
Abstract
Main conclusion
In Brassica rapa, the epigenetic modifier BraA.CLF orchestrates flowering by modulating H3K27me3 levels at the floral integrator genes FT, SOC1, and SEP3, thereby influencing their expression.
Abstract
CURLY LEAF (CLF) is the catalytic subunit of the plant Polycomb Repressive Complex 2 that mediates the trimethylation of histone H3 lysine 27 (H3K27me3), an epigenetic modification that leads to gene silencing. While the function of CURLY LEAF (CLF) has been extensively studied in Arabidopsis thaliana, its role in Brassica crops is barely known. In this study, we focused on the Brassica rapa homolog of CLF and found that the loss-of-function mutant braA.clf-1 exhibits an accelerated flowering together with pleiotropic phenotypic alterations compared to wild-type plants. In addition, we carried out transcriptomic and H3K27me3 genome-wide analyses to identify the genes regulated by BraA.CLF. Interestingly, we observed that several floral regulatory genes, including the B. rapa homologs of FT, SOC1 and SEP3, show reduced H3K27me3 levels and increased transcript levels compared to wild-type plants, suggesting that they are direct targets of BraA.CLF and key players in regulating flowering time in this crop. In addition, the results obtained will enhance our understanding of the epigenetic mechanisms regulating key developmental traits and will aid to increase crop yield by engineering new Brassica varieties with different flowering time requirements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants are complex organisms with intricate developmental processes that evolve in coordination with the surrounding environment. Within this context, the epigenetic mechanisms regulating gene expression play a pivotal role in orchestrating growth, development and differentiation (Schuettengruber et al. 2017; Baile et al. 2022). Polycomb Group (PcG) complexes are epigenetic regulators that transcriptionally repress target genes through the deposition of posttranslational histone modifications (Schuettengruber et al. 2017). The Polycomb Repressive Complex 2 (PRC2) catalyzes the trimethylation of histone H3 lysine 27 (H3K27me3), which eventually leads to the silencing of gene expression (Guo et al. 2021). PRC2 was initially discovered in Drosophila where it was shown to contain four core subunits: Enhancer of Zeste (E(z)), Extra sex combs (Esc), Suppressor of Zeste 12 (Su(z)12), and Nucleosome remodelling factor 55 (Nurf55) (Schuettengruber et al. 2017). In Arabidopsis thaliana, extensive studies have found that PRC2 subunits are conserved although they are encoded by multiple genes: CURLY LEAF (CLF), SWINGER (SWN), and MEDEA (MEA) are homologs of E(z); VERNALIZATION 2 (VRN2), EMBRYONIC FLOWERING 2 (EMF2) and FERTILIZATION-INDEPENDENT SEED 2 (FIS2) are homologs of Su(z)12; FERTILIZATION-INDEPENDENT ENDOSPERM 1 (FIE1) is the homolog of ESC; and there are five MULTI-SUBUNIT SUPPRESSOR OF IRA proteins (MSI1–5) homologs of Nurf55 (Vijayanathan et al. 2022; Godwin and Farrona 2022).
The A. thaliana PRC2 E(z)-like catalytic subunits CLF, SWN and MEA have different expression profiles and specialized functions (Kinoshita et al. 1999; Chanvivattana et al. 2004). The MEA gene exhibits imprinting and it is expressed in the female gametophyte and endosperm, being required to silence the paternal MEA allele after fertilization (Grossniklaus et al. 1998; Kinoshita et al. 1999). CLF acts as the primary H3K27 methyltransferase in the sporophyte, and is partially redundant with SWN (Chanvivattana et al. 2004; Shu et al. 2020). Mutations in CLF lead to pleiotropic developmental defects, whereas swn mutants exhibit subtle developmental abnormalities (Goodrich et al. 1997; Chanvivattana et al. 2004). Notably, SWN mutations considerably enhance clf phenotypic alterations, and the clf swn double mutant displays callus-like structures and somatic embryos (Chanvivattana et al. 2004).
The strong developmental defects displayed by clf mutants include small plant size, curved leaves and accelerated flowering time. The leaf curling phenotype is caused by the misregulation of the floral homeotic genes AGAMOUS (AG) and SEPALLATA3 (SEP3) (Goodrich et al. 1997; Lopez-Vernaza et al. 2012). Meanwhile, the early-flowering phenotype is at least partially due to the upregulation of the floral integrator gene FLOWERING LOCUS T (FT) (Farrona et al. 2011). A number of studies have also highlighted the critical role of CLF in the regulation of cell proliferation and meristematic activity (Shu et al. 2020). For example, CLF represses the expression of differentiation-promoting genes in the shoot apical meristem, thereby sustaining the undifferentiated state of root and floral stem cells (Liu et al. 2011). However, the function of CLF extends beyond developmental processes and encompasses plant immunity and lipid metabolism (Liu et al. 2016; Singkaravanit‐Ogawa et al. 2021). In fact, genome-wide analyses show direct binding of CLF protein to hundreds of A. thaliana target genes involved in a number of biologic processes (Wang et al. 2016; Shu et al. 2019).
The Brassica genus comprises a variety of vegetables, condiments, and economically significant oilseed crops that are closely related to A. thaliana. Within the Brassica genus, Brassica rapa holds economic significance globally. This species displays remarkable variations in morphology resulting in different agricultural uses including leafy vegetables like Chinese cabbage, enlarged roots in turnip, and oilseed varieties such as yellow sarson. The genome of B. rapa is fully sequenced and contributes to half of the genomes of the allotetraploid crops B. juncea (Indian mustard) and B. napus (oilseed rape) (Wang et al. 2011; Zhang et al. 2018). Recent advances in B. rapa epigenomics have provided valuable insights into the regulatory mechanisms governing gene expression in this important crop species. For example, the repressive histone modification H3K27me3 has been proposed to play a key role in the regulation of the floral transition (Payá-Milans et al. 2019; Poza-Viejo et al. 2022). Furthermore, there is a distinct distribution of H3K36me3 and H3K27me3 among homoeologous paired genes, which has been proposed to lead to variations in gene-expression levels or tissue specificity (Mehraj et al. 2021).
Despite the significant progress made in understanding the function of PRC2 in A. thaliana, the role of this gene in Brassica crops is only beginning to be understood. We previously isolated braA.clf-1, a mutant line in B. rapa R-o-18 with abnormalities that resemble some of the classic A. thaliana clf-mutant phenotypes (Payá-Milans et al. 2019). One of the most conspicuous A. thaliana clf phenotypic alterations is an early flowering. However, the flowering time of braA.clf-1 has not been described yet. Here, we report that braA.clf-1 shows an acceleration of flowering time in comparison to wild-type plants. In addition, to unveil the underlying genomic defects, we performed transcriptomics and genome-wide analysis of H3K27me3 in braA.clf-1 leaves. To our knowledge, this is the first genome-wide study of an epigenetic mark of a Polycomb mutant in a Brassica crop. Our data show that a number of floral integrator genes, including the B. rapa homologs of FT and SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1), display reduced H3K27me3 occupancy and higher transcript levels in braA.clf-1 that are consistent with its early-flowering phenotype.
Materials and methods
Plant materials and growth conditions
We worked with B. rapa R-o-18, an inbred variety of Brassica rapa subsp. trilocularis (Yellow Sarson) that has been widely studied as a model oilseed crop (Stephenson et al. 2010). The TILLING (Targeting Induced Local Lesions In Genomes) mutant line braA.clf-1 (JI32391-A) was obtained from RevGenUK. Plants were grown in controlled-environment growth chambers under long day conditions of 16 h of light with day/night temperatures of 21/19 °C and mix of cool-white and wide-spectrum FLOURA fluorescent lights (100 µE/m2s).
Flowering time analysis
B. rapa flowering time experiments were performed in controlled-environment chambers using 12 cm diameter pots. Flowering time was quantified using three metrics: the number of days from germination to bolting, the number of days until the first flower opened, and the number of leaves present at bolting.
Chromatin immunoprecipitation and sequencing
The braA.clf-1 plants used in our genomic experiments were grown in parallel and collected together with the plant materials described in Poza-Viejo et al. 2022. We used primary leaves of B. rapa plants grown for 14 days collected at the end of the light period (Zeitgeber time ZT16).
Chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) experiments were performed using an anti-H3K27me3 antibody (Diagenode C15410195) as described in Poza-Viejo et al. (2019). Two biologic ChIP replicates (one leaf from 8 independent plants) were processed (Table S1). Input contained a pool of DNA from all genotypes and it was sequenced at greater depth to improve peak-calling identification. ChIP-seq libraries were prepared using NEBNext Ultra DNA Library Prep kit (New England BioLabs) and sequenced at 2 × 50 bp paired-end reads by the Genomics Unit of the Centro Nacional de Análisis Genómico CNAG-CRG (Barcelona, Spain).
For the ChIP-qPCR assays, DNA was amplified using real-time quantitative PCR (qPCR) with the primers listed in Table S2. The enrichment of ChIP DNA was calculated as the percentage of immunoprecipitated DNA relative to the input DNA normalized to the concentration of total DNA content determined by QUBIT fluorometer (ThermoFisher Scientific).
Transcriptomic and gene-expression analyses
Transcriptome analysis was performed by RNA sequencing (RNA-seq). Total RNA was extracted from the same plants used for the ChIP-seq using E.Z.N.A. Plant RNA Kit (Omega Bio-tek). At least three biologic replicates (one leaf from eight independent plants) were processed for each genotype (Table S3). RNA-seq libraries were prepared and sequenced at 2 × 100 bp paired-end reads by MACROGEN Inc. (Korea).
For reverse transcription quantitative PCR (RT-qPCR) expression analyses, RNA extraction and cDNA synthesis were performed using the E.Z.N.A. Plant RNA Kit (Omega Bio-tek) and the Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific), respectively, following the manufacturers’ guidelines. The RT-qPCR data were presented as relative mRNA levels, calculated using the 2−ΔΔCT method, with BraA.TUBULIN (BraA10g026070.3C) serving as the housekeeping gene (Xu et al. 2014). The primers used for RT-qPCR can be found in Table S2.
ChIP-seq and RNA-seq computational analyses
Transcriptomic and epigenomic data were analyzed using our previously described analytical workflow (Payá-Milans et al. 2019; Poza-Viejo et al. 2022) at the Supercomputing Galician Centre (CESGA) high-throughput computing server. The specific code can be found at https://github.com/mpaya/epigenomics_scripts. Briefly, raw reads were trimmed with Skewer v0.2.2 (Jiang et al. 2014), then mapped to the B. rapa Chiifu v3.0 genome with the fast aligner Bowtie2 v2.3.5 (Langmead and Salzberg 2012), and mapping metrics were collected with Picard v2.21.1 (2018). For RNA-seq samples, counts were obtained with htseq-count v0.11.2 (Anders et al. 2015) and differentially expressed genes (DEG) were determined with DESeq2 (Love et al. 2014). The correlation of gene-expression profiles between RNA-seq samples is shown in Fig. S1. Analysis of ChIP-seq data was performed using Bowtie2 v2.3.5 as read aligner, Epic2 (Stovner and Sætrom 2019) as peak caller, and a quantitative comparison of mutant vs wild-type ChIP-seq signal was performed with MAnorm v1.2.0 (Shao et al. 2012). Peaks were annotated to overlapping genes of B. rapa at 500 bp distance using ChIPpeakAnno R package (Zhu et al. 2010). ChIP-seq replicates yielded consistent results (Fig. S2).
Other bioinformatic analyses
Custom annotation of gene models was obtained by comparing B. rapa genome v3.0 coding sequences against Arabidopsis (TAIR10 proteins, blastx of B. rapa coding sequences with an E-value cutoff of 1e − 25) using BLAST (Basic Local Alignment Search Tool). The phylogenetic tree was obtained using the web service Phylogeny.fr (http://www.phylogeny.fr/index.cgi) (Dereeper et al. 2008). Sequences were aligned using MUSCLE, and ambiguous regions were eliminated with Gblocks post-alignment; the PhyML program was utilized for maximum likelihood-based reconstruction of the phylogenetic tree, and TreeDyn program was employed for graphical representation (Dereeper et al. 2008). Singular Enrichment Analysis (SEA) of Gene Ontology (GO) terms analyses were conducted utilizing agriGO v2.0 employing Fisher statistical test approach, Yekutieli Multi-test adjustment technique (p ≤ 0.05), and Plant GO slim ontology category (Du et al. 2010). To reduce the complexity and redundant GO terms we used REVIGO (Supek et al. 2011) with default parameters (allowed similarity = 0.7; semantic similarity measure = SimRel). Graphs and statistical analyses were performed using GraphPad Prism 9 (www.graphpad.com). Venn diagrams were drafted using the free web tool DeepVenn (Hulsen 2022). The hypergeometric test was calculated using the Hypergeometric P value calculator from the Graeber Lab (https://systems.crump.ucla.edu/hypergeometric/). The Integrative Genomic Viewer (IGV) software (Thorvaldsdottir et al. 2013) was used to visualize H3K27me3 ChIP-seq peaks.
Results
CURLY LEAF is a single-copy gene in Brassica rapa
To investigate the functional role of H3K27me3 methyltransferases in Brassica crops, we looked for homologs of the A. thaliana CLF (AT2G23380) protein using BLAST and performed a phylogenetic analysis. Consistent with the findings reported by Huang et al. (2011) we determined that BraA.CLF (BraA04g017190.3C) is the sole CLF homolog, and identified one homolog to SWN (BraA09g002170.3C) as well as two homologs for MEA (BraA09g065690.3C and BraA10g001320.3C) within the updated B. rapa genome assembly V3.0 (Zhang et al. 2018) (Fig. 1A). Our analysis of the most recent B. rapa genome assembly V4.0 yielded similar results (Zhang et al. 2023).

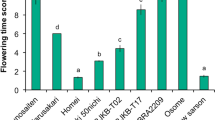
Homologs of the A. thaliana H3K27me3 methyltransferases in B. rapa. A Phylogenetic tree showing the relationship between A. thaliana H3K27me3 methyltransferases (CLF AT2G23380, SWN AT4G02020 and MEA AT1G02580) and its B. rapa homologs (BraA.CLF BraA04g017190.3C, SWN homolog BraA09g002170.3C and MEA homologs BraA10g001320.3C and BraA09g065690.3C). B Number of transcripts (cpm, counts per million) of B. rapa H3K27me3 methyltransferases-encoding genes found in leaves and inflorescences. Data obtained from Payá-Milan et al. 2019
In A. thaliana, the genes encoding the three histone H3K27me3 methyltransferases are differentially expressed. CLF and MEA genes are expressed in a variety of tissues, with MEA expression being highest in the endosperm (Kinoshita et al. 1999; Chanvivattana et al. 2004; Shu et al. 2020). We studied the expression of the four B. rapa H3K27 methyltransferase genes using our published RNA-seq data (Payá-Milans et al. 2019). We found that BraA.CLF and BraA.SWN genes were expressed in B. rapa R-o-18 leaves and inflorescences, whereas the expression of B. rapa MEA homologs was not detected in these experiments (Fig. 1B). We then investigated B. rapa MEA homologs in public transcriptomic databases at the Brassicaceae Database (BRAD) (Cheng et al. 2011) and found that BraA10g001320.3C was not detected in any tissue or growth condition, while BraA09g065690.3C was only expressed in siliques. This suggested that B. rapa MEA gene functions would be restricted to the gametophyte, whereas CLF and SWN may have a prominent role during the sporophyte phase. Although further research is needed, all these data are consistent with a conserved expression pattern of the H3K27 methyltransferases-encoding genes in B. rapa and A. thaliana.
BraA.CLF contributes to the repression of the floral transition
We previously isolated braA.clf-1 (Payá-Milans et al. 2019), a TILLING mutant line that carries a stop-codon (Q615*) upstream of the CXC and catalytic SET domain in the BraA.CLF gene (Fig. S3), although its flowering time phenotype was not evaluated at that time. Prior to the flowering phenotypic analysis, the mutant braA.clf-1 was backcrossed twice to the wild-type parental line to reduce the load of secondary TILLING mutations. Homozygous plants carrying the braA.clf-1 mutation derived from these backcrosses exhibited pleiotropic phenotypic alterations as previously observed (Payá-Milans et al. 2019). For instance, the stem of braA.clf-1 plants was thinner than that of the wild-type plants (Figs. 2 and 3A). The leaf length and size was also altered in the mutant (Fig. 2B) with the first five leaves of braA.clf-1 mutants displaying on average a 50% length reduction compared to the wild-type leaves (Fig. 2C). However, we could not observe any abnormally elongated pistil or severe reduction in plant height as described by Nugroho et al. (2023). In our study, both braA.clf-1 and wild-type plants attained similar heights at the flowering stage (Fig. 3A). These discrepancies may be attributed either to different growth conditions, or the use of backcrossed mutant lines in our study.

Phenotypic alterations of braA.clf-1. A Picture of braA.clf-1 and wild-type plants at 12-, 17-, 21- and 26-days after germination (DAG); scale bar = 1 cm. B Picture showing severe abnormalities in the leaf shape and size of braA.clf-1 compared to wild-type plants at 26 DAG; scale bar = 1 cm. C Quantification of leaf length measured from the tip to the end of the petiole of the first five leaves of braA.clf-1 and wild-type plants. Mean values ± SD (n = 10). Statistical significance was calculated using one-way analysis of variance (ANOVA) followed by Sidak’s multiple comparison test (****P value < 0.0001)

BraA.CLF acts as a floral repressor. A Picture of braA.clf-1 and wild-type plants at the flowering stage; scale bar = 13 cm. B–D Flowering time of braA.clf-1 and wild-type plants was measured as days from germination to bolting (B), days from germination to the opening of the first flower (C), and the number of primary leaves generated by the plant until the first flower opens (D). Mean values ± SD (n = 15). Statistical significance was calculated using the Student´s t-test (****P value < 0.0001)
In A. thaliana, the expression of many floral regulatory genes is controlled by H3K27me3 methylation (Baile et al. 2022; Vijayanathan et al. 2022). We have also observed a similar scenario in B. rapa (Payá-Milans et al. 2019; Poza-Viejo et al. 2022). Thus, we decided to characterize the flowering time of the braA.clf-1 mutant. For that, we grew plants in controlled-climate chambers and observed that braA.clf-1 plants exhibited an early-flowering phenotype compared to wild-type B. rapa R-o-18 plants. (Fig. 3B–D). This flowering time acceleration was statistically significant when quantified as the number of days before reaching the bolting stage or the number of days prior to opening of the first flower (Fig. 3B–C). In addition, we also observed that the number of leaves developed by the braA.clf-1 mutant before flowering, a commonly used parameter to estimate flowering time in A. thaliana, was reduced compared to wild-type plants (Fig. 3D). All these observations demonstrate that braA.clf-1 is an early-flowering mutant, and suggest that BraA.CLF could be repressing the expression of key flowering promoting genes.
The braA.clf-1 mutant displays genome-wide alterations in H3K27me3 levels
To investigate the functional role of BraA.CLF as a histone methyltransferase in B. rapa, we performed H3K27me3 ChIP-seq experiments on braA.clf-1 mutants and wild-type leaves. Immunoprecipitated DNA samples were sequenced using high-throughput next-generation sequencing (see Methods and Supplementary Table S1). After the ChIP-seq bioinformatic analysis, we identified 7385 genes with altered levels of H3K27me3 in the braA.clf-1 mutant compared to the wild type (Fig. 4A and Supplementary Data S1; |M value|≥ 0.5 and P value ≤ 0.1). Among these genes, 4219 were hypomethylated and represented likely targets of BraA.CLF methyltransferase activity. This number accounts for over 30% of the H3K27me3-marked genes in leaves of B. rapa (Payá-Milans et al. 2019). Remarkably, there was also an increase of H3K27me3 in a number of genes in braA.clf-1. This phenomenon has also been observed in previous studies in other plant and animal species, and it is likely due to an indirect effect (Wang et al. 2016).

H3K27me3 epigenomic analysis of braA.clf-1. A MA plot showing the differential accumulation of H3K27me3 on ChIP‐seq peaks determined with MAnorm. Significance is indicated with color scale, red indicates − log10 (P value) > 50. Normalized read densities of braA.clf-1 relative to the wild type were compared to represent the average signal strength of samples (A value) against their log2 fold‐change (M value) of each peak. B Semantic clustering of enriched GO terms of H3K27me3 hypomethylated genes in braA.clf-1 mutant. SEA was performed with agriGO and the significant GO terms were clustered with the REVIGO tool. The size of the bubble indicates the frequency of the GO term and P values are indicated by a color scale
To unravel the functional implications of BraA.CLF activity, we performed a SEA-GO analysis of the set of braA.clf-1 hypomethylated genes using AgriGO (Supplementary Data S2). The GO term list obtained was condensed and represented using the REVIGO tool (Fig. 4B). We found that among the most overrepresented biological process GO categories were: regulation of gene expression (GO:0010468), biological regulation (GO:0065007) and regulation of metabolic process (GO:0019222). There were also several GO categories related to regulation of cellular process (GO:0050794) and response to stimulus (GO:0050896). Consistent with a key role of BraA.CLF repressing floral homeotic genes in leaves (Payá-Milans et al. 2019), we also found the GO category of developmental process (GO:0032502) including floral development (GO:0009908) enriched in the list of hypomethylated genes (Supplementary Data S2 and Fig. 4B).
To compare our data with A. thaliana clf data sets available, we first identified homologs of our list of hypomethylated genes in B. rapa using BLAST, and removed any duplicated A. thaliana gene terms. Next, we compared the set of hypomethylated and hypermethylated genes in braA.clf-1 with different clf datasets in A. thaliana, which varied in the number of identified hypomethylated genes (Wang et al. 2016; Carter et al. 2018; Shu et al. 2019). We found a statistically significant overlap in all cases (Fig. S4). We then performed a comparative enrichment analysis of GO terms between the datasets of hypomethylated from Wang et al. (2016) and our braA.clf-1 mutant using the SEACOMPARE tool from AgriGO. Despite comparing data from different sample materials (2 weeks old A. thaliana plants vs B. rapa leaves) and experiments performed in different laboratories, we found an extensive coincidence between the enriched GO terms categories between hypomethylated genes in B. rapa braA.clf-1 and A. thaliana clf (Fig. 5). These data suggest that BraA.CLF, as a key component of the PRC2 complex, plays a role in the regulation of several biological processes that are conserved between B. rapa and A. thaliana.

Comparison of enriched GO categories between hypomethylated genes from braA.clf-1 and A. thaliana clf-29. Comparison between common and exclusive enriched GO terms from A. thaliana clf-29 (Wang et al. 2016) and our braA.clf-1 mutant datasets using the SEACOMPARE tool from AgriGO. The color indicates the FDR value
The braA.clf-1 mutant exhibits a large number of misregulated genes
In combination with our ChIP-seq, we performed a transcriptomic study by RNA-seq of braA.clf-1 mutant and wild-type leaves (see Methods and Supplementary Table S3 for details). Following the bioinformatic analysis, we identified 2159 genes that were upregulated, and 2116 genes that were downregulated in comparison to the wild type. (Fig. 6A and Supplementary Data S3; DESeq2 |log2(FC)|≥ 1; P-adj ≤ 0.1). This large number of misregulated genes was consistent with the pleiotropic phenotype of braA.clf-1 mutant, which may be due to direct and indirect effects of the methyltransferase activity of CLF in B. rapa.

Transcriptomic analysis of the braA.clf-1. A Volcano plot showing the differentially expressed genes in braA.clf‐1 compared to wild-type plants. Upregulated genes (red) and downregulated genes (blue) are shown (DESeq2 |Log2(FC)|≥ 1; P-adj ≤ 0.1). B–C SEA-GO analysis of upregulated (B) and downregulated (C) genes in braA.clf-1. Data were represented using REVIGO to reduce complexity
To gain insight into the biological processes affected in the mutant, we then performed a SEA-GO analysis of the braA.clf-1 upregulated and downregulated set of genes (Fig. 6B and C, and Supplementary Data S4 and S5). The top-enriched Gene Ontology (GO) categories in the set of upregulated genes in braA.clf-1 included regulation of biological processes (GO:0050789), regulation of gene expression (GO:0010468), and regulation of macromolecule metabolic processes (GO:0060255) (Supplementary Data S4). In addition, the list of upregulated genes included several GO terms related to cellular processes (GO:0050794), response to stimulus (GO:0050896), and developmental processes (GO:0032502), such as flower development (GO:0009908). The set of downregulated genes was enriched in several terms related to response to stimulus (GO:0050896 and GO:0009628), cellular processes (GO:0009987, GO:0044237, GO:0044249), and metabolism (GO:0008152 and GO:0019748) (Supplementary Data S5). Notably, we observed several GO categories related to the regulation of development or metabolism that were enriched in both gene sets.
To sum up, our transcriptomic analyses suggest that while BraA.CLF plays a prominent role in regulating developmental genes, it also directly or indirectly regulates a large number of genes related to other biological processes, including metabolism.
BraA.CLF modulates the expression of a number of floral integrator genes
To explain the accelerated flowering time of the braA.clf-1 mutant, we compared the genes with reduced levels of H3K27me3 (hypomethylated) and increased expression (upregulated) in braA.clf-1. We identified a set of 332 genes (Fig. 7A), which likely includes the direct target genes of BraA.CLF contributing to the mutant phenotype. Then, we determined the A. thaliana homologs by BLAST analysis and performed a cross-comparison with the FLOweRing Interactive Database (FLOR-ID), a curated list of more than 306 floral regulatory genes in A. thaliana (Bouché et al. 2016). Following this reasoning, we discovered 23 B. rapa genes hypomethylated and upregulated in braA.clf-1 directly associated with the floral transition (Table 1).

BraA.CLF modulates the expression of floral regulatory genes. A Venn diagram showing the overlap between hypomethylated and upregulated genes of braA.clf-1 mutant. The overlap is over enriched 1.54 fold compared to expectations (hypergeometric test, P value = 2.21e-16). B–C RT-qPCR data showing the relative expression of BraA.FTa locus (B) and BraA04g031640.3C (BraA.SOC1) (C) in leaves of wild-type and braA.clf-1 mutant plants. Bars represent the average of two replicates for each experiment (n = 2), and error bars represent the standard deviation error. Mean values ± SD (n = 2). D–E Representation of BraA.FTa (D) and BraA.SOC1 (BraA04g031640.3C) (E) loci showing the regions analyzed by ChIP-qPCR. F–G ChIP-qPCR data showing the H3K27me3 enrichment over BraA.FTa locus (F) and BraA04g031640.3C (SOC1) (G) in leaves of wild-type and braA.clf-1 mutant plants. Mean values ± SD (n = 2)
The early-flowering phenotype observed in the A. thaliana clf mutant is caused by the upregulation of the floral integrator gene FT and the homeotic gene SEP3 (Jiang et al. 2008; Lopez-Vernaza et al. 2012). We found several homologs of the AGAMOUS-like MADS-box transcription factor family that have been implicated in flowering time regulation in A. thaliana. Among them, we identified three B. rapa SEP3 homologs that were hypomethylated and upregulated in braA.clf-1 (Table 1). We also found the three homologs of B. rapa SOC1 and the main B. rapa FT, BraA.FT.a or FT1 (Table 1), to be hypomethylated and upregulated in the braA.clf-1 mutant compared to the wild type. The increased mRNA expression levels and decreased H3K27me3 levels in braA.clf-1 compared to the wild type for BraA.FT.a and BraA04g031640.3C (BraA.SOC1) were confirmed through independent RT-qPCR (Fig. 7B–C) and ChIP-qPCR experiments (Fig. 7D–G). All these floral integrator genes have been shown to promote flowering in B. rapa (del Olmo et al. 2019; Calderwood et al. 2021; Wang et al. 2022) and their upregulation is likely responsible for the early-flowering phenotype observed in the braA.clf-1 mutant.
On the other hand, in A. thaliana CLF also regulates the floral repressor FLOWERING LOCUS C (FLC), but its effects on flowering time are masked by the increased FT expression in the clf mutant (Doyle and Amasino 2009; Lopez-Vernaza et al. 2012). However, there was no B. rapa FLC-like gene in our braA.clf-1 ChIP-seq or RNA-seq datasets. Further research will be required to define whether BraA.CLF regulates the expression of FLC-like genes in other B. rapa cultivars.
Discussion
Orthologs of CLF are found throughout the green lineage, and are the only E(z) homologs in bryophytes, lycopodiophytes, and gymnosperms (Vijayanathan et al. 2022). In A. thaliana, CLF is the catalytic subunit of the main PRC2 in the sporophyte and it is required for the proper development of the plant (Baile et al. 2022; Godwin and Farrona 2022). While A. thaliana clf mutants display severe phenotypes including dwarfism, CURLY LEAF, and early flowering, the swn mutants only show subtle changes during vegetative phase transition (Chanvivattana et al. 2004). To determine the function of BraA.CLF, the only B. rapa homolog of CLF (Fig. 1A), we studied the loss-of-function mutant braA.clf-1, which exhibited severe developmental alterations, including curved leaves (Fig. 2). While we cannot rule out the presence of secondary mutations in braA.clf-1, these phenotypes closely resemble the classic clf-mutant phenotypes observed in A. thaliana. In addition, we found that braA.clf-1 displayed early flowering, which is consistent with recent reports indicating that mutations in the Chinese cabbage (B. rapa ssp. pekkinensis) CLF homolog promotes premature bolting (Huang et al. 2020; Tan et al. 2021). However, the early-flowering phenotype of braA.clf-1 was not as conspicuous as the Chinese cabbage and A. thaliana mutant alleles, due to R-o-18 being already an early-flowering B. rapa variety.
The braA.clf-1 epigenomic profile of H3K27me3 performed in this work (Fig. 4) confirmed the crucial role of BraA.CLF activity in regulating the levels of H3K27m3 in B. rapa. However, most of B. rapa H3K27me3-marked genes were not hypomethylated in braA.clf-1, which is consistent with published genome-wide studies of clf alleles in A. thaliana (Wang et al. 2016; Carter et al. 2018; Shu et al. 2019). This suggests functional redundancy with other H3K27me3 methyltransferases, likely the B. rapa SWN homolog (Fig. 1). In the near future, it will be interesting to explore the function exerted by the other H3K27 methyltransferases in B. rapa. On the other hand, our transcriptomic analysis (Fig. 6A) revealed a large number of differentially expressed genes between braA.clf-1 and the wild-type plants. H3K27me3 is an epigenetic mark associated with gene silencing (Guo et al. 2021). Interestingly, not all the hypomethylated genes were upregulated in braA.clf-1 (Fig. 7A). These findings are consistent with previous reports indicating that gene expression is not initiated by default in the absence of H3K27me3, but rather requires the coordinated action of multiple chromatin regulators and transcription factors (Schuettengruber et al. 2017; Guo et al. 2021).
Taking advantage of our combined ChIP-seq and RNA–seq analyses, we focused on the set of genes that appeared hypomethylated and upregulated in braA.clf-1 compared to wild type. These loci very likely include the direct targets of BraA.CLF. The analysis on this dataset showed a significant enrichment on GO terms related to development and gene expression (Supplementary Data S4 and S5). However, we also found a large number of upregulated and downregulated genes related to metabolism in braA.clf-1 (Supplementary Data S3). These results are consistent with a recent transcriptomic report suggesting that BraA.CLF may be regulating the expression of stress-response and metabolic genes (Nugroho et al. 2023). These authors also proposed that BraA.CLF suppresses the expression of genes involved in the glucosinolate metabolism (Nugroho et al. 2023). However, our analysis only revealed five upregulated genes in braA.clf-1 related to the glucosinolate-pathway, compared to the 27 genes described by Nugroho and colleagues. Actually, only BraA03g054450.3C, a homolog of the A. thaliana UDP-GLUCOSYL TRANSFERASE 74B1 (AT1G24100), and BraA07g015340.3C and BraA07g015330.3C, two homologs of the A. thaliana INDOLE GLUCOSINOLATE O-METHYLTRANSFERASE 1 (AT1G21100), were upregulated and hypomethylated in braA.clf-1 leaves. Further research will be needed to define the precise role of BraA.CLF on glucosinolate metabolism.
Our genomic results are consistent with the broad role of CLF and the PRC2 complex previously described in the model plant A. thaliana (Shu et al. 2020). For example, as observed in A. thaliana, we found that BraA.CLF activity is required to repress the expression of a number of genes related to the floral transition and floral meristem identity. Specifically, the expression of the homologs of floral homeotic genes AG, PISTILLATA (PI), SEP3 and SEP4 were strongly upregulated in leaves, likely contributing to the pleiotropic clf phenotype. In addition, all the SOC1 homologs and the main FT gene in B. rapa were upregulated and showed reduced H3K27me3 levels in braA.clf-1 leaves (Fig. 7). The upregulation of these floral integrator genes is in agreement with the early-flowering time phenotype of braA.clf-1. The FLC gene is a well-known target of CLF in A. thaliana (Jiang et al. 2008; Lopez-Vernaza et al. 2012). However, no B. rapa FLC-like gene was misregulated in the braA.clf-1 mutant. These data confirm that certain floral regulators exhibit differences in gene-expression patterns between A. thaliana and B. rapa (Calderwood et al. 2021). Further research is necessary to fully understand the complexities of chromatin-dependent regulation of flowering time in Brassica crops.
Our data indicate that BraA.CLF regulates flowering time by affecting the expression of the floral integrator genes FT and SOC1 in B. rapa. These observations suggest that, to a certain extent, the methyltransferase activity of CLF is evolutionarily conserved between A. thaliana and B. rapa following their divergence from a shared ancestor. We believe that the study of the intricate interplay of CLF and other chromatin-modifying factors in crops may offer novel approaches for modulating flowering time to enhance agricultural productivity and sustainability.
Data availability
All ChIP‐seq and RNA‐seq genomic data have been submitted to NCBI Sequence Read Archive under the submission number SUB13676337.
References
Anders S, Pyl PT, Huber W (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. https://doi.org/10.1093/bioinformatics/btu638
Baile F, Gómez-Zambrano Á, Calonje M (2022) Roles of Polycomb complexes in regulating gene expression and chromatin structure in plants. Plant Commun. https://doi.org/10.1016/j.xplc.2021.100267
Bouché F, Lobet G, Tocquin P, Périlleux C (2016) FLOR-ID: an interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res 44:D1167–D1171. https://doi.org/10.1093/nar/gkv1054
Calderwood A, Hepworth J, Woodhouse S et al (2021) Comparative transcriptomics reveals desynchronisation of gene expression during the floral transition between Arabidopsis and Brassica rapa cultivars. Quant Plant Biol. https://doi.org/10.1017/qpb.2021.6
Carter B, Bishop B, Ho KK et al (2018) The Chromatin remodelers PKL and PIE1 act in an epigenetic pathway that determines H3K27me3 homeostasis in arabidopsis. Plant Cell 30:1337–1352. https://doi.org/10.1105/tpc.17.00867
Chanvivattana Y, Bishopp A, Schubert D et al (2004) Interaction of Polycomb-group proteins controlling flowering in Arabidopsis. Development 131:5263–5276. https://doi.org/10.1242/dev.01400
Cheng F, Liu S, Wu J et al (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11:136–141. https://doi.org/10.1186/1471-2229-11-136
del Olmo I, Poza-Viejo L, Piñeiro M et al (2019) High ambient temperature leads to reduced FT expression and delayed flowering in Brassica rapa via a mechanism associated with H2A.Z dynamics. Plant J 100:343–356. https://doi.org/10.1111/tpj.14446
Dereeper A, Guignon V, Blanc G et al (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. https://doi.org/10.1093/nar/gkn180
Doyle MR, Amasino RM (2009) A single amino acid change in the Enhancer of Zeste ortholog CURLY LEAF results in vernalization-independent, rapid-flowering in Arabidopsis. Plant Physiol 151:1688–1697. https://doi.org/10.1104/pp.109.145581
Du Z, Zhou X, Ling Y et al (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70. https://doi.org/10.1093/nar/gkq310
Farrona S, Thorpe FL, Engelhorn J et al (2011) Tissue-specific expression of FLOWERING LOCUS T in Arabidopsis is maintained independently of polycomb group protein repression. Plant Cell 23:3204–3214. https://doi.org/10.1105/tpc.111.087809
Godwin J, Farrona S (2022) The importance of networking: plant polycomb repressive complex 2 and its interactors. Epigenomes 6:8. https://doi.org/10.3390/epigenomes6010008
Goodrich J, Puangsomlee P, Martin M et al (1997) A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386:44–51. https://doi.org/10.1038/386044a0
Grossniklaus U, Vielle-Calzada J-P, Hoeppner MA, Gagliano WB (1998) Maternal control of embryogenesis by MEDEA, a polycomb group gene in arabidopsis. Science (-80) 280:446–450. https://doi.org/10.1126/science.280.5362.446
Guo Y, Zhao S, Wang GG (2021) Polycomb gene silencing mechanisms: PRC2 chromatin targeting, H3K27me3 “readout”, and phase separation-based compaction. Trends Genet 37:547–565. https://doi.org/10.1016/j.tig.2020.12.006
Huang Y, Liu C, Shen W-H, Ruan Y (2011) Phylogenetic analysis and classification of the Brassica rapa SET-domain protein family. BMC Plant Biol 11:175. https://doi.org/10.1186/1471-2229-11-175
Huang S, Hou L, Fu W et al (2020) An insertion mutation in Bra032169 encoding a histone methyltransferase is responsible for early bolting in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Front Plant Sci 11:1–9. https://doi.org/10.3389/fpls.2020.00547
Hulsen T (2022) DeepVenn: a web application for the creation of area-proportional Venn diagrams using the deep learning framework Tensorflow.js. arXiv. https://doi.org/10.48550/arXiv.2210.04597 Focus to learn more
Jiang D, Wang Y, Wang Y, He Y (2008) Repression of FLOWERING LOCUS C and FLOWERING LOCUS T by the arabidopsis polycomb repressive complex 2 components. PLoS ONE. https://doi.org/10.1371/journal.pone.0003404
Jiang H, Lei R, Ding S-W, Zhu S (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. https://doi.org/10.1186/1471-2105-15-182
Kinoshita T, Yadegari R, Harada JJ et al (1999) Imprinting of the MEDEA Polycomb gene in the arabidopsis endosperm. Plant Cell 11:1945–1952. https://doi.org/10.1105/tpc.11.10.1945
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Liu X, Kim YJ, Müller R et al (2011) AGAMOUS terminates floral stem cell maintenance in arabidopsis by directly repressing WUSCHEL through recruitment of Polycomb Group proteins. Plant Cell 23:3654–3670. https://doi.org/10.1105/tpc.111.091538
Liu J, Deng S, Wang H et al (2016) CURLY LEAF regulates gene sets coordinating seed size and lipid biosynthesis in arabidopsis. Plant Physiol. https://doi.org/10.1104/pp.15.01335
Lopez-Vernaza M, Yang S, Müller R et al (2012) Antagonistic roles of SEPALLATA3, FT and FLC genes as targets of the polycomb group gene CURLY LEAF. PLoS ONE. https://doi.org/10.1371/journal.pone.0030715
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Mehraj H, Takahashi S, Miyaji N et al (2021) Characterization of Histone H3 Lysine 4 and 36 Tri-methylation in Brassica rapa L. Front Plant Sci. https://doi.org/10.3389/fpls.2021.659634
Nugroho ABD, Kim S, Lee SW, Kim D (2023) Transcriptomic and epigenomic analyses revealed that polycomb repressive complex 2 regulates not only developmental but also stress responsive metabolism in Brassica rapa. Front Plant Sci 14:1–19. https://doi.org/10.3389/fpls.2023.1079218
Payá-Milans M, Poza-Viejo L, San Martín-Uriz P et al (2019) Genome-wide analysis of the H3K27me3 epigenome and transcriptome in Brassica rapa. Gigascience 8:1–13. https://doi.org/10.1093/gigascience/giz147
(2018) Picard tools. In: Broad Institute, GitHub Repos. https://github.com/broadinstitute/picard
Poza-Viejo L, Payá-Milans M, San Martín-Uriz P et al (2022) Conserved and distinct roles of H3K27me3 demethylases regulating flowering time in Brassica rapa. Plant Cell Environ. https://doi.org/10.1111/pce.14258
Poza-Viejo L, Del Olmo I, Crevillén P (2019) Plant chromatin immunoprecipitation V.2. In: protocols.io. https://www.protocols.io/view/plant-chromatin-immunoprecipitation-444gyyw
Schuettengruber B, Bourbon H-M, Di CL et al (2017) Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171:34–57. https://doi.org/10.1016/j.cell.2017.08.002
Shao Z, Zhang Y, Yuan G et al (2012) MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13:R16. https://doi.org/10.1186/gb-2012-13-3-r16
Shu J, Chen C, Thapa RK et al (2019) Genome-wide occupancy of histone H3K27 methyltransferases CURLY LEAF and SWINGER in Arabidopsis seedlings. Plant Direct. https://doi.org/10.1002/pld3.100
Shu J, Chen C, Li C, Cui Y (2020) The complexity of PRC2 catalysts CLF and SWN in plants. Biochem Soc Trans 48:2779–2789. https://doi.org/10.1042/BST20200660
Singkaravanit-Ogawa S, Kosaka A, Kitakura S et al (2021) Arabidopsis CURLY LEAF functions in leaf immunity against fungal pathogens by concomitantly repressing SEPALLATA3 and activating ORA59. Plant J 108:1005–1019. https://doi.org/10.1111/tpj.15488
Stephenson P, Baker D, Girin T et al (2010) A rich TILLING resource for studying gene function in Brassica rapa. BMC Plant Biol 10:62. https://doi.org/10.1186/1471-2229-10-62
Stovner EB, Sætrom P (2019) epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz232
Supek F, Bošnjak M, Škunca N et al (2011) Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE. https://doi.org/10.1371/journal.pone.0021800
Tan C, Ren J, Wang L et al (2021) A single amino acid residue substitution in BraA04g017190.3C, a histone methyltransferase, results in premature bolting in Chinese cabbage (Brassica rapa L. ssp. Pekinensis). BMC Plant Biol 21:373. https://doi.org/10.1186/s12870-021-03153-9
Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. https://doi.org/10.1093/bib/bbs017
Vijayanathan M, Trejo-Arellano MG, Mozgová I (2022) Polycomb repressive complex 2 in eukaryotes—an evolutionary perspective. Epigenomes 6:3. https://doi.org/10.3390/epigenomes6010003
Wang X, Wang H, Wang J et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039. https://doi.org/10.1038/ng.919
Wang H, Liu C, Cheng J et al (2016) Arabidopsis flower and embryo developmental genes are repressed in seedlings by different combinations of polycomb group proteins in association with distinct sets of cis-regulatory elements. PLOS Genet. https://doi.org/10.1371/journal.pgen.1005771
Wang Y, Huang X, Huang X et al (2022) BcSOC1 promotes bolting and stem elongation in flowering Chinese cabbage. Int J Mol Sci. https://doi.org/10.3390/ijms23073459
Xu X, Yang Z, Sun X et al (2014) Selection of reference genes for quantitative real-time PCR during flower bud development in CMS7311 of heading Chinese cabbage (Brassica rapa L. ssp. pekinensis). Acta Physiol Plant 36:809–814. https://doi.org/10.1007/s11738-013-1437-0
Zhang L, Cai X, Wu J et al (2018) Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic Res 5:50. https://doi.org/10.1038/s41438-018-0071-9
Zhang L, Liang J, Chen H et al (2023) A near-complete genome assembly of Brassica rapa provides new insights into the evolution of centromeres. Plant Biotechnol J 21:1022–1032. https://doi.org/10.1111/pbi.14015
Zhu LJ, Gazin C, Lawson ND et al (2010) ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11:237. https://doi.org/10.1186/1471-2105-11-237
Acknowledgements
We are grateful to Abdul Baten and Graham King (SCU, Australia) for sharing information about B. rapa R-o-18 genome. The authors thank CESGA for high-throughput computing and CNAG-CRG for assistance with ChIP sequencing. This work was funded by grants BIO2015‐68031‐R, RTI2018‐097749‐B‐I00 and PID2021-122241OB-I00 to PC, and PID2019‐104899GB‐I00 and PID2022-137131NB-I00 to JAJ and MP from MCIN/AEI/10.13039/ 501100011033 and FEDER; and fellowships BES‐2016‐078939 to LP and RYC‐2013‐14689 to PC from MINECO of Spain. MPM was supported by a Postdoctoral contract associated to the “Severo Ochoa Program for Centres of Excellence in R&D” grant SEV‐2016‐0672 (2017–2021) to CBGP from MCIN/AEI/https://doi.org/10.13039/501100011033.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Contributions
Laura Poza-Viejo: conceptualization, investigation, visualization and writing—review and editing. Miriam Payá-Milans: formal analysis, data curation and writing—review and editing. Mark D. Wilkinson: resources, supervision and writing—review and editing. Manuel Piñeiro: resources, supervision and writing—review and editing. José A. Jarillo: resources, supervision and writing—review and editing. Pedro Crevillén: conceptualization, resources, supervision, project administration and writing—original draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Stefan de Folter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poza-Viejo, L., Payá-Milans, M., Wilkinson, M.D. et al. Brassica rapa CURLY LEAF is a major H3K27 methyltransferase regulating flowering time. Planta 260, 27 (2024). https://doi.org/10.1007/s00425-024-04454-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00425-024-04454-7