EBV detection limit of this assay is approximately ten copies per reaction for whole blood and the linear measuring range of the assay is 102 to 106 per reaction.
Molecular studies
For PCR amplification of the TCR-γ locus, DNA was prepared by standard proteinase K digestion and phenol/chloroform extraction. PCR followed by single-stranded conformational polymorphism analysis was performed as previously described [18].
Detection of chromosomal abnormalities
Unstimulated isolated bone marrow cells were cultured for 24 h and G-banded according to standard procedures. Metaphases were analyzed and karyotyped according to the nomenclature system proposed by the International System for Human Cytogenetic Nomenclature, 1995.
Results
Clinical findings
The clinical characteristics of the patients are summarized in Tables 1 and 2. All patients were Korean, with eight male and five female patients, ranging in age from 10 to 84 years (mean 35.4 years). The elderly group consisted of five patients (four males and one female), ranging from 50 to 84 years (mean 62 years), and the children and young adult group consisted of eight patients (four males and four females), ranging in age from 10 to 34 years (mean 16.7 years).
The clinical presentations of the children and young adult group and the elderly group were similar, but with some differences. In the elderly patients, three of the five patients presented with generalized lymphadenopathy as well as fever, cytopenia, and liver dysfunction (cases 1, 2, and 5). One patient had fever, liver dysfunction, pancytopenia, and gastrointestinal symptoms (case 3). Biopsies from the duodenum and colon revealed atypical cells. One patient presented with abdominal pain and a retroperitoneal mass (case 4). Anemia and thrombocytopenia were observed in all patients. Bone marrow biopsy revealed hemophagocytosis in one patient (case 3). A history of hypersensitivity to mosquito bite or CAEBV infection was not observed.
Among the children and young adults, four of eight patients presented with acute onset of fever, general malaise, and hepatosplenomegaly (cases 7–9 and 11). Laboratory testing showed pancytopenia and liver function abnormalities. Among the other four patients, three had a history compatible with chronic EBV infection. One of them had a history of liver dysfunction and enlarged cervical lymph nodes for 2 years (case 6). Other two patients had intermittent fever of unknown origin for 6 months and 1 year respectively (cases 12 and 13). One patient presented with hydroa vacciniforme-like skin lesions on the light-exposed areas such as the face and arms occurring over several years, along with IgA nephropathy (case 10). At admission, the larynx was involved by EBV+ T-cell lymphoma. Generalized lymphadenopathy was identified in only one case (case 6). Bone marrow was initially infiltrated by hemophagocytic histiocytes or atypical tumor cells in the majority of cases.
All patients were treated with steroids or chemotherapy, with various regimens depending on the patient’s general condition; however, the clinical course was fulminant or aggressive, with all patients dying within 10 days to 14 months of diagnosis. Causes of death for the elderly patients were septic shock (four patients) and systemic mucormycosis (one patient). The children and young adults died of sepsis with disseminated intravascular coagulation (two patients), pneumocystis pneumonia with disseminated intravascular coagulation (one patient), multi-organ failure (one patients), liver failure and tumor lysis syndrome (one patient), and renal failure(one patient). Cause of death was not known in one children.
Histological findings
Four out of five patients with lymphadenopathy had lymph node biopsies. The lymph nodes in three patients (cases 1, 2, and 5) showed diffuse effacement of the normal nodal architecture by infiltration of relatively monotonous small, medium, or large lymphocytes with hyperchromatic nuclei and irregular nuclear contours. Inflammatory cells such as eosinophils and plasma cells were frequently found. The lymph nodes from case 6 showed preserved but partially effaced lymph node architecture and intact capsules and sinuses. The interfollicular T zone was widened and infiltrated by small lymphocytes lacking significant cytological atypia. Some of the lymphocytes were large. Epithelioid histiocytes or small granulomas were scattered.
The bone marrow was involved in three patients. Tumor cells massively infiltrated the bone marrow in case 11, whereas case 7 and case 9 showed scattered pleomorphic atypical cells with or without hemophagocytosis. Five patients showed an infiltration of hemophagocytic histiocytes and increased small T lymphocytes without cytological atypia.
Skin biopsies were performed in three cases (cases 1, 3, and 10). Erythematous papules with or without ulceration were documented clinically and showed perivascular and periappendageal lymphocytic infiltration in the upper dermis, with or without necrosis. One case showed a subcutaneous infiltration of lymphocytes (case 3). The patient with the hydroa vacciniforme-like eruption (case 10) showed characteristic skin lesions. A biopsy of necrotic vesicles demonstrated necrosis of the epidermis and subjacent dermis. Perivascular and periappendageal lymphocytic infiltration of small lymphocytes with angioinvasion and mild atypia was observed.
Endoscopic biopsies from the stomach, duodenum, and colon were performed for suspicious lesions from elderly patients (cases 2 and 3). One patient showed mucosal erosion with many T cells showing mild atypia and villous atrophy (case 3) and no obvious atypical infiltrates were observed in the other patient.
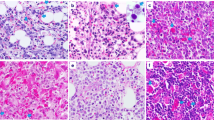
To compare the degree of histological change between the elderly group and the children and young adult group, we classified the 13 cases into three grades according to histology (Figs. 1, 2 and 3). We defined grade 1 as cases with polymorphic infiltrates including plasma cells, histiocytes, and small lymphocytes without significant atypia; grade 2 as cases with monomorphic infiltrates with predominantly small to medium lymphocytes showing mild atypia; grade 3 as cases with overt lymphoma showing monomorphic infiltrates with medium to large lymphocytes showing marked atypia. The cytological atypia of grade 1 cases was minimal or equivocal, so if EBER in situ hybridization is not performed, the lesion can be easily overlooked as chronic inflammation. The elderly group and the children and young adult group showed no significant difference in the histological changes. The elderly group showed one grade 1, two grade 2, and two grade 3 cases, and the children and young adult group showed two grade 1, four grade 2, and two grade 3 cases.

Microscopic finding of grade 1. There are polymorphic infiltrates of plasma cells, histiocytes, and small lymphocytes without significant atypia (case 6)

Microscopic finding of grade 2. Monotonous infiltrates with predominantly small to medium lymphocytes showing mild atypia (case 4)

Microscopic finding of grade 3. Overt lymphoma showing monomorphic infiltrates of large lymphocytes with marked atypia (case 11)
Immunophenotypic findings
Immunophenotypic features are summarized in Table 1. The infiltrate in all cases was composed of CD3+ T cells. The CD4/8 phenotype was evaluated in four cases from the elderly group and six cases from the children and young adults group. Nine cases were evaluated by immunohistochemistry and one case by flow cytometry. The infiltrate was predominantly cytotoxic CD8+ αβ T cells in eight cases (3/4 in the elderly group and 5/6 in the children and young adults group). CD4+ and CD8+ T cells were admixed with slight CD4 predominance in one case (1/6 in the children and young adult group). One elderly patient was regarded of γ/δ phenotype due to the lack of staining with the βF1 antibody in the proper reactivity with internal control. Three cases did not have specimens available for CD4 and CD8 study. All cases were negative for CD56 and positive for granzyme B.
EBV analysis
EBV serology was available in eight patients. The EBV-related antibody of each patient was detected, as shown in Table 1. In all the cases examined, EBV-VCA IgG was positive in the absence of EBV-VCA IgM. EBV-EA was positive in two of five cases in the children and young adult patients, but was consistently negative in the elderly patients.
In situ hybridization (EBER-1) showed the presence of EBV-infected cells in the tissues. All cases showed numerous EBER-1 positive cells (Fig. 4).

EBER in situ hybridization showing numerous positive cells (case 7)
Peripheral blood was obtained from five patients and the viral load was determined by quantitative real-time PCR. In four patients, the viral loads were measured serially over observation periods ranging from 5 to 9 months (mean, 6.5 months). The highest results were shown in Table 1.
Gene rearrangement study
Analysis by PCR of paraffin-embedded tissue was performed in nine cases—four from the elderly patient group and five from the children and young adult patient group—and showed clonal rearrangements of TCR-γ genes in all cases.
Chromosomal study
Cytogenetic studies were successfully performed using isolated bone marrow cells from six patients (five children and young adult cases and one elderly patient case). Only two patients from the children and young adults group showed chromosomal abnormalities. The karyotypes of those cases were 46XY, inv(14)(q11.2q32)[7]/46,XY[13] (case 7) and 47,Y,-X,+19,+mar[1]/47,idem,add(12)(p12)[2] (case 9).
Discussion
EBV-positive systemic T-cell lymphoma has been clinically recognized as EBV-associated hemophagocytic lymphohistiocytosis or EBV-associated hemophagocytic syndrome, severe CAEBV infection, and fatal IM. Even though this disease entity is rare, there have been many sporadic reports describing fulminant T-cell LPD following acute EBV infection and CAEBV infection in Asian countries especially Japan and Taiwan [15, 28]. The patients were mainly children or young adults, with cases occurring in elderly patients hard to find in the literature.
In this study, we compared the clinicopathological features and immunohistochemical and molecular findings of five elderly patients and eight children or young adult patients. Although the study has limitations because of the small number of elderly patients, the data suggested some differences in the clinicopathological features. The cases in the children and young adult group usually presented with chronic intermittent or acute onset of fever, general malaise, and hepatosplenomegaly. The bone marrow or liver was the initial site of involvement and generalized lymphadenopathy was unusual. Laboratory tests showed pancytopenia and liver function abnormalities. The elderly patients more commonly presented with generalized lymphadenopathy. Hemophagocytosis or the involvement of bone marrow by tumor cells was not common at initial presentation.
Regarding the underlying disease, a hydroa vacciniforme-like skin lesion was seen in one patient and chronic active EBV infection in three patients in the children and young adults group. Therefore, about 50% of the systemic T-cell lymphomas in the children and young adult group seem to develop through a prodromal period of chronic EBV infection. This incidence of preceding chronic EBV infection is higher than those reported previously by Quintanilla-Martinez et al. [23]. Our five elderly patients had no history of either CAEBV infection or hydroa vacciniforme-like eruptions. Instead, three of five patients were hepatitis B virus carriers or had chronic hepatitis C virus infection. Although the patients had no proven immune defects, the common association with hepatitis virus in these patients suggests an apparently ineffective antiviral T-cell response.
It is intriguing that IgA nephropathy was present in the girl with the hydroa vacciniforme-like skin lesions and subsequent development of T-cell lymphoma. A previous study demonstrated the EBV genome in seven of 12 patients with IgA nephropathy (58%) and suggested that EBV contributes to glomerular mesangial injury [9].
The histological features of 13 cases showed broad cytological appearances ranging from a reactive appearance to overt lymphoma, and there were no significant differences between the children and the elderly patients. Grade 1 histology was identified in two cases that revealed monoclonality according to TCR gene rearrangement and numerous EBV-positive cells by in situ hybridization. The differential diagnosis between reactive change and lymphoma was difficult on morphologic grounds alone. EBER in situ hybridization is essential to avoid misdiagnosis.
We analyzed EBV-related antibodies in eight patients. In primary EBV infection in healthy hosts, IgM antibodies to VCA rise first, followed by VCA IgG and EBV-EA. EBV IgM and EBV-EA will eventually disappear, and lastly, an antibody to EBNA arises at least 1 month after the onset of symptoms [30]. EBV IgG and EBNA will remain for life. In many people, detection of antibody to the early antigen is a sign of active infection. In the absence of EBNA, EBV-EA indicates primary infection. Considering this principle, the serologic profiles of the members of the children and young adult group were more compatible with primary and active EBV infection while those of the elderly patients were compatible with past infection. However, high viral loads and acute onset of disease in cases 1 and 3 in the elderly group indicate active EBV infection. A recent study demonstrated low frequencies of EBV-specific CD8+ T cells in patients with chronic active EBV infection [29]. Failure to produce an antibody against EBV in elderly patients seems to be associated with underlying antiviral T-cell dysfunction.
The immunophenotype of the proliferating cells in the children and young adults group in our study were predominantly CD8-positive cytotoxic αβ T cells. Only one case developed from a CAEBV infection and consisted of mixed CD4+ and CD8+ cells with a predominance of CD4+ cells. The elderly group showed similar results, with a case with a γδ T-cell phenotype. Sporadic reports of fulminant EBV+ T-cell LPD showed a predominantly CD8+ phenotype with a few mixed CD4 and CD8 phenotype cells [2–4, 13, 19, 20, 27, 32]. However, other studies could not find any subset predominance in EBV-infected cells. Quintanilla-Martinez et al. [23] reported six cases of fulminant EBV-positive clonal T-cell LPD after acute or chronic EBV infection. The cells in two cases were CD4+, two CD8+, and two had admixed CD4+ and CD8+ cells. All were CD56-negative. In a recent retrospective study of 43 children and young adult patients with EBV-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection), eight cases were compatible with fulminant or subacute EBV+ T-cell LPD following CAEBV infection, one case with a CD8+ phenotype, two with a CD4+ phenotype, three with an admixture of CD8+ and CD4+ cells, and two showed a CD4−/CD8− phenotype. Six cases had αβ T cells and two γδ T cells [16]. Kasahara et al. analyzed the frequency of EBV-infected cells in circulating lymphocyte subpopulations from four patients with acute EBV-HLH and four with CAEBV infection [12], and they showed that EBV-infected cells in the acute phase of EBV-HLH were mainly an activated CD8+ T-cell population, whereas the EBV-infected populations in CAEBV infection were heterogeneous, including CD4, CD8, B, and NK cells.
In conclusion, fulminant EBV+ systemic T-cell lymphoma of elderly patients is a rare disease that shares similar clinical and pathologic findings with those seen in children and young adults, but which has some differences in clinical features. Although those elderly patients showed no obvious immune defect, a common association with chronic hepatitis virus infection suggests an underlying derangement of T-cell immunity and failure to eradicate infected virus. In these patients, additional factors related to senility may play a role in the disruption of homeostasis between the virus and the host’s immune system, eventually leading to neoplastic transformation of EBV-infected T lymphocytes.
References
Callan MF, Steven N, Krausa P et al (1996) Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med 2:906–911
Chan LC, Srivastava G, Pittaluga S, Kwong YL, Liu HW, Yuen HL (1992) Detection of clonal Epstein–Barr virus in malignant proliferation of peripheral blood CD3+ CD8+ T cells. Leukemia 6:952–956
Craig FE, Clare CN, Sklar JL, Banks PM (1992) T-cell lymphoma and the virus-associated hemophagocytic syndrome. Am J Clin Pathol 97:189–194
Gaillard F, Mechinaud-Lacroix F, Papin S et al (1992) Primary Epstein–Barr virus infection with clonal T-cell lymphoproliferation. Am J Clin Pathol 98:324–333
Gulley ML, Fan H, Elmore SH (2006) Validation of Roche LightCycler Epstein–Barr virus quantification reagents in a clinical laboratory setting. J Mol Diagn 8:589–597
Henter JI, Horne A, Arico M et al (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48:124–131
Imai S, Sugiura M, Oikawa O et al (1996) Epstein–Barr virus (EBV)-carrying and -expressing T-cell lines established from severe chronic active EBV infection. Blood 87:1446–1457
Ishihara S, Okada S, Wakiguchi H, Kurashige T, Hirai K, Kawa-Ha K (1997) Clonal lymphoproliferation following chronic active Epstein–Barr virus infection and hypersensitivity to mosquito bites. Am J Hematol 54:276–281
Iwama H, Horikoshi S, Shirato I, Tomino Y (1998) Epstein–Barr virus detection in kidney biopsy specimens correlates with glomerular mesangial injury. Am J Kidney Dis 32:785–793
Jones JF, Shurin S, Abramowsky C et al (1988) T-cell lymphomas containing Epstein–Barr viral DNA in patients with chronic Epstein–Barr virus infections. N Engl J Med 318:733–741
Kanegane H, Bhatia K, Gutierrez M et al (1998) A syndrome of peripheral blood T-cell infection with Epstein–Barr virus (EBV) followed by EBV-positive T-cell lymphoma. Blood 91:2085–2091
Kasahara Y, Yachie A, Takei K et al (2001) Differential cellular targets of Epstein–Barr virus (EBV) infection between acute EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Blood 98:1882–1888
Kawaguchi H, Miyashita T, Herbst H et al (1993) Epstein–Barr virus-infected T lymphocytes in Epstein–Barr virus-associated hemophagocytic syndrome. J Clin Invest 92:1444–1450
Kawa-Ha K, Ishihara S, Ninomiya T et al (1989) CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein–Barr viral DNA. J Clin Invest 84:51–55
Kikuta H, Sakiyama Y, Matsumoto S et al (1993) Fatal Epstein–Barr virus-associated hemophagocytic syndrome. Blood 82:3259–64
Kikuta H, Taguchi Y, Tomizawa K et al (1988) Epstein–Barr virus genome-positive T lymphocytes in a boy with chronic active EBV infection associated with Kawasaki-like disease. Nature 333:455–457
Kimura H, Hoshino Y, Kanegane H et al (2001) Clinical and virologic characteristics of chronic active Epstein–Barr virus infection. Blood 98:280–286
Ko YH, Ree HJ, Kim WS, Choi WH, Moon WS, Kim SW (2000) Clinicopathologic and genotypic study of extranodal nasal-type natural killer/T-cell lymphoma and natural killer precursor lymphoma among Koreans. Cancer 89:2106–2116
Mori M, Kurozumi H, Akagi K, Tanaka Y, Imai S, Osato T (1992) Monoclonal proliferation of T cells containing Epstein–Barr virus in fatal mononucleosis. N Engl J Med 327:58
Noma T, Kou K, Yoshizawa I et al (1994) Monoclonal proliferation of Epstein–Barr virus-infected T-cells in a patient with virus-associated haemophagocytic syndrome. Eur J Pediatr 153:734–738
Ohga S, Takada H, Honda K et al (1999) Central nervous system T-cell lymphoproliferative disorder in a patient with chronic active Epstein–Barr virus infection. J Pediatr Hematol Oncol 21:42–46
Okano M, Matsumoto S, Osato T, Sakiyama Y, Thiele GM, Purtilo DT (1991) Severe chronic active Epstein–Barr virus infection syndrome. Clin Microbiol Rev 4:129–135
Quintanilla-Martinez L, Kumar S, Fend F et al (2000) Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood 96:443–451
Rickinson AB (1986) Chronic, symptomatic Epstein–Barr virus infections. Immunol Today 7:13–14
Schooley RT, Carey RW, Miller G et al (1986) Chronic Epstein–Barr virus infection associated with fever and interstitial pneumonitis. Clinical and serologic features and response to antiviral chemotherapy. Ann Intern Med 104:636–643
Straus SE, Cohen JI, Tosato G, Meier J (1993) NIH conference. Epstein–Barr virus infections: biology, pathogenesis, and management. Ann Intern Med 118:45–58
Su IJ, Lin KH, Chen CJ et al (1990) Epstein–Barr virus-associated peripheral T-cell lymphoma of activated CD8 phenotype. Cancer 66:2557–2562
Su IJ, Wang CH, Cheng AL, Chen RL (1995) Hemophagocytic syndrome in Epstein–Barr virus-associated T-lymphoproliferative disorders: disease spectrum, pathogenesis, and management. Leuk Lymphoma 19:401–406
Sugaya N, Kimura H, Hara S et al (2004) Quantitative analysis of Epstein–Barr virus (EBV)-specific CD8+ T cells in patients with chronic active EBV infection. J Infect Dis 190:985–988
Sumaya CV, Ench Y (1985) Epstein–Barr virus infectious mononucleosis in children. I. Clinical and general laboratory findings. Pediatrics 75:1003–1010
Suzuki K, Ohshima K, Karube K et al (2004) Clinicopathological states of Epstein–Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection) of children and young adults. Int J Oncol 24:1165–1174
Tazawa Y, Nishinomiya F, Noguchi H et al (1993) A case of fatal infectious mononucleosis presenting with fulminant hepatic failure associated with an extensive CD8-positive lymphocyte infiltration in the liver. Hum Pathol 24:1135–1139
Conflict of interest statement
We declare that we have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Park, S., Kim, K., Kim, W.S. et al. Systemic EBV+ T-cell lymphoma in elderly patients: comparison with children and young adult patients. Virchows Arch 453, 155–163 (2008). https://doi.org/10.1007/s00428-008-0640-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-008-0640-7