
Abstract
The natriuretic peptides (NPs) ANP (atrial natriuretic peptide) and BNP (B-type natriuretic peptide) mediate their widespread effects by activating the natriuretic peptide receptor-A (NPR-A), while C-type natriuretic peptide (CNP) acts via natriuretic peptide receptor-B (NPR-B). NPs are removed from the circulation by internalization via the natriuretic peptide clearance receptor natriuretic peptide receptor-C (NPR-C). In addition to their well-known functions, for instance on blood pressure, all three NPs confer significant cardioprotection and renoprotection. Since neither the NP-mediated renal functions nor the renal target cells of renoprotection are completely understood, we performed systematic localization studies of NP receptors using in situ hybridization (RNAscope) in mouse kidneys. NPR-A mRNA is highly expressed in glomeruli (mainly podocytes), renal arterioles, endothelial cells of peritubular capillaries, and PDGFR-receptor β positive (PDGFR-β) interstitial cells. No NPR-A mRNA was detected by RNAscope in the tubular system. In contrast, NPR-B expression is highest in proximal tubules. NPR-C is located in glomeruli (mainly podocytes), in endothelial cells and PDGFR-β positive cells. To test for a possible regulation of NPRs in kidney diseases, their distribution was studied in adenine nephropathy. Signal intensity of NPR-A and NPR-B mRNA was reduced while their spatial distribution was unaltered compared with healthy kidneys. In contrast, NPR-C mRNA signal was markedly enhanced in cell clusters of myofibroblasts in fibrotic areas of adenine kidneys. In conclusion, the primary renal targets of ANP and BNP are glomerular, vascular, and interstitial cells but not the tubular compartment, while the CNP receptor NPR-B is highly expressed in proximal tubules. Further studies are needed to clarify the function and interplay of this specific receptor expression pattern.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The group of natriuretic peptides (NPs) includes the structurally related hormone atrial (ANP), B-type (BNP), and C-type natriuretic peptide (CNP) that possess various cardiovascular, renal, and endocrine effects. ANP and BNP are circulating heart-derived peptides, secreted in response to wall stress in the atrium and ventricles, whereas CNP acts in a paracrine manner and, in the cardiovascular system, is produced mainly in the endothelium [33]. ANP, CNP, and urodilatin, which is an alternative cleavage product of the ANP precursor, are also synthesized in the kidney itself, presumably forming locally acting NP systems [40, 57, 69]. Besides their crucial role in maintaining blood pressure, NPs also have strong protective potential, especially with regard to the cardiovascular system and the kidney [8, 27, 41].
NPs exert their biological actions by binding to their specific natriuretic peptide receptors (NPR). While ANP and BNP are ligands for natriuretic peptide receptor-A (NPR-A), CNP acts via natriuretic peptide receptor-B (NPR-B). NPR-A and NPR-B are membrane-bound guanylyl cyclases (guanylyl cyclase-A and -B, respectively), and their activation results in the formation of cyclic guanosine monophosphate (cGMP) [33]. Since, as discussed below, activation of NPR-B by CNP does not induce natriuresis but plays a fundamental role in bone formation and blood pressure regulation, the nomenclature NPR-B, for “natriuretic” peptide receptor, can be misleading and the use of “guanylyl cyclase B” (GC-B) instead of NPR-B has been suggested [1]. In line, the term “guanylyl cyclase A (GC-A)” can be used instead of “NPR-A”. The receptor natriuretic peptide receptor-C (NPR-C) lacks guanylyl cyclase-activity and acts mainly as clearance receptor for NPs [39]. In addition, recent data suggest that NPR-C is coupled to inhibitory G proteins hereby mediating some of the cardiovascular and metabolic effects of CNP [42, 48]. Due to the widespread distribution of NPRs in various tissues and cell types, the biological effects of NPs are very complex. In the kidney, activation of NPR-A increases glomerular filtration rate (GFR) and renal plasma flow (RPF), induces diuresis and natriuresis, and suppresses renin release [2, 4, 17, 32, 33, 35, 38, 64]. CNP does not have significant diuretic and natriuretic properties and its renal effects are less clear [33]. While the suppression of the renin-angiotensin system and the hypotensive effects most likely contribute to cardiorenal protection by NPs, also, a direct interaction with cardiomyocytes or with components of the immune system has been described [11, 24, 71]. NPs ameliorated renal injury in aldosterone- and high-salt-induced hypertension models, and the beneficial effects appeared to be independent of their antihypertensive mode of action [46]. In addition, NPs confer renoprotection in acute renal failure after ischemia/reperfusion, inflammatory renal injury, and diabetic nephropathy [9, 11, 13, 28, 37, 65]. In line, genetic deletion of NPR-A results in higher levels of proinflammatory cytokines, fibrosis, and mesangial proliferation in the kidney [14, 21, 47]. Whether this anti-inflammatory effect is due to a direct effect on immune cells or mediated indirectly, e.g., via inhibition of the renin system, is unclear.
Despite of profound effects of NPs in the control of renal function and on renal integrity, the underlying mechanisms or involved cell types are only incompletely understood. This is particularly true for the contribution of individual NPR-expressing cell types to the complex regulation of renal function in vivo. Cell type-specific deletion of individual components of the NP system helped to fill this knowledge gap partially. For instance, using podocyte-specific NPR-A knockout mice, we have shown previously that NPR-A signaling in podocytes is dispensable for the effects of ANP and BNP on GFR and natriuresis, but, unexpectedly, exerts direct renoprotection [64]. An essential prerequisite for a better understanding of the renal effects of NPs is knowledge of the exact localization of their receptors. Accordingly, studies on the spatial distribution of NPRs in the kidney have already been performed. However, because these studies either did not include all three NPRs, focused only on individual components of the kidney, or did not allow precise assignment to individual cell types, a detailed and comprehensive overview of the distribution of NPRs is lacking to date [19, 51, 59]. We therefore examined the mRNA expression of NPR-A, NPR-B, and NPR-C using high-resolution RNAscope technology. Our studies extend and complement a recent work on the localization of NPRs in human kidney [19], as the use of murine kidneys allows optimal sample preparation and sufficient material for studies on the exact cellular assignment of the receptors. To identify potential target cells for the renoprotective effects of natriuretic peptides, the distribution of NPRs in diseased kidneys was also investigated. Since NPs have anti-inflammatory properties and their renoprotective effects might therefore be related to direct effects on immune cells [14, 21, 47], the disease model chosen was the adenine nephropathy model characterized by severe tubulointerstitial inflammation and fibrosis.
Methods
Animals
Male and female wildtype mice on a C57BL/6 background were used and had free access to standard rodent chow and tap water. All animal experiments were conducted according to Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health and approved by the local authorities (Regierung von Unterfranken, 55.2.2–2532.2–1107).
Adenine-induced nephropathy
Male mice aged 16 to 22 weeks were fed 0.2% adenine containing diet. Control animals received standard rodent chow. After 3 weeks of treatment mice were anesthetized with buprenorphine (0.05 mg/kg BW), xylazine (10 mg/kg BW) and ketamine (80 mg/kg BW) and kidneys and adrenal glands were perfusion-fixed with 4% paraformaldehyde via the abdominal aorta.
Excess intake of adenine results in the formation of 2,8 hydroxyadenine, which is excreted via the kidneys. Due to its low solubility, 2,8 hydroxyadenine precipitates, resulting in the deposition of hydroxyadenine crystals in the tubular lumen and the renal interstitium, in tubulointerstitial inflammation, fibrosis, and progressive deterioration of kidney function with a three- to fourfold increase in serum creatinine after 3 weeks [67].
In situ hybridization using RNAscope
Detection of mRNA in mouse kidneys was performed using either the RNAscope® Multiplex Fluorescent v2 kit or the chromogenic RNAscope® 2.5 HD Reagent Kit-BROWN (Advanced Cell Diagnostics ACD, Cat. No 322300/323100), according to the manufacturer’s protocol. Fixed kidneys and adrenal glands were dehydrated in concentration ascending ethanol series and embedded in paraffin. Hybridization protocol was performed on 5 μm tissue sections, pre-treated for 15 min with 1 × target retrieval solution and 30 min with protease plus reagent (contained in RNAscope kit). If in situ hybridization was followed by immunofluorescence staining, protease plus treatment was reduced to 20 min. Multiplex RNAscope signals were detected using the AKOYA biosciences Opal™ fluorophores Opal690 (FP1497001KT), Opal570 (FP1488001KT), and Opal Polaris 480 (FP1500001KT) diluted in Tyramide Signal Amplification (TSA) buffer (1:750 for Cy3 and Cy5, 1:500 for Cy2). Slices were mounted with ProLong™ Gold Antifade Mountant mounting medium (Thermo Fisher Scientific) and stored at 4 °C. Negative controls were routinely enclosed and evaluated.
The RNAscope probes used are listed in Table1 below.
In situ hybridization followed by immunofluorescence staining
In situ hybridization was performed as described above. Before mounting, slices were washed with PBS, blocked for 30 min with 1% BSA and 10% horse serum solution and incubated with primary antibodies diluted in blocking solution overnight at 4 °C. Slices were incubated with fluorescent-labelled secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) for 90 min at room temperature and mounted with Dako Mounting medium. Slides were stored at 4 °C until further use. Antibodies are listed in Table 2.
Microdissection of single tubular segments
Microdissection was performed on four female and male C57BL/6 wildtype mice. The different nephron segments were identified by their morphology and manually isolated from perfused mouse kidney following a well-established protocol [74]. Briefly, the kidneys were perfused with 10 ml incubation solution (140 mmol/l NaCl, 0.4 mmol/l KH2PO4, 1.6 mmol/l K2HPO4, 1 mmol/l MgSO4, 10 mmol/l sodium acetate, 1 mmol/l α-ketoglutaric acid, 1.3 mmol/l calcium gluconate, 37.5 mg/100 ml glycine, 48 mg/100 ml trypsin inhibitor, pH 7.4 at 37 °C) containing 0.1 mg/ ml collagenase II (Sigma Aldrich, Cat. No. C6885). Sorting solution was prepared by adding 1 mg/ml PVA 4–88 (Sigma Aldrich) to the incubation solution. After perfusion, one kidney was removed, decapsulated, cut into slices, and transferred into prewarmed collagenase-containing incubation solution in a thermo shaker (Eppendorf; 850 rpm, 10 min, 37 °C). After adding prewarmed incubation solution, 1 ml of the digested tubule solution was transferred into a fresh tube. The contained glomeruli and tubular segments were washed and resuspended in 2 ml of sorting solution, and subsequent sorting of glomeruli and tubules was performed at 4 °C using a dissection microscope. Fresh tubule suspension was taken every 5 min from the digestion solution. 300 glomeruli and 100 of each nephron segment (PT S1/S2, PT S3, loops of Henle, CD) were collected. Since whole kidneys were used in the isolation procedure, no discrimination between cortical and medullary collecting ducts was possible. After collection, samples were centrifuged (5 min, 8,500 rpm, 4 °C) and tubule pellets stored at − 80 °C until further use.
Isolation of podocytes and glomerular cells
Podocytes and other glomerular cells were isolated from reporter mice with mG/mT expression as described previously [64]. These mice express green fluorescent GFP exclusively in podocytes while all other cells, including all non-podocyte cells in glomeruli, show red fluorescence. In brief, mice were sacrificed by cervical dislocation and 15 ml magnetic bead suspension (37 °C, 2 × 106 beads/ml; Invitrogen, Carlsbad, CA) was infused at a pressure of 100 mmHg into the renal arteries via the abdominal aorta. Subsequently, kidneys were further processed (digestion, cell strainer) and glomeruli were separated from the tissue suspension by a magnet. Glomeruli were further processed to receive a single cell suspension and podocytes (green fluorescence) and all other glomerular cells (red fluorescence, termed “non-podocytes” in the following) were sorted by FACS. On average, 250,000 podocytes were isolated from each mouse. To test for a possible enrichment of NPR mRNA in podocytes or glomerular non-podocytes compared with total kidney, complete kidneys were harvested from six mG/mT reporter mice and mRNA was isolated from them, as described below.
RNA isolation and cDNA-synthesis
RNA from microdissected nephron segments, isolated podocytes and glomerular non-podocytes were isolated using the Zymo Quick-RNA® Microprep Kit (Zymo Research, Cat. No. R1050). Cell lysis was performed by repetitive freezing/unfreezing of the samples in lysis puffer (provided in the kit). Subsequent isolation of the RNA was performed according to the manufacturer's protocol. 200 ng RNA of glomeruli and tubular segments or 100 ng RNA of podocytes and glomerular non-podocytes was used for cDNA synthesis. Reverse transcription reaction was conducted using the FastGene® Scriptase Basic cDNA Kit (Nippon Genetics) with Oligo dT-primers.
Total RNA of mG/mT reporter mouse kidneys, control, and adenine-nephropathy mouse kidneys was isolated with innuSOLV RNA Reagent (IST innuscreen GmbH). One complete kidney per animal was used in each case. Reverse transcription reaction was carried out using 1 µg of RNA and the moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific) for control and adenine kidneys. For comparability with podocytes, 100 ng of RNA was used for reverse transcription of RNA from the mG/mT reporter mice (FastGene® Scriptase Basic cDNA Kit, Nippon Genetics).
Semi-quantitative real-time-PCR
MRNA expression in different nephron segments, podocytes, and kidneys was determined by real-time PCR using SYBR Green (Roche Diagnostics). For relative quantification, a standard curve was constructed by plotting the threshold cycle values (Ct) against serial dilutions of a standard sample consisting of pooled individual samples from the respective experiment. Subsequently, the Ct values of each sample were referenced to the standard curve to determine its expression relative to the pooled standard sample. All values were related to the housekeeper gene RPL32. Primers are listed in Table 3.
Statistics
Statistical analysis and graphs were generated with GraphPad Prism6. All results are presented as mean values ± standard error of the mean (SEM). Data were tested for normal distribution and homoscedasticity (homogeneity of variance); significances were calculated by t-test or Welch test accordingly. P < 0.05 was considered significant. Statistical significance was marked according to the following scheme: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Results
Expression of NPR-A in murine kidneys
The NPs ANP and BNP mediate their biological effects by activating the Natriuretic Peptide Receptor A (NPR-A). NPR-A mRNA was mainly detected in glomeruli, blood vessels, and interstitial cells, with a clear increase in staining intensity from cortex to papilla. Unexpectedly, no NPR-A signal was visible in the tubular system (Fig. 1). Since the distribution pattern of NPR-A expression was not different between male (Fig. 1) and female (online supplement Fig. 2) mice, the following investigations were performed in male mice only.

NPR-A expression in murine kidneys under physiological conditions and adenine nephropathy. In healthy controls NPR-A mRNA (green) is mainly expressed in glomeruli (dashed lines), renal arterioles (marked as “a”) and interstitial cells (marked with arrows). No mRNA expression was detectable in tubules (marked as “*”). Interstitial NPR-A expression increased from renal cortex to papilla with the strongest RNAscope signal from cells localized in renal papilla. In adenine nephropathy, a reduction of NPR-A mRNA signal was detectable, particularly in the medullary and papillary areas. Nuclei are counterstained with DAPI (grey). Overview scale bars are 1000 µm. Detail scale bars are 100 µm

Identification of NPR-A expressing cell types in murine kidneys. mRNA co-localization studies of NPR-A (green) and various cell markers (red) using co-hybridization a NPR-A mRNA expression in glomeruli. Colocalization of NPR-A with nephrin (podocytes), CD31 (endothelial cells), PDGFR-β (mesangial cells), and renin (juxtaglomerular renin-producing cells). b NPR-A expression in intrarenal arterioles of all sizes, including afferent and efferent arterioles (marked as “a”). Vascular NPR-A mRNA signals predominantly derive from vascular smooth muscle cells (α-SMA). Only few endothelial cells (CD31) showed receptor expression. c Extraglomerular NPR-A expression. The majority of extraglomerular NPR-A signal derived from endothelial cells. PDGFR-β+ interstitial cells express NPR-A, mostly in renal papilla. Also, in adenine nephropathy, renal NPR-A mRNA signal is mainly assigned to endothelial cells of capillaries. d Co-localization of NPR-A and interstitial renin or epo mRNA signals. Nuclei are counterstained with DAPI (grey). Glomeruli are visualized by white dashed lines. Scale bars at lower magnification are 50 µm, at higher magnification 10 µm
In glomeruli, which showed a striking expression of NPR-A mRNA, podocytes were identified as the main NPR-A expressing cell type by duplex RNAscope with nephrin (Fig. 2a). In addition to podocytes, intraglomerular endothelial (co-hybridization with CD31) and mesangial cells (co-hybridization with platelet derived growth factor receptor β, PDGFR-β, a marker for glomerular mesangial cells and interstitial cells) expressed NPR-A (Fig. 2a). Moreover, juxtaglomerular renin-producing cells at the vascular pole of the glomeruli expressed NPR-A (Fig. 2a). Since ANP not only suppresses renal renin release but also the formation of aldosterone in adrenal glands, expression of NPR-A in the adrenal cortex was investigated. In fact, NPR-A is expressed in all cortical zones of adrenal glands including aldosterone-producing cells (online supplement Fig. 3). Intrarenal arterioles of all sizes, including afferent and efferent arterioles, showed clear NPR-A mRNA signals (Fig. 2b). In arterioles, NPR-A signals were predominantly assigned to vascular smooth muscle cells (co-hybridization with alpha smooth muscle actin, α-SMA), whereas endothelial cells (co-hybridization with CD31) had only low NPR-A signal (Fig. 2b). In contrast, endothelial cells of peritubular capillaries and vasa recta showed strong NPR-A signals and were the major extraglomerular cell type expressing NPR-A (Fig. 2c). Additionally, sporadic expression of the receptor was found in cortical PDGFR-β interstitial cells. In the medulla, most signals were assigned to peritubular endothelial cells, but also PDGFR-β cells showed NPR-A expression. In the papilla, NPR-A mRNA was highly detectable in endothelial and PDGFR-β cells. The proportion of PDGFR-β cells that expressed NPR-A continuously increased from the cortex to the papilla, where NPR-A was detectable in almost all PDGFR-β positive interstitial cells (Fig. 2c). The group of PDGFR-β positive interstitial cells contains pericytes, fibroblasts, erythropoietin (epo)-producing cells, but also interstitial cells that express renin [3]. Co-hybridization with epo or renin showed that both cell types expressed NPR-A, but to a lesser extent than endothelial cells (Fig. 2d).

Realtime PCR of NPRs in microdissected tubule segments, glomerular cells, and whole kidneys and in adenine nephropathy. Quantitative analysis of NPR-A, NPR-B, and NPR-C mRNA expression. a–c Left graphs: mRNA abundance in microdissected nephron segments of healthy C57Bl/6 wildtype mice. For comparison, mRNA expression in whole kidneys of C57Bl/6 wildtype mice is shown. Right graphs: Isolated podocytes and glomerular non-podocyte cells (separation by FACS) of mG/mT reporter mouse kidneys. For comparison, mRNA expression in whole kidneys of mG/mT reporter mice is shown. d–f NPR mRNA expression in healthy and adenine kidneys. In adenine kidneys, mRNA expression of all three receptors was numerically reduced, without reaching statistical significance. n (control) = 4, n (adenine) = 7. Abbreviations: PT, proximal tubule; CD, collecting duct
Real-time PCR of microdissected nephron segments and isolated podocytes confirmed that NPR-A mRNA was highly expressed in glomeruli and especially in podocytes (Fig. 3a). In contrast to the RNAscope data, NPR-A was detectable in microdissected tubular segments, especially in the collecting duct (CD), however at very low expression levels.
Mice with adenine-nephropathy exhibited severe tubulointerstitial damage, morphologically detectable by fibrotic areas and massive immune cell infiltration, exemplified in the supplemental Online Resource 1a by the leukocyte marker CD45. NPR-A distribution in the kidney was unaltered compared with healthy controls, but signal intensity was markedly decreased especially in the medulla and in the papilla (Fig. 1). The trend of decreasing NPR-A expression in adenine animals was confirmed by realtime PCR in whole kidney homogenates (Fig. 3d). In order to examine NPR-A expression in inflammatory cells of injured kidneys, we performed co-hybridization with immune cell markers (Online Resource 1b). However, no robust and reproducible NPR-A mRNA expression was observed in CD45 (pan leucocyte marker), Cx3cr1 (fractalkine receptor, dendritic cells, macrophages, monocytes), or F4/80 (macrophages) positive cells (see Online Resource 1b) [72]. Besides immune cell infiltrates, adenine-nephropathy is characterized by α-SMA expressing, collagen-producing interstitial myofibroblasts. While few myofibroblasts showed a faint NPR-A signal, most of these cells were NPR-A negative (Online Resource 1b). As in kidneys of healthy mice, by far the largest proportion of NPR-A expression could be mapped to endothelial cells (Fig. 2c).
In summary, in murine kidneys, NPR-A mRNA is expressed by podocytes, mesangial cells, and endothelial cells in glomeruli and by PDGFR-β-positive cells in the interstitium. In renal arterioles, NPR-A is predominantly detectable in vascular smooth muscle cells and less in endothelial cells. In contrast, endothelial cells in peritubular capillaries and vasa recta have high NPR-A expression levels. In mice with adenine nephropathy, marked reduction of NPR-A signal was observed (Table 4).
Expression of NPR-B in murine kidneys
Strong NPR-B mRNA signal was detectable in tubular cells in renal cortex and medulla, while no NPR-B signal was visible in tubular cells in the papilla. Furthermore, NPR-B was expressed in some interstitial cells, in glomeruli and in straight structures between tubules in medulla and papilla, but with lower signal intensity compared with tubules. (Figs. 4 and 5c).

NPR-B expression in murine kidneys of control animals and in adenine nephropathy. NPR-B mRNA distribution (green) in healthy mouse kidneys and in adenine nephropathy. The NPR-B receptor is expressed in the cortical and medullary tubular system (marked as “*”). At a lower level, mRNA signal was found in interstitial cells and in glomeruli (white dashed lines). In adenine nephropathy NPR-B mRNA signal was reduced, while distribution pattern remained the same as in healthy controls. Renal arterioles are marked as “a.” Nuclei are counterstained with DAPI (grey). Overview scale bars are 1000 µm. Detail scale bars are 100 µm

NPR-B mRNA expression in the murine kidney. a Chromogenic NPR-B RNAscope (black dots, arrows) followed by immunofluorescence staining of the different tubular segments: Megalin (PT, purple), Na–K-2Cl cotransporter (NKCC2, thick ascending loop of Henle, red), calbindin (distal tubule, orange), and aquaporin 2 (CD, green). b Multiplex in-situ hybridization of NPR-B (green) and the sodium-dependent glucose cotransporter 1 (SGLT1, red) plus megalin (purple) immunofluorescence staining. NPR-B mRNA was strongly detectable in the PT, with the highest expression in the S3 segment. c NPR-B mRNA is localized in vasa recta in renal medulla and papilla, but not in the endothelium of renal arterioles. d NPR-B mRNA signal (green) in glomeruli colocalized with mesangial (PDGFR-β) and endothelial (CD31) cell markers. Interstitial signals mainly derived from PDGFR-β+ fibroblasts but also from endothelial cells. Nuclei are counterstained with DAPI (grey). Glomeruli are visualized by white dashed lines. Scale bars are 50 µm, at higher magnification 10 µm
By conducting in situ hybridization followed by immunofluorescence staining of the individual tubular sections, we identified the proximal tubule (PT) as the NPR-B expressing nephron segment (Fig. 5a). Megalin expression in the PT decreases from S1 to S3, while the signal of the sodium-dependent glucose cotransporter 1 (SGLT1) increases from S1 to S3. NPR-B signal intensity increased along the PT and was the highest in SGLT1high/megalinlow S3 segments, mainly located in the medulla (Fig. 5b). In the thick ascending limb of the loop of Henle (NKCC2), distal tubules (calbindin), and collecting ducts (CD, aquaporin-2), only single NPR-B mRNA signals were found in a few tubules (Fig. 5a and b). Intrarenal arteries showed NPR-B signal, but to a lesser extent compared with NPR-A. There was no co-localization with the endothelial marker CD31, suggesting that the expressing cells are smooth muscle cells. The strongly NPR-B positive straight structures were identified as endothelial cells of vasa recta (Fig. 5c). Focusing on glomeruli and the interstitium, endothelial and PDGFR-β+ cells showed rather low NPR-B expression. In the medulla, most of the detected mRNA signal derived from endothelial cells, while in the papilla, PDGFR-β+ cells appeared as the main source of NPR-B expressing cells (Fig. 5d). In juxtaglomerular renin-producing cells, weak mRNA signal was found, while interstitial renin+ cells and epo-producing cells were not consistently NPR-B-positive (data not shown).
As visualized in RNAscope stainings, real-time PCR identified the S3 segment of the PT as a strong NPR-B expressing nephron segment of the murine kidney. Receptor expression was also detectable in the S1/S2 regions and the glomeruli, where the NPR-B signal is apparently not derived from podocytes (Fig. 3b).
The NPR-B mRNA signal was reduced in the kidneys of mice with adenine nephropathy in RNAscope and tended to be lower in real-time PCR data (Fig. 4 + 3e). Since NPR-B expression pattern was not altered in adenine nephropathy and the fibrotic areas in the adenine kidneys did not show increased NPR-B mRNA signals, no detailed investigation on receptor expression in immune cells was performed. A summary of NPR-B mRNA expression in murine kidneys is listed in Table 5.
Expression of NPR-C in murine kidneys
The NPR-C plays a crucial role in the regulation of NP signaling due to its function as a clearance receptor for ANP, BNP, and CNP. Compared with NPR-A and -B, NPR-C mRNA expression was more restricted in healthy control animals, with the strongest signal in glomeruli. In addition, NPR-C mRNA was found sporadically in interstitial cells of the cortex and, to an even lower extent, in the medulla and papilla. As for NPR-A, the tubular system showed no NPR-C mRNA expression, neither did intrarenal blood vessels (Fig. 6).

NPR-C mRNA expression in murine kidneys of control animals and in adenine nephropathy. NP clearance receptor NPR-C mRNA distribution (green) in healthy mouse kidneys and in adenine nephropathy. Under physiological conditions, only weak NPR-C expression was detectable in the mouse kidney, with the strongest signal in the glomeruli (white dashed lines). Additionally, NPR-C is also present in interstitial cells (marked with arrows). Renal tubules do not express NPR-C (marked with “*”). In adenine nephropathy, NPR-C signal was significantly increased and appeared mainly in cell clusters located in fibrotic areas, especially within the subcapsular cortex. Nuclei are counterstained with DAPI (grey). Overview scale bars are 1000 µm. Detail scale bars are 100 µm
In the glomerulus, podocytes were identified to highly express NPR-C, but also mesangial cells showed a weak signal (Fig. 7a). NPR-C is also synthesized in capillaries and fibroblasts in the interstitium, where co-localization with CD31 was stronger than with PDGFR-β (Fig. 7b). Renin- and epo-producing cells did not show NPR-C mRNA signal (not shown). Realtime PCR largely confirmed in-situ hybridization data since NPR-C mRNA expression was most abundant in FACS sorted podocytes (Fig. 3c). However, PCR further detected weak NPR-C expression in the tubular system, where expression levels were similar between the different segments but much lower compared to glomeruli.

NPR-C mRNA expression in glomeruli and the interstitium of mouse kidneys. a NPR-B mRNA expression (green) in glomeruli is highly colocalized with nephrin (podocytes), and to a lower extend with PDGFR-β (mesangial cells). b Interstitial receptor distribution is relatively low, but some endothelial cells as well as single PDGFR-β-positive interstitial cells showed hybridization signal. Nuclei are counterstained with DAPI (grey). Glomeruli are visualized by white dashed lines. Scale bars are 50 µm, at higher magnification 10 µm
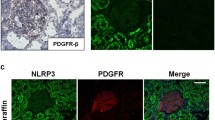
RNAscope in kidneys with adenine nephropathy showed a marked increase in NPR-C mRNA expression in cell clusters within fibrotic areas, especially in the subcapsular region, but also in the medulla and papilla, where only little expression was found in healthy animals (Fig. 6). PDGFR-β and α-SMA co-expressing myofibroblasts in the renal interstitium were identified as the main NPR-C expressing cell population in adenine nephropathy (Fig. 8a). Adenine-induced fibrotic remodelling of renal tissue is shown by significantly increased production of extracellular matrix proteins α-SMA, collagen 1a1 and fibronectin (Fig. 8b). Macrophages, DCs and leukocytes showed no NPR-C signal. However, it was noticeable that the immune cells were mostly located in the immediate vicinity of highly NPR-C expressing cells within fibrotic areas (Fig. 8c). In contrast to the visual impression, semi-quantitative realtime PCR revealed no increase of NPR-C mRNA expression in adenine kidneys but rather indicated decreasing receptor mRNA levels under inflammatory conditions (Fig. 3f).

NPR-C mRNA expression in adenine nephropathy. Investigation of highly NPR-C mRNA expressing (green) cell types in kidneys with adenine nephropathy. a Most NPR-C signal derives from PDGFR-β-and α-SMA-positive interstitial myofibroblasts in fibrotic areas. b Fibrotic changes in adenine nephropathy shown by the significantly increased production of the extracellular matrix proteins α-SMA, collagen 1a1 and fibronectin. n(control) = 4, n(adenine) = 6. c Leukocytes, macrophages, and dendritic cells showed no NPR-C signal, but immune cells were located within the areas with the highest receptor expression. Nuclei are counterstained with DAPI (grey). Glomeruli are visualized by white dashed lines. Scale bars are 50 µm. Statistical significance is indicated by *p < 0.05, **p < 0.01, and ***p < 0.001
In summary, under control conditions, NPR-C was primarily found in podocytes of the glomeruli, but also in mesangial, endothelial, and PDGFR-β+ interstitial cells. In mice with adenine nephropathy, there was a massive increase in NPR-C expression, particularly in fibrotic areas in the subcapsular cortex, which was mainly derived from PDGFR-β+ myofibroblasts. No NPR-C mRNA signal was detectable in immune cells. An overview of NPR-C distribution is listed in Table 6.
Discussion
The NPR-A receptor (guanylyl cyclase A), which mediates the effects of ANP and BNP, has a broad tissue distribution in murine kidneys, since it is expressed in glomeruli, intrarenal arterioles, renin- and epo-producing cells, PDGFR-β-positive interstitial cells, and endothelial cells of peritubular capillaries and vasa recta. Unexpectedly, but in line with results in human kidneys, no NPR-A mRNA signal was detected by RNAscope in the tubular system [19]. This result, which is in contrast to previous expression studies and functional studies showing direct effects of ANP on tubular cells (for review see [69]), might be explained by expression levels below the limit of detection by RNAscope. In fact, realtime PCR of microdissected nephron segments revealed NPR-A mRNA expression in tubules, especially in collecting ducts that, however, was very low compared with NPR-A expression in glomeruli (Fig. 3). It should be mentioned that the weak NPR-A expression in isolated tubules could be due to contamination with adherent interstitial cells and therefore does not necessarily prove expression in tubular cells. In summary, our data indicate that, if present at all, NPR-A mRNA expression in tubular cells is very low, calling into question a direct tubular effect of ANP in vivo. Further functional studies in tubule-specific knockout mice may clarify this issue. While our RNAscope data do not definitely exclude that tubular cells express NPR-A, it direct special interest to those cell types with high NPR-A expression. First and foremost is the prominent expression of NPR-A in podocytes that has already been described in different species including humans [19, 64]. Podocyte-restricted deletion of NPR-A did not attenuate the effects of ANP and BNP on kidney perfusion, GFR and natriuresis but markedly aggravated glomerular damage in the hypervolemic UNx/DOCA-salt model, indicating a direct protective effect of NP/NPR-A signaling [64]. Vascular smooth muscle cells of renal arterioles of all diameters, including afferent and efferent arterioles, express NPR-A which is the basis for ANP mediated increase in renal perfusion and GFR. Early studies indicated that ANP dilates afferent arterioles while it constricts efferent arterioles, hereby increasing intraglomerular filtration pressure [38]. Moreover, renin-producing cells in afferent arterioles express NPR-A, which is a prerequisite for direct inhibition of renin release by ANP [35]. Recently, renal interstitial cells have been shown to express renin mRNA, without a detailed characterization of the regulation and function of interstitial renin [3]. Our data show expression of NPR-A in these interstitial renin-producing cells, suggesting that ANP and BNP may have a regulatory role here. In addition, epo-producing interstitial cells express NPR-A. Acute infusion of ANP resulted in a rapid increase of hematocrit, which, however, was due to ANP-mediated intravascular volume contraction and cannot be induced by epo-dependent stimulation of hematopoiesis in the short observational period [53]. Evidence for an influence of ANP or BNP on epo formation and subsequent changes in hematocrit is not yet available, and the fact that NPR-A knockout mice have normal hematocrit levels under control conditions argues against this relationship [36, 56]. However, a possible role of NPs in pathologic conditions, in which ANP and BNP concentrations increase significantly, should be considered. Of note, the BNP promotor contains a hypoxia response element which is activated by the hypoxia inducible factor-1 (HIF1) and hypoxia markedly stimulates BNP mRNA expression in cardiomyocytes, so that a link between hypoxia, BNP and epo might exist [73]. In addition to podocytes, we found the highest NPR-A expression in endothelial cells of peritubular capillaries and vasa recta. In agreement, it was shown previously, that selective deletion of NPR-A in endothelial cells reduces total NPR-A mRNA abundance in the kidney by more than 50% [53]. Activation of NPR-A increases endothelial permeability resulting in extravasation of albumin and an accompanying fluid shift into the interstitium [53]. Accordingly, mice with endothelium-specific deletion of NPR-A have hypervolemic hypertension. Under control conditions urine and sodium excretion of these mice are not significantly different compared with control mice, but more detailed analyses of kidney function under challenging conditions were not performed [53]. Accordingly, the biological function of renal endothelial NPR-A remains unclear. It might be speculated that ANP-mediated plasma extravasation, which occurs in several organs including the kidney, changes osmotic driving forces in the renal tubulointerstitium and affects tubular reabsorption [53]. Moreover, endothelium-derived relaxation of vasa recta could increase medullary perfusion, resulting in a washout of interstitial osmolytes and consequently reduced water reabsorption, a well-known effect of NPs. Finally, ANP attenuates inflammatory activation of endothelial cells and expression of adhesion molecules, hereby controlling extravasation of immune cells [20, 71]. Since endothelial cells are considered mediators of renal inflammation, ANP/BNP might exert renoprotection via endothelial NPR-A activation [45]. Finally, significant NPR-A expression was detected in PDGFR-β-positive interstitial cells that include pericytes, fibroblasts, and epo-producing cells (see above). Pericytes are regulators of vasa recta tone and renal medullary perfusion [29]. Deletion of NPR-A in PDGFR-β-positive cells completely abolishes the vasodilating effects of ANP on capillaries in the cremaster muscle and the retinal microcirculation and results in arterial hypertension [63]. While no alterations of urine and sodium excretion under control conditions were found in vivo, the ANP-induced increase in renal perfusion of isolated perfused kidneys was significantly attenuated in mice with PDGFR-β-cell specific deletion of NPR-A. Noteworthy, this effect was only observed in kidneys in which blood vessels had been pre-constricted with endothelin-1 and not in untreated control kidneys [63]. Thus, further investigations on the role of NPR-A in renal PDGFR-β-positive cells are needed.
Several studies have already demonstrated the nephroprotective properties of NPs, which are at least partially independent of their hypotensive and RAAS antagonizing effects [11, 44, 64]. To test for a possible regulation of NPRs in kidney disease, we examined their distribution in adenine nephropathy. While RNAscope signals of NPR-A and -B were markedly reduced in adenine nephropathy, their spatial distribution remained unaltered. It is unclear whether the downregulation of NPR-A and -B occurs as a direct physiological response to adenine-induced processes or as a negative feedback to increased NP levels seen in cardiorenal disease [54]. Only little NPR-A and -B mRNA signal was detectable in the fibrotic areas heavily infiltrated by immune cells and co-hybridization with immune cell-specific markers did not show any overlap. These observations do not support the hypothesis of a direct effect of NPs on these cells, but rather suggest that the previously observed high levels of proinflammatory cytokines in kidneys of NPR-A knockout mice indirectly result from hypertensive kidney damage in this mouse strain [21, 70]. Since NPR expression was shown previously in different cell types of the innate and adaptive immune system of humans and rodents [5, 15, 30, 43], it is also possible that the expression level was below the detection limit of RNAscope in our study.
RNAscope for NPR-B (guanylyl cyclase B), which is the receptor for CNP, showed significant overlap with NPR-A, although at different signal intensities. This was shown for vascular smooth muscle cells, endothelial cells of vasa recta and PDGFR-β-positive cells. Since both receptors are membrane-bound guanylyl cyclases synthesizing cGMP, similar cellular effects can be assumed upon stimulation. In fact, similar to NPR-A, cell specific deletion of NPR-B in PDGFR-β-positive cells prevented the dilatory effects of CNP on pre-capillary arterioles and capillaries of the cremaster muscle and the retinal circulation and induced arterial hypertension [62, 63]. Urine and sodium excretion was unaltered under control conditions in these mice [62]. The endowment of the aforementioned cells with two NP receptor types that activate the same intracellular signaling cascade, appears redundant at first glance. However, the activating hormones differ considerably in their characteristics. While ANP and BNP are classical circulating hormones whose release is stimulated for instance by cardiac wall tension, CNP is a paracrine hormone from different sources which is regulated by completely different factors. Besides the endothelium, CNP is synthesized in the kidney. Since renal CNP is low under normal conditions but is elevated in several form of renal diseases such as nephrotic syndrome, unilateral ureteral obstruction or diabetes mellitus in patients or rodent models, studies on the functional role of CNP and NPR-B mainly focused on disease conditions [7, 25, 50, 61].
In contrast to NPR-A, RNAscope as well as real-time PCR results showed that murine NPR-B is markedly expressed in proximal tubules, with the strongest signal in the S3 segment. Moreover, strong expression was found in the vasa recta. The presence of NPR-B in the murine vasa recta has already been reported by other groups [68]. In contrast, the detection of NPR-B mRNA in the tubular system is surprising, as CNP lacks natriuretic and diuretic potential in experimental animals and healthy volunteers at least at physiological concentrations [10, 12, 49]. Moreover, this observation contrasts the experiments conducted in human tissue, where no renal NPR-B mRNA expression was detectable in tubular cells [19]. While the RNAscope probe used in our experiments covers all NPR-B protein-coding variants, the one available for human tissue does not detect at least one alternative protein-coding mRNA construct starting with exon 13 [19]. By using primers binding exon 18/exon 19, Hirsch et al. detected NPR-B expression in human tubules. Further, they reported variant-dependent expression of the receptor and located normal NPR-B in the glomeruli and proximal tubules, which agrees with our observations [22]. CNP has been shown to be highly protective on glomerular health by mediating anti-fibrotic and anti-proliferative processes, such as inhibition of mesangial cell proliferation and of the accumulation of extracellular matrix proteins [6]. It is also possible that the controversial results regarding NPR-B expression are due to species-dependent differences. Experiments with labeled CNP and immunohistochemical stainings have identified binding sites in renal tissue whose localization is also consistent with our NPR-B in-situ hybridization results [16, 75].
NPR-B expression in the PT suggests CNP as a regulator of tubular transport in this nephron segment, which is compatible with previous results in NPR-B knockout mice that have impaired diuresis and developed salt-dependent renal injury [28]. Because of the nephroprotective potential of CNP, observed in various animal experiments, the expression of NPR-B in the PT might exert a primary protective function [9, 28, 77]. Interestingly, marked protective effects of CNP treatment was observed in models with primary damage of proximal tubules, such as cisplatin-induced nephrotoxicity, ischemia/reperfusion or hemorrhagic shock [8, 28, 31]. The PT has enormous transport activity and therefore is particularly susceptible to damage. Especially in the S3-segment a disproportion between oxygen consumption and supply exists. Accordingly, this segment is usually particularly vulnerable, especially in the case of reduced blood supply [60]. It would be conceivable that expression of the NPR-B receptor in this nephron segment primarily serves nephroprotection.
Under physiological conditions, the expression of NPR-C is relatively restricted in murine kidneys. It is responsible for the degradation of circulating NPs but may also exert biological effects itself, mediated mainly through the adenylate cyclase/cAMP signaling pathway. Instead of an intracellular guanylylcyclase, NPR-C receptor is presumably coupled to a pertussis toxin-sensitive inhibitory G protein domain [52]. In our study, the strongest expression of NPR-C in the kidney occurred in podocytes, where also NPR-A mRNA is highly concentrated, followed by endothelial cells. The presence of NPR-C in podocytes has already been described before [64]. Real-time PCR revealed low receptor expression in the tubular system. As discussed for NPR-A, the different detection limits of RNAscope technique and PCR or contamination of microdissected tubules with adherent cells may be the explanation for the inconsistent results.
In adenine nephropathy, we observed a strong increase of NPR-C mRNA signal, which was mainly derived from PDGFR-β-positive myofibroblasts in fibrotic areas. It has already been shown that stimulation of renal PDGFR-β induces tubulointerstitial cell proliferation, formation of myofibroblasts, and fibrotic tissue remodeling under disease conditions [18]. This observation makes the strong expression of the NPR-C receptor in fibrotic areas interesting. On the one hand, the NPR-C receptor could act in its classical function as NP clearance receptor under the given inflammatory conditions. In doing so, it would degrade circulating and locally produced NPs and thereby attenuate their effects on the target cells. In the tubulointerstitial damage model of the unilateral ureteral obstruction (UUO), in addition to tubular cells, interstitial myofibroblasts synthesize CNP [66]. On the other hand, NPR-C might directly activate intracellular signaling as a negative regulator of the adenylate cyclase/cAMP system [26]. Since the cAMP pathway induces anti-fibrotic processes, the NPR-C-induced reduction of cAMP levels would promote fibrosis in this case [55]. In contrast to this possibility, NPR-C signaling is considered to promote anti-proliferative and -inflammatory processes [26, 52, 58]. Further, the receptor appeared to have a protective role in ischemia/reperfusion injury model [23]. An increase in NPR-C receptor expression in renal and cardiovascular disease is already reported in the literature [34, 52], but comparing our realtime PCR data with the results of in situ hybridization, the increased expression of the NPR-C receptor in the fibrotic areas is not evident at the whole organ level. This discrepancy may be a consequence from the upregulation of NPR-C in myofibroblasts on the one hand and a downregulation of NPR-C in other cells in adenine nephropathy, which compensates high NPR-C expression in myofibroblasts at the whole organ level.
In conclusion, all NPRs are expressed in murine renal tissue and are differentially regulated in renal injury. By identifying new NPR expressing cells types in the kidney, this work provides the basis for further investigations on the renal effects of natriuretic peptides in health and disease.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Alexander SP, Fabbro D, Kelly E et al (2021) The concise guide to pharmacology 2021/22: Catalytic receptors. Br J Pharmacol 178(Suppl 1):S264–S312. https://doi.org/10.1111/bph.15541
Bie P, Wang BC, Leadley RJ et al (1988) Hemodynamic and renal effects of low-dose infusions of atrial peptide in awake dogs. Am J Physiol 254:R161–R169. https://doi.org/10.1152/ajpregu.1988.254.2.R161
Broeker KAE, Fuchs MAA, Schrankl J et al (2022) Prolyl-4-hydroxylases 2 and 3 control erythropoietin production in renin-expressing cells of mouse kidneys. J Physiol 600:671–694. https://doi.org/10.1113/JP282615
Burnett JC, Opgenorth TJ, Granger JP (1986) The renal action of atrial natriuretic peptide during control of glomerular filtration. Kidney Int 30:16–19. https://doi.org/10.1038/ki.1986.144
Cabiati M, Sabatino L, Caruso R et al (2012) Gene expression of C-type natriuretic peptide and of its specific receptor NPR-B in human leukocytes of healthy and heart failure subjects. Peptides 37:240–246. https://doi.org/10.1016/j.peptides.2012.07.026
Canaan-Kühl S, Ostendorf T, Zander K et al (1998) C-type natriuretic peptide inhibits mesangial cell proliferation and matrix accumulation in vivo. Kidney Int 53:1143–1151. https://doi.org/10.1046/j.1523-1755.1998.00895.x
Cataliotti A, Giordano M, de Pascale E et al (2002) CNP production in the kidney and effects of protein intake restriction in nephrotic syndrome. Am J Physiol Renal Physiol 283:F464–F472. https://doi.org/10.1152/ajprenal.00372.2001
Chen Y, Burnett JC (2018) Particulate guanylyl cyclase A/cGMP signaling pathway in the kidney: physiologic and therapeutic indications. Int J Mol Sci 19(4):1006. https://doi.org/10.3390/ijms19041006
Chen G, Song X, Yin Y et al (2017) C-type natriuretic peptide prevents kidney injury and attenuates oxidative and inflammatory responses in hemorrhagic shock. Amino Acids 49:347–354. https://doi.org/10.1007/s00726-016-2367-0
Chen Y, Zheng Y, Iyer SR et al (2019) C53: A novel particulate guanylyl cyclase B receptor activator that has sustained activity in vivo with anti-fibrotic actions in human cardiac and renal fibroblasts. J Mol Cell Cardiol 130:140–150. https://doi.org/10.1016/j.yjmcc.2019.03.024
Choi MR, Fernández BE (2021) Protective renal effects of atrial natriuretic peptide: where are we now? Front Physiol 12:680213. https://doi.org/10.3389/fphys.2021.680213
Clavell AL, Stingo AJ, Wei CM et al (1993) C-type natriuretic peptide: a selective cardiovascular peptide. Am J Physiol 264:R290–R295. https://doi.org/10.1152/ajpregu.1993.264.2.R290
Conger JD, Falk SA, Hammond WS (1991) Atrial natriuretic peptide and dopamine in established acute renal failure in the rat. Kidney Int 40:21–28. https://doi.org/10.1038/ki.1991.174
Das S, Au E, Krazit ST et al (2010) Targeted disruption of guanylyl cyclase-A/natriuretic peptide receptor-A gene provokes renal fibrosis and remodeling in null mutant mice: role of proinflammatory cytokines. Endocrinology 151:5841–5850. https://doi.org/10.1210/en.2010-0655
de Bold MLK, Etchepare A, Martinuk A et al (2010) Cardiac hormones ANF and BNP modulate proliferation in the unidirectional mixed lymphocyte reaction. J Heart Lung Transplant 29:323–326. https://doi.org/10.1016/j.healun.2009.07.008
Dugbartey GJ, Quinn B, Luo L et al (2019) The protective role of natriuretic peptide receptor 2 against high salt injury in the Renal Papilla. Am J Pathol 189:1721–1731. https://doi.org/10.1016/j.ajpath.2019.05.020
Dunn BR, Ichikawa I, Pfeffer JM et al (1986) Renal and systemic hemodynamic effects of synthetic atrial natriuretic peptide in the anesthetized rat. Circ Res 59:237–246. https://doi.org/10.1161/01.res.59.3.237
Floege J, Eitner F, Alpers CE (2008) A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol 19:12–23. https://doi.org/10.1681/ASN.2007050532
Frees A, Assersen KB, Jensen M et al (2021) Natriuretic peptides relax human intrarenal arteries through natriuretic peptide receptor type-A recapitulated by soluble guanylyl cyclase agonists. Acta Physiol (Oxf) 231:e13565. https://doi.org/10.1111/apha.13565
Fu H, Zhang J, Cai Q et al (2022) Pleiotropic roles of atrial natriuretic peptide in anti-inflammation and anti-cancer activity. Cancers (Basel) 14(16):3981. https://doi.org/10.3390/cancers14163981
Gogulamudi VR, Mani I, Subramanian U et al (2019) Genetic disruption of Npr1 depletes regulatory T cells and provokes high levels of proinflammatory cytokines and fibrosis in the kidneys of female mutant mice. Am J Physiol Renal Physiol 316:F1254–F1272. https://doi.org/10.1152/ajprenal.00621.2018
Hirsch JR, Skutta N, Schlatter E (2003) Signaling and distribution of NPR-Bi, the human splice form of the natriuretic peptide receptor type B. Am J Physiol Renal Physiol 285:F370–F374. https://doi.org/10.1152/ajprenal.00049.2003
Hobbs A, Foster P, Prescott C et al (2004) Natriuretic peptide receptor-C regulates coronary blood flow and prevents myocardial ischemia/reperfusion injury: novel cardioprotective role for endothelium-derived C-type natriuretic peptide. Circulation 110:1231–1235. https://doi.org/10.1161/01.CIR.0000141802.29945.34
Holtwick R, van Eickels M, Skryabin BV et al (2003) Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest 111:1399–1407. https://doi.org/10.1172/JCI17061
Hu P, Wang J, Zhao XQ et al (2013) Overexpressed C-type natriuretic peptide serves as an early compensatory response to counteract extracellular matrix remodeling in unilateral ureteral obstruction rats. Mol Biol Rep 40:1429–1441. https://doi.org/10.1007/s11033-012-2186-7
Jansen HJ, Mackasey M, Moghtadaei M et al (2019) NPR-C (natriuretic peptide receptor-C) modulates the progression of angiotensin II-mediated atrial fibrillation and atrial remodeling in mice. Circ Arrhythm Electrophysiol 12:e006863. https://doi.org/10.1161/CIRCEP.118.006863
Jeson Sangaralingham S, Kuhn M, Cannone V et al. (2022) Natriuretic peptide pathways in heart failure - further therapeutic possibilities. Cardiovasc Res. Epub ahead of print. https://doi.org/10.1093/cvr/cvac125
Jin X, Zhang Y, Li X et al (2014) C-type natriuretic peptide ameliorates ischemia/reperfusion-induced acute kidney injury by inhibiting apoptosis and oxidative stress in rats. Life Sci 117:40–45. https://doi.org/10.1016/j.lfs.2014.09.023
Kennedy-Lydon TM, Crawford C, Wildman SSP et al (2013) Renal pericytes: regulators of medullary blood flow. Acta Physiol (Oxf) 207:212–225. https://doi.org/10.1111/apha.12026
Kiemer AK, Vollmar AM (1997) Effects of different natriuretic peptides on nitric oxide synthesis in macrophages. Endocrinology 138:4282–4290. https://doi.org/10.1210/endo.138.10.5459
Kimura T, Nojiri T, Hosoda H et al (2015) Protective effects of C-type natriuretic peptide on cisplatin-induced nephrotoxicity in mice. Cancer Chemother Pharmacol 75:1057–1063. https://doi.org/10.1007/s00280-015-2734-7
Kishimoto I, Dubois SK, Garbers DL (1996) The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: acute handling of sodium and water in response to volume expansion. Proc Natl Acad Sci U S A 93:6215–6219. https://doi.org/10.1073/pnas.93.12.6215
Kuhn M (2016) Molecular physiology of membrane guanylyl cyclase receptors. Physiol Rev 96:751–804. https://doi.org/10.1152/physrev.00022.2015
Kuhn M, Voss M, Mitko D et al (2004) Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end-stage heart failure. Cardiovasc Res 64:308–314. https://doi.org/10.1016/j.cardiores.2004.07.004
Kurtz A, Della Bruna R, Pfeilschifter J et al (1986) Atrial natriuretic peptide inhibits renin release from juxtaglomerular cells by a cGMP-mediated process. Proc Natl Acad Sci U S A 83:4769–4773. https://doi.org/10.1073/pnas.83.13.4769
Lopez MJ, Wong SK, Kishimoto I et al (1995) Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 378:65–68. https://doi.org/10.1038/378065a0
Makino H, Mukoyama M, Mori K et al (2006) Transgenic overexpression of brain natriuretic peptide prevents the progression of diabetic nephropathy in mice. Diabetologia 49:2514–2524. https://doi.org/10.1007/s00125-006-0352-y
Marin-Grez M, Fleming JT, Steinhausen M (1986) Atrial natriuretic peptide causes pre-glomerular vasodilatation and post-glomerular vasoconstriction in rat kidney. Nature 324:473–476. https://doi.org/10.1038/324473a0
Matsukawa N, Grzesik WJ, Takahashi N et al (1999) The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A 96:7403–7408. https://doi.org/10.1073/pnas.96.13.7403
Mattingly MT, Brandt RR, Heublein DM et al (1994) Presence of C-type natriuretic peptide in human kidney and urine. Kidney Int 46:744–747. https://doi.org/10.1038/ki.1994.329
McFarlane SI, Winer N, Sowers JR (2003) Role of the natriuretic peptide system in cardiorenal protection. Arch Intern Med 163:2696–2704. https://doi.org/10.1001/archinte.163.22.2696
Moyes AJ, Chu SM, Aubdool AA et al (2020) C-type natriuretic peptide co-ordinates cardiac structure and function. Eur Heart J 41:1006–1020. https://doi.org/10.1093/eurheartj/ehz093
Nakagawa M, Naruko T, Sugioka K et al (2017) Enhanced expression of natriuretic peptide receptor A and B in neutrophils of culprit lesions in patients with acute myocardial infarction. Mol Med Rep 16:3324–3330. https://doi.org/10.3892/mmr.2017.7034
Nigwekar SU, Navaneethan SD, Parikh CR et al (2009) Atrial natriuretic peptide for management of acute kidney injury: a systematic review and meta-analysis. Clin J Am Soc Nephrol 4:261–272. https://doi.org/10.2215/CJN.03780808
Oates JC, Russell DL, van Beusecum JP (2022) Endothelial cells: potential novel regulators of renal inflammation. Am J Physiol Renal Physiol 322:F309–F321. https://doi.org/10.1152/ajprenal.00371.2021
Ogawa Y, Mukoyama M, Yokoi H et al (2012) Natriuretic peptide receptor guanylyl cyclase-A protects podocytes from aldosterone-induced glomerular injury. J Am Soc Nephrol 23:1198–1209. https://doi.org/10.1681/ASN.2011100985
Pandey KN, Nguyen HT, Li M et al (2000) Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells: role of cGMP-dependent protein kinase. Biochem Biophys Res Commun 271:374–379. https://doi.org/10.1006/bbrc.2000.2627
Perez-Ternero C, Aubdool AA, Makwana R et al (2022) C-type natriuretic peptide is a pivotal regulator of metabolic homeostasis. Proc Natl Acad Sci U S A 119:e2116470119. https://doi.org/10.1073/pnas.2116470119
Pham I, Sediame S, Maistre G et al (1997) Renal and vascular effects of C-type and atrial natriuretic peptides in humans. Am J Physiol 273:R1457–R1464. https://doi.org/10.1152/ajpregu.1997.273.4.R1457
Prickett TCR, Lunt H, Warwick J et al (2019) Urinary amino-terminal Pro-C-type natriuretic peptide: a novel marker of chronic kidney disease in diabetes. Clin Chem 65:1248–1257. https://doi.org/10.1373/clinchem.2019.306910
Ritter D, Dean AD, Gluck SL et al (1995) Natriuretic peptide receptors A and B have different cellular distributions in rat kidney. Kidney Int 48:5758–5766. https://doi.org/10.1038/ki.1995.474
Rubattu S, Sciarretta S, Morriello A et al (2010) NPR-C: a component of the natriuretic peptide family with implications in human diseases. J Mol Med (Berl) 88:889–897. https://doi.org/10.1007/s00109-010-0641-2
Sabrane K, Kruse MN, Fabritz L et al (2005) Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest 115:1666–1674. https://doi.org/10.1172/JCI23360
Santos-Araújo C, Leite-Moreira A, Pestana M (2015) Clinical value of natriuretic peptides in chronic kidney disease. Nefrologia 35:227–233. https://doi.org/10.1016/j.nefro.2015.03.002
Schinner E, Wetzl V, Schlossmann J (2015) Cyclic nucleotide signalling in kidney fibrosis. Int J Mol Sci 16:2320–2351. https://doi.org/10.3390/ijms16022320
Schreier B, Börner S, Völker K et al (2008) The heart communicates with the endothelium through the guanylyl cyclase-A receptor: acute handling of intravascular volume in response to volume expansion. Endocrinology 149:4193–4199. https://doi.org/10.1210/en.2008-0212
Schulz-Knappe P, Forssmann K, Herbst F et al (1988) Isolation and structural analysis of “urodilatin”, a new peptide of the cardiodilatin-(ANP)-family, extracted from human urine. Klin Wochenschr 66:752–759. https://doi.org/10.1007/BF01726570
Scotland RS, Cohen M, Foster P et al (2005) C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc Natl Acad Sci U S A 102:14452–14457. https://doi.org/10.1073/pnas.0504961102
Sexton PM, Zhuo J, Mendelsohn FA (1992) Localization and regulation of renal receptors for angiotensin II and atrial natriuretic peptide. Tohoku J Exp Med 166:41–56. https://doi.org/10.1620/tjem.166.41
Shanley PF, Brezis M, Spokes K et al (1986) Transport-dependent cell injury in the S3 segment of the proximal tubule. Kidney Int 29:1033–1037. https://doi.org/10.1038/ki.1986.103
Shin SJ, Wen JD, Lee YJ et al (1998) Increased C-type natriuretic peptide mRNA expression in the kidney of diabetic rats. J Endocrinol 158:35–42. https://doi.org/10.1677/joe.0.1580035
Špiranec K, Chen W, Werner F et al (2018) Endothelial C-type natriuretic peptide acts on pericytes to regulate microcirculatory flow and blood pressure. Circulation 138:494–508. https://doi.org/10.1161/CIRCULATIONAHA.117.033383
Špiranec Spes K, Chen W, Krebes L et al (2020) Heart-microcirculation connection: effects of ANP (atrial natriuretic peptide) on pericytes participate in the acute and chronic regulation of arterial blood pressure. Hypertension 76:1637–1648. https://doi.org/10.1161/HYPERTENSIONAHA.120.15772
Staffel J, Valletta D, Federlein A et al (2017) Natriuretic peptide receptor guanylyl cyclase-A in podocytes is renoprotective but dispensable for physiologic renal function. J Am Soc Nephrol 28:260–277. https://doi.org/10.1681/ASN.2015070731
Suganami T, Mukoyama M, Sugawara A et al (2001) Overexpression of brain natriuretic peptide in mice ameliorates immune-mediated renal injury. J Am Soc Nephrol 12:2652–2663. https://doi.org/10.1681/ASN.V12122652
Surendran K, Simon TC (2003) CNP gene expression is activated by Wnt signaling and correlates with Wnt4 expression during renal injury. Am J Physiol Renal Physiol 284:F653–F662. https://doi.org/10.1152/ajprenal.00343.2002
Tamura M, Aizawa R, Hori M et al (2009) Progressive renal dysfunction and macrophage infiltration in interstitial fibrosis in an adenine-induced tubulointerstitial nephritis mouse model. Histochem Cell Biol 131:483–490. https://doi.org/10.1007/s00418-009-0557-5
Terada Y, Tomita K, Nonoguchi H et al (1994) PCR localization of C-type natriuretic peptide and B-type receptor mRNAs in rat nephron segments. Am J Physiol 267:F215–F222. https://doi.org/10.1152/ajprenal.1994.267.2.F215
Theilig F, Wu Q (2015) ANP-induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol 308:F1047–F1055. https://doi.org/10.1152/ajprenal.00164.2014
Vellaichamy E, Kaur K, Pandey KN (2007) Enhanced activation of pro-inflammatory cytokines in mice lacking natriuretic peptide receptor-A. Peptides 28:893–899. https://doi.org/10.1016/j.peptides.2006.12.009
Vollmar AM (2005) The role of atrial natriuretic peptide in the immune system. Peptides 26:1086–1094. https://doi.org/10.1016/j.peptides.2004.08.034
von Vietinghoff S, Kurts C (2021) Regulation and function of CX3CR1 and its ligand CX3CL1 in kidney disease. Cell Tissue Res 385:335–344. https://doi.org/10.1007/s00441-021-03473-0
Weidemann A, Klanke B, Wagner M et al (2008) Hypoxia, via stabilization of the hypoxia-inducible factor HIF-1alpha, is a direct and sufficient stimulus for brain-type natriuretic peptide induction. Biochem J 409:233–242. https://doi.org/10.1042/BJ20070629
Willmann JK, Bleich M, Rizzo M et al (1997) Amiloride-inhibitable Na+ conductance in rat proximal tubule. Pflugers Arch 434:173–178. https://doi.org/10.1007/s004240050380
Woodard GE, Li X, Rosado JA (2004) Water deprivation enhances the inhibitory effect of natriuretic peptides on cAMP synthesis in rat renal glomeruli. Am J Physiol Renal Physiol 287:F418–F426. https://doi.org/10.1152/ajprenal.00069.2004
Wotus C, Levay-Young BK, Rogers LM et al (1998) Development of adrenal zonation in fetal rats defined by expression of aldosterone synthase and 11beta-hydroxylase. Endocrinology 139:4397–4403. https://doi.org/10.1210/endo.139.10.6230
Zhang DD, Wu YF, Chen WX et al (2019) C-type natriuretic peptide attenuates renal osteodystrophy through inhibition of FGF-23/MAPK signaling. Exp Mol Med 51:1–18. https://doi.org/10.1038/s12276-019-0265-8
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project number 387509280, SFB 1350.
Author information
Authors and Affiliations
Contributions
E. S. H. wrote the paper, interpreted the data, and conducted in situ hybridization; R. H., K. K., and K. O. performed microdissection and helped generating real-time-PCR data; K. A.-E. B. established RNAscope protocol and provided material; C. L. conducted adenine-nephropathy experiments and provided material; P. T. supervised microdissection and interpreted the data. F. S. conceived the study, interpreted the data and wrote the paper with E.S.H.. All authors edited and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All animal experiments were conducted according to Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health and approved by the local ethics committee (Regierung von Unterfranken, 55.2.2–2532.2–1107).
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heinl, ES., Broeker, K.AE., Lehrmann, C. et al. Localization of natriuretic peptide receptors A, B, and C in healthy and diseased mouse kidneys. Pflugers Arch - Eur J Physiol 475, 343–360 (2023). https://doi.org/10.1007/s00424-022-02774-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02774-9