
Abstract
Despite numerous studies which have explored the pathogenesis of pain disorders in preclinical models, there is a pronounced translational gap, which is at least partially caused by differences between the human and rodent nociceptive system. An elegant way to bridge this divide is the exploitation of human-induced pluripotent stem cell (iPSC) reprogramming into human iPSC-derived nociceptors (iDNs). Several protocols were developed and optimized to model nociceptive processes in health and disease. Here we provide an overview of the different approaches and summarize the knowledge obtained from such models on pain pathologies associated with monogenetic sensory disorders so far. In addition, novel perspectives offered by increasing the complexity of the model systems further to better reflect the natural environment of nociceptive neurons by involving other cell types in 3D model systems are described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The discriminative component of pain sensations includes the ability to identify a stimulus as originating from somatic or visceral tissue, to inform about the physical properties of the stimulus along a continuum of intensities, and to localize it in space and time. Of particular importance for the extraction of information on the stimulus is the primary nociceptive neuron, which serves transduction and transformation processes and transmits the information to the next level of integrating neurons in the CNS [1]. The first recordings from nociceptive nerve fibers were pioneered by Yngve Zotterman [2] and Ainsley Iggo [3] in nonhuman species followed by microneurography recordings from human nociceptive afferents by Hagbarth and Vallbo [4]. Since then, nociceptors have been classified according to conduction velocities of their axons and responsiveness to specific natural physical, chemical, and electrical stimuli. Sir Charles Sherrington introduced these important functions as the main tasks of nociceptors [5]. In addition, it is generally accepted that nociceptors act as chemosensors for cytokines, prostaglandins, kinins, bioactive sphingolipids, and other mediators or chemical irritants [6, 7]. Based on these findings, the concept of Sherrington has been questioned, and the idea emerged that these primary afferents, rather than functioning simply as Sherringtonian nociceptors, may have more general and long-term roles in signaling and more recently even in controlling the microenvironment and components of the immune system in their respective target tissue [8,9,10]. Specifically, peptidergic nociceptors release neuropeptides in response to noxious stimuli not only by volume transmission to modify spinal pain circuits but also at their peripheral terminals in a reaction causing vasodilation and plasma extravasation in their target tissue, which has been long known as axon reflex (alias neurogenic inflammation, Lewis reaction – [11,12,13]). Of the eleven different sensory neuron subtypes emerging from recent unbiased transcriptomic studies [14], peptidergic and non-peptidergic small neurons serve different roles: peptidergic nociceptors control specific immune cell types but non-peptidergic neurons are equally important for balancing of the local immune reaction via the release of the fast neurotransmitter glutamate from nerve terminals in their target tissues [15, 16].
To date, nociceptor excitation and sensitization processes have mainly been explored and seminal findings made in nonhuman model systems, but increasing numbers of studies are exploiting the human dorsal root ganglion (DRG) as a model system [17,18,19,20,21]. The studies available to date, however, suggest fundamental differences between mice and men. In particular, there are human nociceptor populations that are not found in mice [21]. Also, transcripts for transient receptor potential channels, cholinergic receptors, potassium channels, sodium channels, and other markers/targets are differentially expressed, suggesting human-specific spatial and functional organization of neuronal cell subpopulations and supporting the idea that sensory system organizational principles are different between both species [22].
A major drawback, however, is the restricted availability of live human nociceptive neurons for transcriptomic and functional in vitro studies for ethical and logistic reasons. The results from nonhuman models turn out to be only partially transferable to healthy humans and even less to patients suffering from pain disorders arising from nociceptor pathologies [23]. Primary cultures of human or mouse DRG neurons also suffer from drawbacks as they adopt a neuropathic phenotype under culture conditions [24]. To better elaborate on human nociceptor (patho)physiology towards patient-centered mechanistic insight and the development of mechanism-based treatments, rigidly controlled human model systems are largely favorable over nonhuman models and offer major advantages to develop personalized medicines tailored to the individual needs of each patient. This review article compares nociceptor phenotypes in mice and humans and genetic pain disorders and summarizes additional cellular components interacting with nociceptors as well as recent approaches towards improved modeling of human nociception to provide an overview on the current state of the art and identify methodological needs.
Exploring molecular sensory neuron phenotypes
The cell bodies of primary afferent neurons serving different functionalities are located within dorsal root ganglia and trigeminal ganglia (TG). In general, large diameter neurons give rise to low-threshold mechanosensitive fibers whereas small diameter neurons are slowly conducting, unmyelinated C-fibers or thinly myelinated Aδ-fibers implicated in thermosensation or the transduction of noxious stimuli. Immunoreactivity to specific cell markers is generally used to distinguish these neuron classes since, for example, neurofilament heavy chain (NEFH) is abundant in large, myelinated neurons, and the intermediate filament protein type 3 (peripherin) is typically found in small diameter sensory neurons [25,26,27]. These small diameter sensory neurons are further subclassified into C-fiber nociceptors expressing the high-affinity neurotropic growth factor (NGF) receptor TrkA, Aδ-fiber nociceptors expressing the TrkB receptor for brain-derived neurotrophic factor (BDNF), and mechanoreceptors or pruriceptors expressing TrkC for neurotrophin-3 (NT-3) [28]. While the expression of TrkA in nociceptors is highly correlated with a peptidergic sensory neuron phenotype, the absence of TrkA and the expression of the Runt-related transcription factor-1 (RUNX1) and RET more likely drive sensory neurons towards a non-peptidergic fate [29]. Electrophysiological classification of nociceptors is usually based on the presence of TTX-sensitive and resistant voltage-gated sodium channels or endogenous temperature transducers belonging to the class of transient receptor potential channels (TRP channels) [30,31,32,33,34,35,36,37]. These classifications were the basis for a broad range of studies in rodents investigating the different roles and distinct synaptic connections in the spinal dorsal horn of peptidergic nociceptors that express neuropeptides such as calcitonin gene–related peptide (CGRP)/somatostatin (SST) or substance P (TAC1) and non-peptidergic nociceptors that bind the isolectin B4 [38,39,40,41,42]. Although sensory neurons largely share transcriptional signatures, specific molecular genetic profiles and a high-resolution atlas of sensory neuron subtypes illustrate cell-type–specific functionalities such as mechanoreceptors expressing TrkB, the two-pore potassium channel KCNK4, and the mechanotransducer channel PIEZO2 or proprioceptors expressing TrkC and parvalbumin (PVALB) [43, 44].
Bulk RNA sequencing and bulk proteomics reveal a strong overlap of DRG developmental transcription factors such as PRDM12, BRN3A, and DRGX, suggesting conserved developmental signatures of sensory neurons between humans and rodents [45, 46]. However, with the recent rise of single-cell RNA (scRNA) sequencing technologies, increasing evidence documents that even nociceptor subtypes are much more complex than initially thought: Five clusters of larger diameter sensory neurons are tied together by the expression of NEFH and separated by the abundance of TrkC, TrkB, PVALB, and RET, revealing mechanoreceptors (TrkBhigh, REThigh) as well as proprioceptors (TrkChigh, Pvalbhigh) [14]. In addition, two subclusters of peptidergic sensory neurons and three subclusters of non-peptidergic neurons can be distinguished by the unique transcriptomic signatures of the P2X purinoceptor 3 (P2RX3, non-peptidergic nociceptors) and tachykinin precursor 1 (TAC1, peptidergic nociceptors) [14]. More recent studies further increased the resolution of cell type clustering by enhancing the read coverage, adding different species (i.e., primates), and extending the number of cells as well as adding disease phenotypes such as neuropathic pain, which led to more refined clusters of sensory neurons [47,48,49]. These subclusters are defined by the expression of SST and PVALB, the nociceptor-specific voltage-gated sodium channels Nav1.7 (SCN9A) and Nav1.8 (SCN10A), and the unique expression pattern of cytokine receptors such as IL31RA [49, 50]. Thus, 11 different clusters with unique gene expression signatures were associated with mechano-heat sensory neurons, which are predominantly distinguished by the expression of the neuropeptide galanin (GAL), claudin-9 (CLDN9), zinc finger CCHC-type containing 12 (ZCCHC12), lysophosphatidic acid receptor 2 (LPAR2), TRPM8 or TRPA1, and mechanoceptive markers such as PIEZO2, KCNK2, as well as MRGPRB4.
In contrast to the extensive literature on rodent nociceptors, to date only two scRNA transcriptomic studies address human DRG (hDRG) neurons [51, 52]. In general, expression signatures and cell type clusters appear badly conserved between rodents, nonhuman primates, and humans, and several neuronal subpopulations were identified (Fig. 1A, [47, 52]). For example, the most abundant marker NEFH used to distinguish rodent small diameter nociceptors from larger diameter sensory neurons is gradually expressed in the majority of hDRG sensory neurons and cannot be used as a reliable discriminator in human DRGs [22, 51, 53, 54]. To overcome this hurdle, recent advances in the field of spatial transcriptomics allow for a more precise correlation of cell soma sizes with RNA expression signatures, thereby efficiently separating small from large-diameter sensory neurons [52]. Cell clustering and marker expression suggest that the majority of hDRG sensory neurons adopt a peptidergic phenotype, and non-peptidergic human DRG neurons expressing the oncostatin M receptor (OSMR) and SST are implicated in the perception of itch rather than pain [51]. Five nociceptor-related neuron types can be distinguished from non-nociceptive neurons based on the conserved expression of TrkA and Nav1.8 [51]. Human nociceptors responding to different modalities such as cold or heat show specific expression of the marker molecules TRPA1, proenkephalin (PENK), TRPM8, the alpha 3 subunit of nicotinic acetylcholine receptor (CHRNA3), or TrkC+ nociceptors. Species differences were especially evident for putative silent nociceptors that are usually activated following inflammation [52].

A Tree view of hDRG sensory neuron subpopulations, derived from Tavares-Ferreira et al. [52]. B Mapping of iPSC-derived sensory neuron equivalents to hDRG single-nuclei data derived from Nguyens (2021)
Human-derived sensory neuron differentiation
Since acquisition of human sensory neuron tissue is challenging and mostly relies on postmortem material from elderly patients, research started to adopt recent advances in induced pluripotent stem cell (iPSC) models. Human iPSCs are generated from somatic cells such as fibroblasts obtained from skin biopsies or blood samples [55,56,57]. Since their discovery, the Yamanaka factors Oct4, SOX2, c-Myc, and KLF4/NANOG are used to initialize the reprogramming of somatic cells into iPSCs that exhibit pluripotent properties [56]. This was the initial kickoff to establish and develop differentiation protocols that enable the differentiation of iPSCs into virtually every cell type, such as cardiomyocytes, immune cells, as well as neurons [58, 59].
In contrast to cortical neurons, differentiation protocols for sensory neurons were established relatively late [60,61,62,63]. Induced sensory neurons are established via intermediate neural crest cells, which express SOX9, SNAIL, and dHAND [64, 65]. Subsequent studies extended these protocols by selectively blocking activin/TGF-β and BMP signaling, also called dual-SMAD inhibition, and cells that express markers comparable to mouse neural crest cells (which can differentiate into sensory neuron-like cells), such as the SRY-box transcription factor 9/10 (SOX9, SOX10) and the paired box 3 gene (PAX3), which are established efficiently [60, 66]. In 2012, Chambers et al. identified a set of three small molecule inhibitors (CHIR99021, SU5402, and DAPT,called 3i for “three inhibitors”) that efficiently induce sensory neuron-like cell differentiation following dual-SMAD inhibition [61]. While the administration of CHIR99021 is indispensable for the differentiation process, SU5402 and DAPT enhance the efficiency and the speed of the differentiation process [61]. The resulting “mature” differentiated cell types are positive for POU domain class 4 transcription factor 1 (POU4F1, BRN3A) and islet-1 (ISL1) and exhibit neuronal morphology indicated by neurite outgrowth and synapse formation [67, 68]. In addition, TrkA and neurogenin-1 (NEUROG1) suggest peripheral neuronal induction and expression of Nav1.7, Nav1.8, and TRPV1 specialization into nociceptor-like sensory neurons at D12 [61, 68]. Maturation of neurons is induced by co-administration of the nerve growth factor (NGF), the glial cell–derived neurotrophic factor (GDNF), BDNF, and NT-3. These more mature iPSC-derived sensory neurons fire single as well as trains of action potentials and are immune-positive for the peptide hormone precursors TAC1 (substance P, neurokinin A, neuropeptide K, and neuropeptide gamma) and CALCA (CGRP, calcitonin, and katacalcin), indicating that they adopt a peptidergic sensory neuron phenotype [61, 68, 69].
Although this protocol is considered the “gold standard” for iPSC-derived sensory neuron and nociceptor differentiation, several studies introduced modifications by inhibiting 3i until day 12 instead of day 10 of differentiation, replating cells before passaging, neglecting NT-3 as growth factor, adding ascorbic acid, changing media gradients, or adding proliferation inhibitors (such as AraC or MitoC), which overall can lead to an increased efficiency of differentiation [69,70,71]. Different timings of 3i inhibition, changes in growth factor administration (NGF, BDNF, GDNF, and NT-3), and addition of BMP-4 or cAMP produce different subtypes of sensory neurons such as proprioceptors, mechanoreceptors, as well as pruriceptors [72,73,74] (for an overview of the available differentiation protocols, see Table 1). Neuronal induction media composed of DMEM, N2, NEAA, and heparin, followed by administration of retinoic acid and BMP4, successfully induce TrkA+ nociceptors from human embryonic stem cells [75]. However, the effect on the cell phenotype of these adjustments has not yet been sufficiently resolved.
In addition, alternative approaches are emerging such as trans-differentiation to generate TrkA+/CGRP+ peptidergic sensory neurons from human and mouse embryonic fibroblasts overexpressing a fusion construct of BRN3A with neurgenin-1/2 [76] or to differentiate human iPSCs into mechanoreceptors [77]. A chemically defined differentiation strategy followed by immunopanning invokes differentiation into functional nociceptors, proprioceptors, and mechanoceptors simultaneously [78]. Even iPSC-derived DRG organoids can be generated with iPSCs differentiating into sensory nociceptors, mechanoreceptors, and proprioceptors [79].
Mapping iDN phenotypes to the landscape of human DRGs
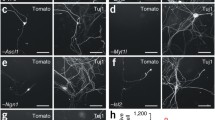
Although iPSC-derived sensory neurons are increasingly used as a model system, phenotypic characterization of the retrieved cells and comparison to human sensory neurons are only insufficiently explored. Recently published hDRG scRNA datasets offer the opportunity to phenotype the iPSC-derived neurons against expression data derived from human DRG sensory neuron subpopulations (Fig. 1A, [52]). Mapping signatures of marker genes such as neuropeptides (CGRP, TAC1, SST), voltage-gated sodium channels (Nav1.7, Nav1.8), TRP channels (TRPV1, TRPM8, TRPA1), and transcription factors (DRGX, MAF) suggest that most iPSC-derived nociceptor-like neurons represent an immature state of peptidergic nociceptors that express substance P, Nav1.8, TRPV1, NEFH, and peripherin but also the P2X-purinoreceptor 3, which is a common marker for non-peptidergic sensory neurons (Fig. 1, [22, 67, 69]). In addition, a trajectory analysis of all relevant and enriched human transcription factors (DRGX, PIRT, BRN3A, and TLX3) reveals that most of these factors are also enriched in iPSC-derived sensory neurons and abundant in human peptidergic sensory neurons. Furthermore, iPSC-derived nociceptor-like neurons transcriptionally approach to hDRG throughout differentiation [69]. Likewise, iPSC-derived low-threshold mechanoreceptors (LTMRs), proprioceptors, as well as pruriceptors can be mapped to their human DRG orthologs, due to the abundant expression of TrkC and TrkB. While proprioceptors show strong expression of TrkC, mechanoreceptors are either positive for TrkC, TrkB, parvalbumin, and the transcription factor short stature homeobox 2 (SHOX2) [72]. iPSC-derived pruriceptor expression of itch-related molecules such as IL31R, IL-4R, and the histamine receptor HRH1 is highly correlated with non-peptidergic itch-related human DRG neurons [74].
In general, it is difficult to estimate the affiliation of different iPSC-derived sensory neurons to specific sensory neuron clusters, since these cells represent an immature developmental state in contrast to mature human DRG sensory neurons and only a minor fraction of nociceptor-like neurons are truly mature [80] [79]. Thus, there is a considerable need for protocols to improve the maturity state of iPSC-derived sensory neurons, as already shown for cortical neurons [81]. This further suggests the necessity for more comparative studies between hDRGs and iPSC-derived sensory neurons, to elucidate the actual maturity state of the derived neurons as well as meta-analyses and deconvolution studies to compare the different iPSC-derived sensory neuron subtypes and identify differences as well as similarities with hDRG sensory neurons.
Modeling hereditary disorders affecting nociceptors
With the increase of routinely performed genomic diagnostic testing of patients, our knowledge about hereditary disorders affecting nociceptors has enormously increased in the last decades. Not only have different hereditary sensory neuropathies been described based on the particular clinical symptom presentation, but also their genetic predisposition has been revealed for the majority of them [82]. Likewise, familial hemiplegic subtypes of migraine – a common primary headache disorder that presents with moderate to severe unilateral pain attacks with several typical variants with or without aura – are associated with specific disease-associated mutations in the voltage-gated calcium channel CACNA1A (FHM1), the sodium/potassium-transporting ATPase subunit ATP1A2 (FHM2), and the voltage-gated sodium channel SCN1A (FHM3) genes [83]. This knowledge about disease-causing mutations in the affected genes opens new possibilities to investigate the underlying pathogenetic mechanisms and to generate personalized treatment strategies based on patient-derived iPSC models (Table 2).
Currently, nine different subtypes of hereditary sensory neuropathies (HSN; if the autonomic nervous system is involved: HSAN; 1–9) are listed in the National Center for Biotechnology Information (NCBI) database MedGen for human medical genetics (NCBI 2022; [84]. They are generally classified based on age of onset, clinical features, type of inheritance, and genetic background [82, 85]. To date, iPSC-derived nociceptor models offer particularly promising opportunities to explore personalized pharmacological treatments for those HSNs with a hypernormal pain phenotype such as gain-of-function mutations in the SCN9A gene, coding for the DRG, and sympathetic neuron-specific voltage-gated sodium channel Nav1.7 [86, 87].
Primary erythromelalgia patients suffer from unbearable pain episodes and redness, mainly in the distal extremities, impaired distal temperature sensation, and itching. This is caused by a gain-of-function mutation of SCN9A [88]. Erythromelalgia patient–derived nociceptors have recently been generated that exhibit decreased firing thresholds together with a hyperpolarizing shift of Nav activation as compared to control neurons [70]. This model system will be helpful for further investigations of the disease-causing mechanisms as well as for the screening of potential treatment-effective compounds [89].
Paroxysmal extreme pain disorder (PEPD) is similar to erythromelalgia also caused by a gain-of-function mutation of SCN9A. Symptoms include skin redness and flushing as well as severe pain attacks in different parts of the body [90]. To date, no iPSC models for PEPD-specific SCN9A mutations are available.
Familial hemiplegic migraine 1 (FHM1) is in the majority of cases caused by a missense mutation of the CACNA1A gene that codes for the pore-forming subunit of the neuronal voltage-gated calcium channel Cav2.1, leading to gain-of-function effects that result in hyperexcitability [83, 91]. No studies on iPSC-derived neurons from FHM1 patients have been performed yet. Nevertheless, a number iPSC model systems derived from a different patient group of spinocerebellar ataxia 6, which is based on loss-of-function mutations in the CACNA1A gene, were generated and in part functionally investigated [92,93,94, 94, 95, 95].
Familial hemiplegic migraine 2 (FHM2) has been associated with mutations in the ATP1A2 gene that encodes a catalytic subunit of the Na+/K+-ATPase ion transport pump [83]. It is therefore generally important for the regulation of electrochemical gradients across cell membranes but specifically relevant for the function of excitable cells, including neurons. No functional investigations of patient-derived iPSCs carrying ATP1A2 mutations are available to date.
Familial hemiplegic migraine 3 (FHM3) is caused by mutations in the voltage-gated sodium channel gene SCN1A coding for Nav1.1. This ion channel is important for the sodium ion permeability of excitable membranes and was shown to be mainly expressed in inhibitory GABAergic interneurons [83]. Due to this prominent role in the modulation of network excitability, Nav1.1 is also highly associated with different epilepsy syndromes. And while in epilepsies, the associated mutations mainly lead to loss-of-function effects provoking seizure activity, FHM3 mutations usually cause gain-of-function effects. Therefore, functional investigation of the plethora of iPSC-derived model systems (e.g., [96,97,98]) from epilepsy patients can only partly be used to decipher the mechanisms behind migraine pain.
Migraine with or without aura 13 (MGR13) has been directly linked to a specific gene, the potassium channel KCNK18. This gene codes for the two-pore potassium channel TRESK and is important for regulating the excitability of neurons in the pain pathway [83]. Pettingill et al. [71] could show in nociceptors differentiated from patient-derived iPSCs that a CRISPR-Cas9–mediated repair of a TRESK frameshift mutation normalized neuronal excitability. This and a few other studies point toward exciting novel possibilities for gene therapeutic treatments of neuropathies associated with nociceptor dysfunction, which are currently still in their infancy.
Modeling complex pathologies
Healthy nociceptors serve important roles as an important alarm system and in host defense by sensing physical and chemical stimuli including noxious heat, cold, pressure, and danger signals [99]. Nociceptive transduction, transformation, or transmission may be affected by monogenetic pathologies, and these can be sufficiently modeled in nociceptors derived from patient iPSC clones [70, 86, 100, 101]. However, the pathogenesis of acquired pain disorders such as painful arthritis, complex regional pain syndrome (CRPS), or postherpetic neuralgia is more complex, and numerous studies have reported the contribution of immune and glia cells to nociceptor dysfunction in preclinical models of pathological pain. For example, inflammatory pain is characterized by immune processes at the nociceptor nerve terminal, which liberates a multitude of different pro-inflammatory mediators [102]. Although the different nociceptor subtypes and even more each individual nociceptor expresses only a certain subset of membrane receptors and ion channels, the nociceptor population as a whole possesses the full equipment to sense these mediators and react to their presence with the discharge of action potentials or by lowering their activation thresholds for physical stimuli [14, 48, 103]. Since Elspeth McLachlan and her team first published on immune cells invading the space between sensory neuron cell bodies and the covering satellite cell layer in the DRG as a consequence of peripheral nerve injury, neuroimmune processes have emerged as critical components for inflammatory and even neuropathic pain disorders [102, 104, 105]. Neuroimmune processes are active following peripheral nerve injuries, and the neuroimmune triad contributes to the generation of neuropathic pain at the lesion site but also at non-injured parts of the axon and even the DRG or the central process connecting the nociceptor to spinal projection neurons [104, 106,107,108]. Modeling the complexity of the nociceptor neuron in 3D organoid models employing human iPSC-derived nociceptors therefore needs to include relevant components reflecting its healthy or diseased environment by co-culturing with relevant interactors at the peripheral terminal, the axon, DRG, as well as the central terminal in the spinal cord such as keratinocytes, macrophages, glia cells, and other types of immune cells (Fig. 2).

Nociceptors and associated cells in the target tissue (skin, left), DRG, and the spinal cord (right) to be implemented in complex model systems (generated with BioRender®)
Skin and other target organs
Starting from a 3D platform of human iPSCs-derived nociceptors for peripheral nerve modeling and tissue innervation with heterologous Schwann cells, organoid models engineered from human cells only are now becoming available, which employ sensory neurons and Schwann cells differentiated in parallel even from the same iPSC donor together with engineered skin [109,110,111]. In the skin, specific non-neuronal cell types are crucial for nociceptor function such as keratinocytes releasing ATP [111,112,113,114] and enterochromaffin cells closely collaborating with nociceptive primary afferents in the gut by the exchange of chemical signals for chemosensation [115]. Within target tissues, the communication between target cells and nociceptors is bilateral whereby nociceptors sense danger signals on the one hand and regulate immunity on the other for example in the lung as well as in the skin. This immunomodulation is mediated by different types of afferents through the release of glutamate or the neuropeptide CGRP [10, 116].
Schwann cells and satellite cells
Bidirectional signaling between axons and the peripheral nerve glia Schwann cells is essential for both the development and maintenance of sensory neuron morphology and function. However, primary human Schwann cells are challenging as a resource for nerve tissue engineering (for a review, see [117]). Co-cultures of iPSC-derived sensory neurons with myelinating Schwann cells were first developed to explore mechanisms of demyelination and neurodegeneration underlying respective disorders such as Guillain–Barre-Syndrome and remyelination therapies [118,119,120]. First, human iPSC–derived sensory neurons were cultured with rat Schwann cells and produced long-term and stable myelinating co-cultures, which developed specialized domains of axonal interaction with myelinating Schwann cells, such as clustered voltage-gated sodium channels at the node of Ranvier and Shaker-type potassium channels (Kv1.2) at the juxtaparanode. Several regulators of myelination were explored in these models such as BACE1 or NRN1 and may be relevant for the function of myelinated nociceptors of the A-fiber type or even nonmyelinating Schwann cells covering C-fiber nociceptive axons [120, 121]. However, specific models for directed iPSC differentiation into human nonmyelinating Schwann cells and co-cultivation with human nociceptive neurons are currently unavailable and modeling the role of Schwann cells for nociceptor function has even received less attention.
Immune cells (macrophages, lymphocytes)
Mutual neuroimmune cross talk thus appears to be important for maintaining tissues such as skin or the gut in a healthy state. Nociceptors are important regulators of resident immune cells in the skin by balancing their activity through the release of neuropeptides and glutamate [16]. However, a disbalance of pro- and anti-inflammatory components characterizes disorders leading to inflammatory or neuropathic pain with respective alterations of nociceptor functions. In particular, neuropathic pain shows relevant features of a neuroimmune disorder and involves not only neuronal components, but also Schwann cells and satellite cells, different cell types of the peripheral immune system at the lesion site or in the DRG, as well as spinal microglia and astrocytes (for a review, see [122]). More recently, more painful disorders with neuroimmune pathologies are emerging. For example, fibromyalgia, which has been enigmatic until very recently, has a striking autoimmune component and metabolic disturbances that can be explored in human model systems [123, 124]. Co-cultivation of iPSC-derived nociceptors with relevant immune cells of the same donor may help to model the causative changes on a more personalized basis for monogenetic disorders or even more complex pathologies.
Concluding remarks
Human nociceptors are different from rodents such as they show specific signatures of gene expression and neuropeptides that are typically found in the majority of native human nociceptors such as CGRP or substance P [110, 125]. These differences are partially preserved in nociceptive neurons derived from human iPSCs [14, 52, 69], and these can be exploited for human-centered mechanistic studies and patient-targeted drug discovery in particular for monogenetic pain disorders, which, however, provide benefit only for a minor subgroup of chronic pain patients.
Furthermore, complex 3D systems model human nociceptors in a more precise manner and their natural environment. Based on these considerations and in light of the individual genetic background, all relevant cell types within the 3D model are to be obtained from the same donor. This approach precisely constructs a platform to explore individual nociceptive processes, and the new organoid models offer intriguing possibilities to study the pathophysiology of nociceptors together with their environment and further develop them into screening assays for novel analgesic pharmacies and even personalized treatments.
However, such organoid models are less easily accessible to controlled read-out systems due to their increased complexity and 3D structure. Neuropeptide release evoked by depolarizing concentrations of KCl or irritants such as capsaicin can serve as a quantitative read-out measure of nociceptor excitation [110, 126]. In addition, individual iPSC-derived cell types are accessible to genetic modifications using CRISPR/Cas9 or introduction of Cre recombinase under respective driver constructs. Optogenetic tools as well as reporter dyes can be introduced into the individual cellular components before implementing them in the engineered organoid. Recent developments of specifically designed multi-electrode array chips for 3D organoids will help to overcome the challenges [127].
Overall, specifically tailored co-cultivation of all relevant cell types as an engineered model for particular disorders such as neuropathic pain, fibromyalgia, or even inflammatory bowel disease offers major advantages for a better understanding of mutual interaction of the involved cells with nociceptors and can be exploited for analgesic drug discovery purposes [89]. Modeling human nociception, in particular by reprogramming patient-derived iPSCs into complex organoids involving all relevant cell types, thus rises great expectations to overcome the translational gap between preclinical research and unmet clinical needs for effective analgesic pharmaceuticals.
Data availability
Not applicable.
Change history
04 September 2022
Missing Open Access funding information has been added in the Funding Note.
References
Casey KL (1982) Neural mechanisms of pain: an overview. Acta Anaesthesiol Scand Suppl 74:13–20
Zotterman Y (1939) Touch, pain and tickling: an electro-physiological investigation on cutaneous sensory nerves. J Physiol 95:1–28
Iggo A (1964) Temperature Discrimination in the Skin. Nature 204:481–483
Hagbarth KE, Vallbo AB (1968) Discharge characteristics of human muscle afferents during muscle stretch and contraction. Exp Neurol 22:674–694
Sherrington, Charles Scott. 1906. The integrative action of the nervous system (C. Scribner’s sons: New York,).
Dubin AE, Patapoutian A (2010) Nociceptors: the sensors of the pain pathway. J Clin Invest 120:3760–3772
Hucho T, Levine JD (2007) Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 55:365–376
McMahon SB, Koltzenburg M (1990) Novel classes of nociceptors: beyond Sherrington. Trends Neurosci 13:199–201
Rang HP, Bevan S, Dray A (1991) Chemical activation of nociceptive peripheral neurones. Br Med Bull 47:534–548
Zhang S, Sumpter TL, Kaplan DH (2022) NeuronMast cell cross-talk in the skin. J Invest Dermatol 142:841–848
Chapman LF (1977) Mechanisms of the flare reaction in human skin. J Invest Dermatol 69:88–97
Janowitz H, Grossman MI (1949) Blocking action of tetraethylammonium on axon reflexes in the human skin. Science 109:16
Langley JN (1900) On axon-reflexes in the pre-ganglionic fibres of the sympathetic system. J Physiol 25:364–398
Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P (2015) Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153
Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, Albers KM, Kaplan DH (2019) Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 178(919–32):e14
Zhang S, Edwards TN, Chaudhri VK, Wu J, Cohen JA, Hirai T, Rittenhouse N, Schmitz EG, Zhou PY, McNeil BD, Yang Y, Koerber HR, Sumpter TL, Poholek AC, Davis BM, Albers KM, Singh H, Kaplan DH (2021) Nonpeptidergic neurons suppress mast cells via glutamate to maintain skin homeostasis. Cell 184(2151–66):e16
Chrysostomidou L, Cooper AH, Weir GA (2021) Cellular models of pain: new technologies and their potential to progress preclinical research. Neurobiol Pain 10:100063
North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, Johansson CA, Zhang H, Kim YH, Zhang B, Dussor G, Kim TH, Price TJ, Dougherty PM (2019) Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142:1215–1226
Rabert D, Xiao Y, Yiangou Y, Kreder D, Sangameswaran L, Segal MR, Hunt CA, Birch R, Anand P (2004) Plasticity of gene expression in injured human dorsal root ganglia revealed by GeneChip oligonucleotide microarrays. J Clin Neurosci 11:289–299
Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM, Hunter JC (1997) A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem 272:14805–14809
Shiers SI, Sankaranarayanan I, Jeevakumar V, Cervantes A, Reese JC, Price TJ (2021) Convergence of peptidergic and non-peptidergic protein markers in the human dorsal root ganglion and spinal dorsal horn. J Comp Neurol 529:2771–2788
Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, Lam T, Kim JY, Kim TH, Zhang MQ, Dussor G, Price TJ (2018) Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 159:1325–1345
Yezierski RP, Hansson P (2018) Inflammatory and neuropathic pain from bench to bedside: what went wrong? J Pain 19:571–588
Wangzhou A, McIlvried LA, Paige C, Barragan-Iglesias P, Shiers S, Ahmad A, Guzman CA, Dussor G, Ray PR, thGereau RW, Price TJ (2020) Pharmacological target-focused transcriptomic analysis of native vs cultured human and mouse dorsal root ganglia. Pain 161:1497–1517
Li CL, Li KC, Wu D, Chen Y, Luo H, Zhao JR, Wang SS, Sun MM, Lu YJ, Zhong YQ, Hu XY, Hou R, Zhou BB, Bao L, Xiao LS, Zhang X (2016) Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 26:967
Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI (2003) Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci 23:10988–10997
Torsney C, Anderson RL, Ryce-Paul KA, MacDermott AB (2006) Characterization of sensory neuron subpopulations selectively expressing green fluorescent protein in phosphodiesterase 1C BAC transgenic mice. Mol Pain 2:17
Ibanez CF, Ernfors P (2007) Hierarchical control of sensory neuron development by neurotrophic factors. Neuron 54:673–675
Ma Q, Fode C, Guillemot F, Anderson DJ (1999) Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev 13:1717–1728
Cesare P, McNaughton P (1996) A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci U S A 93:15435–15439
Fukuoka T, Noguchi K (2011) Comparative study of voltage-gated sodium channel alpha-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci Res 70:164–171
Kirschstein T, Busselberg D, Treede RD (1997) Coexpression of heat-evoked and capsaicin-evoked inward currents in acutely dissociated rat dorsal root ganglion neurons. Neurosci Lett 231:33–36
Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL (2009) TRPA1 modulates mechanotransduction in cutaneous sensory neurons. J Neurosci 29:4808–4819
Mickle AD, Shepherd AJ, Mohapatra DP (2015) Sensory TRP channels: the key transducers of nociception and pain. Prog Mol Biol Transl Sci 131:73–118
Nagy I, Rang H (1999) Noxious heat activates all capsaicin-sensitive and also a sub-population of capsaicin-insensitive dorsal root ganglion neurons. Neuroscience 88:995–997
Rush AM, Cummins TR, Waxman SG (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579:1–14
Sousa-Valente J, Andreou AP, Urban L, Nagy I (2014) Transient receptor potential ion channels in primary sensory neurons as targets for novel analgesics. Br J Pharmacol 171:2508–2527
Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321:702–705
Cavanaugh DJ, Chesler AT, Braz JM, Shah NM, Julius D, Basbaum AI (2011) Restriction of transient receptor potential vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J Neurosci 31:10119–10127
Mathivanan S, Devesa I, Changeux JP, Ferrer-Montiel A (2016) Bradykinin induces TRPV1 exocytotic recruitment in peptidergic nociceptors. Front Pharmacol 7:178
Pinto LG, Souza GR, Kusuda R, Lopes AH, Sant’Anna MB, Cunha FQ, Ferreira SH, Cunha TM (2019) Non-peptidergic nociceptive neurons are essential for mechanical inflammatory hypersensitivity in mice. Mol Neurobiol 56:5715–5728
Zhang J, Cavanaugh DJ, Nemenov MI, Basbaum AI (2013) The modality-specific contribution of peptidergic and non-peptidergic nociceptors is manifest at the level of dorsal horn nociresponsive neurons. J Physiol 591:1097–1110
Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, Mathur J, Begay V, Coste B, Mainquist J, Wilson AJ, Francisco AG, Reddy K, Qiu Z, Wood JN, Lewin GR, Patapoutian A (2014) Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516:121–125
Roudaut Y, Lonigro A, Coste B, Hao J, Delmas P, Crest M (2012) Touch sense: functional organization and molecular determinants of mechanosensitive receptors. Channels (Austin) 6:234–245
Faure L, Wang Y, Kastriti ME, Fontanet P, Cheung KKY, Petitpre C, Wu H, Sun LL, Runge K, Croci L, Landy MA, Lai HC, Consalez GG, de Chevigny A, Lallemend F, Adameyko I, Hadjab S (2020) Single cell RNA sequencing identifies early diversity of sensory neurons forming via bi-potential intermediates. Nat Commun 11:4175
Landy MA, Goyal M, Casey KM, Liu C, Lai HC (2021) Loss of Prdm12 during development, but not in mature nociceptors, causes defects in pain sensation. Cell Rep 34:108913
Kupari J, Usoskin D, Parisien M, Lou D, Hu Y, Fatt M, Lonnerberg P, Spangberg M, Eriksson B, Barkas N, Kharchenko PV, Lore K, Khoury S, Diatchenko L, Ernfors P (2021) Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat Commun 12:1510
Li CL, Li KC, Wu D, Chen Y, Luo H, Zhao JR, Wang SS, Sun MM, Lu YJ, Zhong YQ, Hu XY, Hou R, Zhou BB, Bao L, Xiao HS, Zhang X (2016) Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 26:83–102
Wang K, Wang S, Chen Y, Wu D, Hu X, Lu Y, Wang L, Bao L, Li C, Zhang X (2021) Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain. Cell Res 31:904–918
Hu G, Huang K, Hu Y, Du G, Xue Z, Zhu X, Fan G (2016) Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci Rep 6:31851
Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, Davidson S (2021) Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 10:e71752. https://doi.org/10.7554/eLife.71752
Tavares-Ferreira D, Shiers S, Ray PR, Wangzhou A, Jeevakumar V, Sankaranarayanan I, Cervantes AM, Reese JC, Chamessian A, Copits BA, Dougherty PM, thGereau RW, Burton MD, Dussor G, Price TJ (2022) Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 14:eabj8186
Lopes DM, Denk F, McMahon SB (2017) The molecular fingerprint of dorsal root and trigeminal ganglion neurons. Front Mol Neurosci 10:304
Middleton SJ, Barry AM, Comini M, Li Y, Ray PR, Shiers S, Themistocleous AC, Uhelski ML, Yang X, Dougherty PM, Price TJ, Bennett DL (2021) Studying human nociceptors: from fundamentals to clinic. Brain 144:1312–1335
Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R (2010) Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 7:20–24
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Karakikes I, Ameen M, Termglinchan V, Wu JC (2015) Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res 117:80–88
Nianias A, Themeli M (2019) Induced pluripotent stem cell (iPSC)-derived lymphocytes for adoptive cell immunotherapy: recent advances and challenges. Curr Hematol Malig Rep 14:261–268
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280
Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, Shi SH, Studer L (2012) Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30:715–720
Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhaeghen P (2008) An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455:351–357
Johnson MA, Weick JP, Pearce RA, Zhang SC (2007) Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci 27:3069–3077
Lee G, Chambers SM, Tomishima MJ, Studer L (2010) Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc 5:688–701
Pomp O, Brokhman I, Ben-Dor I, Reubinoff B, Goldstein RS (2005) Generation of peripheral sensory and sympathetic neurons and neural crest cells from human embryonic stem cells. Stem Cells 23:923–930
Milet C, Monsoro-Burq AH (2012) Embryonic stem cell strategies to explore neural crest development in human embryos. Dev Biol 366:96–99
Schoepf CL, Zeidler M, Spiecker L, Kern G, Lechner J, Kummer KK, Kress M (2020) Selected ionotropic receptors and voltage-gated ion channels: more functional competence for human induced pluripotent stem cell (iPSC)-derived nociceptors. Brain Sci 10(6):344
Young GT, Gutteridge A, Fox H, Wilbrey AL, Cao L, Cho LT, Brown AR, Benn CL, Kammonen LR, Friedman JH, Bictash M, Whiting P, Bilsland JG, Stevens EB (2014) Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol Ther 22:1530–1543
Zeidler M, Kummer KK, Schopf CL, Kalpachidou T, Kern G, Cader MZ, Kress M (2021) NOCICEPTRA: gene and microRNA signatures and their trajectories characterizing human ipsc-derived nociceptor maturation. Adv Sci (Weinh) 8:e2102354
Meents JE, Bressan E, Sontag S, Foerster A, Hautvast P, Rosseler C, Hampl M, Schuler H, Goetzke R, Le TKC, Kleggetveit IP, Le Cann K, Kerth C, Rush AM, Rogers M, Kohl Z, Schmelz M, Wagner W, Jorum E, Namer B, Winner B, Zenke M, Lampert A (2019) The role of Nav1.7 in human nociceptors: insights from human induced pluripotent stem cell-derived sensory neurons of erythromelalgia patients. Pain 160:1327–1341
Pettingill P, Weir GA, Wei T, Wu Y, Flower G, Lalic T, Handel A, Duggal G, Chintawar S, Cheung J, Arunasalam K, Couper E, Haupt LM, Griffiths LR, Bassett A, Cowley SA, Cader MZ (2019) A causal role for TRESK loss of function in migraine mechanisms. Brain 142:3852–3867
Dionisi C, Rai M, Chazalon M, Schiffmann SN, Pandolfo M (2020) Primary proprioceptive neurons from human induced pluripotent stem cells: a cell model for afferent ataxias. Sci Rep 10:7752
Guimaraes MZP, De Vecchi R, Vitoria G, Sochacki JK, Paulsen BS, Lima I, Rodrigues da Silva F, da Costa RFM, Castro NG, Breton L, Rehen SK (2018) Generation of iPSC-derived human peripheral sensory neurons releasing substance P elicited by TRPV1 agonists. Front Mol Neurosci 11:277
Umehara Y, Toyama S, Tominaga M, Matsuda H, Takahashi N, Kamata Y, Niyonsaba F, Ogawa H, Takamori K (2020) Robust induction of neural crest cells to derive peripheral sensory neurons from human induced pluripotent stem cells. Sci Rep 10:4360
Boisvert EM, Engle SJ, Hallowell SE, Liu P, Wang ZW, Li XJ (2015) The specification and maturation of nociceptive neurons from human embryonic stem cells. Sci Rep 5:16821
Blanchard JW, Eade KT, Szucs A, Lo Sardo V, Tsunemoto RK, Williams D, Sanna PP, Baldwin KK (2015) Selective conversion of fibroblasts into peripheral sensory neurons. Nat Neurosci 18:25–35
Nickolls AR, Lee MM, Espinoza DF, Szczot M, Lam RM, Wang Q, Beers J, Zou J, Nguyen MQ, Solinski HJ, AlJanahi AA, Johnson KR, Ward ME, Chesler AT, Bonnemann CG (2020) Transcriptional programming of human mechanosensory neuron subtypes from pluripotent stem cells. Cell Rep 30(932–46):e7
Saito-Diaz K, Street JR, Ulrichs H, Zeltner N (2021) Derivation of peripheral nociceptive, mechanoreceptive, and proprioceptive sensory neurons from the same culture of human pluripotent stem cells. Stem Cell Reports 16:446–457
Mazzara PG, Muggeo S, Luoni M, Massimino L, Zaghi M, Valverde PT, Brusco S, Marzi MJ, Palma C, Colasante G, Iannielli A, Paulis M, Cordiglieri C, Giannelli SG, Podini P, Gellera C, Taroni F, Nicassio F, Rasponi M, Broccoli V (2020) Frataxin gene editing rescues Friedreich’s ataxia pathology in dorsal root ganglia organoid-derived sensory neurons. Nat Commun 11:4178
Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, Patel M, Goncalves A, Ferreira R, Benn CL, Wilbrey A, Bictash M, Impey E, Cao L, Lainez S, Loucif AJ, Whiting PJ, Consortium HIPSCI, Gutteridge A, Gaffney DJ. 2018. 'Molecular and functional variation in iPSC-derived sensory neurons', Nat Genet, 50: 54-61
Burke EE, Chenoweth JG, Shin JH, Collado-Torres L, Kim SK, Micali N, Wang Y, Colantuoni C, Straub RE, Hoeppner DJ, Chen HY, Sellers A, Shibbani K, Hamersky GR, Diaz Bustamante M, Phan BN, Ulrich WS, Valencia C, Jaishankar A, Price AJ, Rajpurohit A, Semick SA, Burli RW, Barrow JC, Hiler DJ, Page SC, Martinowich K, Hyde TM, Kleinman JE, Berman KF, Apud JA, Cross AJ, Brandon NJ, Weinberger DR, Maher BJ, McKay RDG, Jaffe AE (2020) Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat Commun 11:462
Auer-Grumbach M (2013) Hereditary sensory and autonomic neuropathies. Handb Clin Neurol 115:893–906
Sutherland HG, Albury CL, Griffiths LR (2019) Advances in genetics of migraine. J Headache Pain 20:72
Louden DN (2020) MedGen: NCBI’s portal to information on medical conditions with a genetic component. Med Ref Serv Q 39:183–191
Houlden H, Blake J, Reilly MM (2004) Hereditary sensory neuropathies. Curr Opin Neurol 17:569–577
Alsaloum M, Waxman SG (2022) iPSCs and DRGs: stepping stones to new pain therapies. Trends Mol Med 28:110–122
Foulkes T, Wood JN (2008) Pain genes. PLoS Genet 4:e1000086
Alsaloum M, Labau JIR, Liu S, Effraim P, Waxman SG (2022) Stem cell-derived sensory neurons modelling inherited erythromelalgia: normalization of excitability. Brain Jan 28:awac031
Namer B, Schmidt D, Eberhardt E, Maroni M, Dorfmeister E, Kleggetveit IP, Kaluza L, Meents J, Gerlach A, Lin Z, Winterpacht A, Dragicevic E, Kohl Z, Schuttler J, Kurth I, Warncke T, Jorum E, Winner B, Lampert A (2019) Pain relief in a neuropathy patient by lacosamide: Proof of principle of clinical translation from patient-specific iPS cell-derived nociceptors. EBioMedicine 39:401–408
Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB (2007) Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 69:586–595
Inchauspe CG, Pilati N, Di Guilmi MN, Urbano FJ, Ferrari MD, van den Maagdenberg AM, Forsythe ID, Uchitel OD (2015) Familial hemiplegic migraine type-1 mutated cav2.1 calcium channels alter inhibitory and excitatory synaptic transmission in the lateral superior olive of mice. Hear Res 319:56–68
Bavassano C, Eigentler A, Stanika R, Obermair GJ, Boesch S, Dechant G, Nat R (2017) Bicistronic CACNA1A gene expression in neurons derived from spinocerebellar ataxia type 6 patient-induced pluripotent stem cells. Stem Cells Dev 26:1612–1625
Ishida Y, Kawakami H, Kitajima H, Nishiyama A, Sasai Y, Inoue H, Muguruma K (2016) Vulnerability of Purkinje cells generated from spinocerebellar ataxia type 6 patient-derived iPSCs. Cell Rep 17:1482–1490
Yang J, Liu H, Sun H, Wang Z, Zhang R, Liu Y, Zhang Q, Zhang S, Zhang J, Shi C, Wang Y, Xu Y (2020) Construction of induced pluripotent stem cell line (ZZUi0017-A) from the fibroblast cells of a female patient with CACNA1A mutation by unintegrated reprogramming approach. Stem Cell Res 48:101946
Yang T, Qin J, Zhang Q, Sun H, Wang Z, Yang J, Liu H, Zhang C, Zhang S, Zhang J, Wang Y, Xu Y (2020) Generation of induced pluripotent stem cell line (ZZUi0018-A ) from a patient with spinocerebellar ataxia type 6. Stem Cell Res 44:101777
Kim HW, Quan Z, Kim YB, Cheong E, Kim HD, Cho M, Jang J, Yoo YR, Lee JS, Kim JH, Kim YI, Kim DS, Kang HC (2018) Differential effects on sodium current impairments by distinct SCN1A mutations in GABAergic neurons derived from Dravet syndrome patients. Brain Dev 40:287–298
Scalise S, Scaramuzzino L, Lucchino V, Esposito C, Malatesta P, Grillone K, Perrotti N, Cuda G, Parrotta EI (2020) Generation of iPSC lines from two patients affected by febrile seizure due to inherited missense mutation in SCN1A gene. Stem Cell Res 49:102083
Schuster J, Laan L, Klar J, Jin Z, Huss M, Korol S, Noraddin FH, Sobol M, Birnir B, Dahl N (2019) Transcriptomes of Dravet syndrome iPSC derived GABAergic cells reveal dysregulated pathways for chromatin remodeling and neurodevelopment. Neurobiol Dis 132:104583
Baral P, Udit S, Chiu IM (2019) Pain and immunity: implications for host defence. Nat Rev Immunol 19:433–447
Alsaloum M, Labau JIR, Liu S, Estacion M, Zhao P, Dib-Hajj F, Waxman SG (2021) Contributions of NaV1.8 and NaV1.9 to excitability in human induced pluripotent stem-cell derived somatosensory neurons. Sci Rep 11:24283
McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ, Ebner D, Tracey I, Serra J, Bennett DL (2019) Defining the functional role of NaV1.7 in human nociception. Neuron 101:905–19 e8
Donnelly CR, Chen O, Ji RR (2020) How do sensory neurons sense danger signals? Trends Neurosci 43:822–838
Wangzhou A, Paige C, Ray PR, Dussor G, Price TJ (2021) Diversity of receptor expression in central and peripheral mouse neurons estimated from single cell RNA sequencing. Neuroscience 463:86–96
Hu P, McLachlan EM (2002) Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 112:23–38
Ji RR, Chamessian A, Zhang YQ (2016) Pain regulation by non-neuronal cells and inflammation. Science 354:572–577
Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR (2018) Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 100:1292–1311
Malcangio M (2019) Role of the immune system in neuropathic pain. Scand J Pain 20:33–37
Sideris-Lampretsas G, Malcangio M (2021) Microglial heterogeneity in chronic pain. Brain Behav Immun 96:279–289
Malheiro A, Harichandan A, Bernardi J, Seijas-Gamardo A, Konings GF, Volders PGA, Romano A, Mota C, Wieringa P, Moroni L (2021) 3D culture platform of human iPSCs-derived nociceptors for peripheral nerve modeling and tissue innervation. Biofabrication 14. https://doi.org/10.1088/1758-5090/ac36bf
Muller Q, Beaudet MJ, De Serres-Berard T, Bellenfant S, Flacher V, Berthod F (2018) Development of an innervated tissue-engineered skin with human sensory neurons and Schwann cells differentiated from iPS cells. Acta Biomater 82:93–101
Moehring F, Cowie AM, Menzel AD, Weyer AD, Grzybowski M, Arzua T, Geurts AM, Palygin O, Stucky CL (2018) Keratinocytes mediate innocuous and noxious touch via ATP-P2X4 signaling. Elife 7:e31684
Stucky CL, Mikesell AR (2021) Cutaneous pain in disorders affecting peripheral nerves. Neurosci Lett 765:136233
Sadler KE, Moehring F, Stucky CL (2020) Keratinocytes contribute to normal cold and heat sensation. Elife 9:e58625
Moehring F, Halder P, Seal RP, Stucky CL (2018) Uncovering the cells and circuits of touch in normal and pathological settings. Neuron 100:349–360
Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, Brierley SM, Ingraham HA, Julius D (2017) Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170(185–98):e16
Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, Wei Y, Zhou Y, Kuchroo VK, Burkett PR, Yipp BG, Liberles SD, Chiu IM (2018) Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 24:417–426
Huang Z, Powell R, Phillips JB, Haastert-Talini K (2020) Perspective on Schwann cells derived from induced pluripotent stem cells in peripheral nerve tissue engineering. Cells 9:2497
Cai S, Han L, Ao Q, Chan YS, Shum DK (2017) Human induced pluripotent cell-derived sensory neurons for fate commitment of bone marrow-derived schwann cells: implications for remyelination therapy. Stem Cells Transl Med 6:369–381
Clark AJ (2020) Establishing myelinating cocultures using human iPSC-derived sensory neurons to investigate axonal degeneration and demyelination. Methods Mol Biol 2143:111–129
Clark AJ, Kaller MS, Galino J, Willison HJ, Rinaldi S, Bennett DLH (2017) Co-cultures with stem cell-derived human sensory neurons reveal regulators of peripheral myelination. Brain 140:898–913
Clark AJ, Kugathasan U, Baskozos G, Priestman DA, Fugger N, Lone MA, Othman A, Chu KH, Blesneac I, Wilson ER, Laura M, Kalmar B, Greensmith L, Hornemann T, Platt FM, Reilly MM, Bennett DL (2021) An iPSC model of hereditary sensory neuropathy-1 reveals L-serine-responsive deficits in neuronal ganglioside composition and axoglial interactions. Cell Rep Med 2:100345
Goebel A, Krock E, Gentry C, Israel MR, Jurczak A, Urbina CM, Sandor K, Vastani N, Maurer M, Cuhadar U, Sensi S, Nomura Y, Menezes J, Baharpoor A, Brieskorn L, Sandstrom A, Tour J, Kadetoff D, Haglund L, Kosek E, Bevan S, Svensson CI, Andersson DA (2021) ’Passive transfer of fibromyalgia symptoms from patients to mice2021 Passive transfer of fibromyalgia symptoms from patients to mice. J Clin Invest 131:e144201
Monzon-Nomdedeu MB, Morten KJ, Oltra E (2021) Induced pluripotent stem cells as suitable sensors for fibromyalgia and myalgic encephalomyelitis/chronic fatigue syndrome. World J Stem Cells 13:1134–1150
Verma V, Drury GL, Parisien M, Ozdag AN Acarli, Al-Aubodah TA, Nijnik A, Wen X, Tugarinov N, Verner M, Klares R, 3rd, Linton A, Krock E, Morado Urbina CE, Winsvold B, Fritsche LG, Fors EA, Piccirillo C, Khoutorsky A, Svensson CI, Fitzcharles MA, Ingelmo PM, Bernard NF, Dupuy FP, Uceyler N, Sommer C, King IL, Meloto CB, Diatchenko L, UNT-All In Pain H (2021) Unbiased immune profiling reveals a natural killer cell-peripheral nerve axis in fibromyalgia. Pain. https://doi.org/10.1097/j.pain.0000000000002498
Davidson S, Copits BA, Zhang J, Page G, Ghetti A, R. W. th Gereau. (2014) Human sensory neurons: Membrane properties and sensitization by inflammatory mediators. Pain 155:1861–1870
Opree A, Kress M (2000) Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 20:6289–6293
Molina-Martinez B, Jentsch LV, Ersoy F, van der Moolen M, Donato S, Ness TV, HeutinkP, Jones PD, Cesare P (2022) A multimodal 3D neuro-microphysiological system with neurite-trapping microelectrodes. Biofabrication 14. https://doi.org/10.1088/1758-5090/ac463b
Zhu PP, Denton KR, Pierson TM, Li XJ, Blackstone C (2014) Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet 23:5638–5648
Jonsson ME, Ludvik Brattas P, Gustafsson C, Petri R, Yudovich D, Pircs K, Verschuere S, Madsen S, Hansson J, Larsson J, Mansson R, Meissner A, Jakobsson J (2019) Activation of neuronal genes via LINE-1 elements upon global DNA demethylation in human neural progenitors. Nat Commun 10:3182
Xiaojing W, Yanyan M, Ruonan D, Xiaolin L, Haiyan Z, Jian M, Yi L, Wenjie S, Qiji L (2020) Generation of a human induced pluripotent stem cell line (SDUBMSi001-A) from a hereditary spastic paraplegia patient carrying kif1a c773C>T missense mutation. Stem Cell Res 43:101727
Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, Sakiyama Y, Yoshimura A, Izumo S, Takashima H (2013) Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology 80:1641–1649
Dor L, Rabinski T, Zlotnik D, Shilian M, Weil M, Vatine GD (2021) Induced pluripotent stem cell (iPSC) lines from two individuals carrying a homozygous (BGUi007-A) and a heterozygous (BGUi006-A) mutation in ELP1 for in vitro modeling of familial dysautonomia. Stem Cell Res 55:102495
Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461:402–406
Lee G, Ramirez CN, Kim H, Zeltner N, Liu B, Radu C, Bhinder B, Kim YJ, Choi IY, Mukherjee-Clavin B, Djaballah H, Studer L (2012) Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol 30:1244–1248
Zeltner N, Fattahi F, Dubois NC, Saurat N, Lafaille F, Shang L, Zimmer B, Tchieu J, Soliman MA, Lee G, Casanova JL, Studer L (2016) Capturing the biology of disease severity in a PSC-based model of familial dysautonomia. Nat Med 22:1421–1427
Nakajima K, Miranda A, Craig DW, Shekhtman T, Kmoch S, Bleyer A, Szelinger S, Kato T, Kelsoe JR (2020) Ntrk1 mutation co-segregating with bipolar disorder and inherited kidney disease in a multiplex family causes defects in neuronal growth and depression-like behavior in mice. Transl Psychiatry 10:407
Klein T, Henkel L, Klug K, Kwok CK, Klopocki E, Uceyler N (2018) Generation of the human induced pluripotent stem cell line UKWNLi002-A from dermal fibroblasts of a woman with a heterozygous c.608 C>T (p.Thr203Met) mutation in exon 3 of the nerve growth factor gene potentially associated with hereditary sensory and autonomic neuropathy type 5. Stem Cell Res 33:171–174
Manganelli F, Parisi S, Nolano M, Tao F, Paladino S, Pisciotta C, Tozza S, Nesti C, Rebelo AP, Provitera V, Santorelli FM, Shy ME, Russo T, Zuchner S, Santoro L (2017) Novel mutations in dystonin provide clues to the pathomechanisms of HSAN-VI. Neurology 88:2132–2140
Romano R, Rivellini C, De Luca M, Tonlorenzi R, Beli R, Manganelli F, Nolano M, Santoro L, Eskelinen EL, Previtali SC, Bucci C (2021) Alteration of the late endocytic pathway in Charcot-Marie-Tooth type 2B disease. Cell Mol Life Sci 78:351–372
Funding
Open access funding provided by Austrian Science Fund (FWF). This work was funded by the Austrian Science Fund FWF (P30809 to KK; DK-SPIN, B16-06 to MK) and the European Commission (ncRNAPain, GA 602133 to MK).
Author information
Authors and Affiliations
Contributions
MZ, KK, and MK have written the manuscript, MZ and MK generated figures, and all authors carefully revised the entire manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is published as part of the Special Issue on Electrophysiology of Nociception as an invited Review.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeidler, M., Kummer, K.K. & Kress, M. Towards bridging the translational gap by improved modeling of human nociception in health and disease. Pflugers Arch - Eur J Physiol 474, 965–978 (2022). https://doi.org/10.1007/s00424-022-02707-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02707-6