
Abstract
Hepatocyte nuclear factor 1β (HNF1β) is a transcription factor essential for the development and function of the kidney. Mutations in and deletions of HNF1β cause autosomal dominant tubule interstitial kidney disease (ADTKD) subtype HNF1β, which is characterized by renal cysts, diabetes, genital tract malformations, and neurodevelopmental disorders. Electrolyte disturbances including hypomagnesemia, hyperuricemia, and hypocalciuria are common in patients with ADTKD-HNF1β. Traditionally, these electrolyte disturbances have been attributed to HNF1β-mediated transcriptional regulation of gene networks involved in ion transport in the distal part of the nephron including FXYD2, CASR, KCNJ16, and FXR. In this review, we propose additional mechanisms that may contribute to the electrolyte disturbances observed in ADTKD-HNF1β patients. Firstly, kidney development is severely affected in Hnf1b-deficient mice. HNF1β is required for nephron segmentation, and the absence of the transcription factor results in rudimentary nephrons lacking mature proximal tubule, loop of Henle, and distal convoluted tubule cluster. In addition, HNF1β is proposed to be important for apical-basolateral polarity and tight junction integrity in the kidney. Interestingly, cilia formation is unaffected by Hnf1b defects in several models, despite the HNF1β-mediated transcriptional regulation of many ciliary genes. To what extent impaired nephron segmentation, apical-basolateral polarity, and cilia function contribute to electrolyte disturbances in HNF1β patients remains elusive. Systematic phenotyping of Hnf1b mouse models and the development of patient-specific kidney organoid models will be essential to advance future HNF1β research.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hepatocyte nuclear factor 1β (HNF1β) is a transcription factor expressed in epithelial tissues including the kidney, pancreas, liver, and genital tract and is essential for the development and function of these tissues [20, 22, 32, 33, 45, 90]. Within the kidney, HNF1β is expressed in all epithelial cells of the nephron and operates in homodimeric or heterodimeric complexes with HNF1α [20].
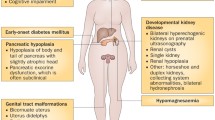
Mutations or deletions in HNF1β are responsible for a dominantly inherited, multisystem disease called autosomal dominant tubulointerstitial kidney disease type HNF1β (ADTKD-HNF1β) [27]. The disease was originally described as renal cysts and diabetes syndrome (RCAD), as kidney cysts (present in 60% of all patients) and maturity-onset diabetes of the young (MODY5) (40%) are common in patients with HNF1β defects [79]. However, the disease has a variable presentation, and not all patients suffer from cysts or diabetes. Kidney anomalies are often present and include renal hypoplasia, unilateral renal agenesis, microcystic dysplasia, and horseshoe kidney. As a consequence, kidney function is impaired in approximately half of the affected children and adults and progresses to end-stage renal disease in 12% of the patients [28, 57, 65]. In contrast to other cystic disorders, electrolyte disturbances are common in ADTKD-HNF1β patients [29, 49, 65]. In particular, the presence of hypomagnesemia is an important predictive criterium to suspect ADTKD-HNF1β [65]. Additionally, hypokalemia, hypocalciuria, hyperparathyroidism, and metabolic alkalosis are present in a minor group of patients [4, 10, 77, 79]. Extrarenal manifestations of ADTKD-HNF1β consist of diabetes, neurodevelopmental disorders, genital and urinary tract malformations, gout, and elevated liver enzymes [10, 12, 79].
The incidence of HNF1β defects is estimated to be 1:200,000 [91]. Approximately 150 different mutations have been reported [18]. These mutations can be familial with a dominant inheritance pattern (60%) or de novo (40%). The majority of the mutations are located in the first four exons encoding the dimerization domain and DNA-binding domains, which are required for binding of HNF1β to the genomic sequence 5′-TTAATNTTTAAC-3′ in promoter or enhancer elements [18, 86]. In addition to intragenic mutations, a 17q12 deletion spanning 15 genes, including HNF1β, accounts for 50% of the cases [19, 26]. Consequently, it is essential to perform an analysis of structural variants in the HNF1β gene, for instance by multiplex ligation-dependent probe amplification (MLPA).
Several groups have attempted to formulate diagnostic criteria to select patients for genetic HNF1β screening. Faguer and colleagues created an HNF1β score based on the clinical presentation [29]. However, several groups demonstrated that patients can be missed using the HNF1β score due to the variability in clinical presentation [18, 65]. The current KDIGO guidelines, therefore, use much simpler diagnostic criteria mainly based on the presence of kidney anomalies [27]. However, these criteria are often not specific for the HNF1β subtype of ADTKD and bear the risk of not identifying the patients that initially present with diabetes or electrolyte phenotype [26, 77]. Several groups have demonstrated that the presence of hypomagnesemia may be particularly predictive of HNF1β mutations [6, 65, 77].
In this review, we present the current knowledge on the electrolyte disturbances in ADTKD-HNF1β patients and discuss the possible mechanisms underlying these disturbances.
Electrolyte disturbances in ADTKD-HNF1β patients
The introduction of next-generation sequencing in standard genetic diagnostic pipelines has resulted in the identification of thousands of ADTKD-HNF1β patients worldwide. Although ADTKD-HNF1β is a rare Mendelian disorder, these technological advances have allowed the formation of large cohorts of HNF1β patients [6, 26, 48, 55, 57]. Careful phenotyping of these cohorts has demonstrated that hypomagnesemia, hyperparathyroidism, hyperuricemia, and hypocalciuria are common in patients with HNF1β defects [5, 6, 30, 55, 92]. Only a minority of the patients have electrolyte disturbances including hypokalemia, metabolic alkalosis, and polyuria [6].
Hypomagnesemia (serum magnesium (Mg2+) < 0.7 mM) is the most common electrolyte disturbance in ADTKD-HNF1β patients. The penetrance of this symptom is estimated to range between 25 and 75% [5, 6, 29, 65, 77]. Several groups have aimed to explain the variability of reported hypomagnesemia cases among cohorts. Prospective cohort studies tend to report the presence of hypomagnesemia more often than retrospective analyses, indicating the poor implementation of Mg2+ measurements in the standard clinical blood biochemistry panels [77]. Several reports noted that young children have generally higher serum Mg2+ concentrations [6, 18, 77]. It was therefore proposed that hypomagnesemia developed later in childhood [6]. However, this notion was recently challenged by Kolbuc and colleagues [92]. Their detailed analysis demonstrated that serum Mg2+ levels are higher in early childhood in both HNF1β patients and healthy controls. Consequently, the reference range of 0.7–1.1 mmol/L is not applicable for young children, resulting in an underestimation of hypomagnesemia in early childhood. Studies establishing age- and gender-specific reference ranges are, therefore, needed.
Hyperparathyroidism (serum parathyroid hormone (PTH) > 6.5 pmol/L) was initially only described in single patients [5, 28]. However, systematic PTH measurements in small cohort studies demonstrated the presence of increased PTH levels in 80% of patients [30, 55]. Because PTH is not reported in many cohort studies, the exact percentage of ADTKD-HNF1β patients suffering from hyperparathyroidism is unknown. Especially, because small cohort studies bare the risk of selection bias, resulting in an overestimation of hyperparathyroidism [30, 55]. Of note, chronic kidney disease may contribute to elevated PTH levels on top of direct HNF1β effects.
Hyperuricemia (serum uric acid > 8 mg/dL) is present in 20–30% of all patients with ADTKD-HNF1β [48, 55, 57, 65]. Reduced kidney function is considered the main mechanism explaining hyperuricemia in ADTKD-HNF1β. Additionally, serum uric acid is independently associated with PTH levels, suggesting that PTH contributes to the molecular mechanism [92]. Indeed, PTH is known to inhibit uric acid secretion by downregulation of ATP-binding cassette transporter G2 (ABCG2) [74]. Interestingly, HNF1β also regulates the expression of renal urate transporter URAT1 [39]. Nevertheless, hyperuricemia and hyperparathyroidism are poor predictors of HNF1β defects as it is also common in other forms of end-stage renal disease [65, 92].
Hypocalciuria is common in patients with ADTKD-HNF1β. The exact penetrance of hypocalciuria is unknown because the reference range for renal calcium (Ca2+) excretion has no generally established lower limit. Nevertheless, several studies demonstrated that urinary Ca2+ levels are significantly lower in patients with HNF1β defects compared to controls [5, 6].
Although serum potassium (K+) and bicarbonate (HCO3−) levels are poorly reported in ADTKD-HNF1β cohorts, Adalat and colleagues demonstrated that HNF1β patients have decreased serum K+ and increased serum HCO3− levels, especially in late childhood [6]. Indeed, case reports have reported K+ values close to the lower border of the reference range (serum K+ 3.5–5.0 mM) [6, 28, 77]. Although these patients are not strictly hypokalemic, their serum K+ concentration is lower than in the general population.
The presence of hypomagnesemia, hypokalemia, metabolic alkalosis, and hypocalciuria is reminiscent of the phenotype of Gitelman syndrome [93, 94]. Indeed, the initial diagnosis of some patients has been Gitelman syndrome, until genetic investigations revealed mutations in the HNF1β gene [7]. However, it should be noted that renin–angiotensin–aldosterone system (RAAS) activation is scarce in patients with HNF1β defects, whereas it is a cardinal symptom of Gitelman patients. Moreover, hypertension is present in 22% of children with ADTKD-HNF1β, whereas Gitelman patients are generally hypotensive compared to healthy family members [69, 95]. Although it should be noted that chronic kidney disease in ADTKD-HNF1β patients may contribute to the hypertension phenotype.
Mechanisms of disturbed electrolyte transport in ADTKD-HNF1β patients
The disturbed electrolyte transport caused by defects in HNF1β has classically been attributed to direct transcriptional regulation of key transporter genes along the nephron [79, 96]. In this review, we will provide an overview of the main transport mechanisms that are determined by HNF1β function. Moreover, we will consider additional mechanisms beyond direct transcriptional regulation, which may contribute to the ADTKD-HNF1β disease phenotype.
Transcriptional control of transporters and channels
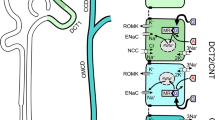
The hypomagnesemia, hypokalemia, and hypocalciuria observed in ADTKD-HNF1β patients are generally assigned to distal tubule dysfunction. In the first description of electrolyte defects in ADTKD-HNF1β patients by Adalat and colleagues, FXYD2 was identified as a transcriptional target in the distal convoluted tubule (DCT) (Fig. 1) [5]. FXYD2 encodes the γ subunit of the Na+-K+-ATPase, and FXYD2 mutations are causative for hypomagnesemia [23, 51]. In recent years, the cardinal role of the Na+-K+-ATPase was further demonstrated by the identification of ATP1A1 mutations, encoding the α subunit of the Na+-K+-ATPase, as a cause of hypomagnesemia [67]. It has been hypothesized that reduced Na+-K+-ATPase activity in the DCT will result in depolarization of the basolateral membrane, resulting in an increased intracellular chloride (Cl−) concentration. Indeed, a high intracellular Cl− concentration has been established to inhibit WNK kinases and thereby the phosphorylation and activity of the thiazide-sensitive Na+-Cl− co-transporter (NCC). Clinical studies confirmed that ADTKD-HNF1β patients have a diminished response to thiazide, confirming lower NCC activity in patients [8]. Interestingly, NCC expression is also decreased in Hnf1b knock-out (KO) mice [41].

HNF1β regulates expression of channels, and transporters in all segments of the nephron. HNF1β regulates target genes involved in electrolyte handling in the PT including TMEM27 encoding the amino acid transport regulator (Collectrin); SLC17A1 encoding the Na-phosphate transporter 1 (NPT1); SLC22A6, SLC22A8, and SLC22A11 encoding the organic anion transporters (OAT1, OAT3, OAT4); and SLC22A12 encoding the renal urate transporter (URAT1); in the TAL including SLC12A1 encoding the Na+-K+-2Cl− co-transporter (NKCC2); UMOD encoding uromodulin (UMOD); CASR encoding the calcium sensing receptor (CaSR); and CLDN16 encoding Claudin 16; in the DCT including KCNJ16 encoding the subunit of the inward rectifier K+ channel (Kir5.1) and FXYD2 encoding the Na+-K+-ATPase subunit gamma; in the CD including TMEM27 and NR1H4 encoding the farnesoid X nuclear receptor (FXR). In return, transcription factor FXR regulates expression of AQP2 in the CD. PT proximal tubules, DCT distal convoluted tubule, TAL thick ascending loop of Henle, CD collecting duct, OA− organic anion, DC− dicarboxylate
Moreover, HNF1β regulates the transcription of KCNJ16, which codes for the Kir5.1 subunit of the basolateral K+ channel in the DCT (Fig. 1) [41]. This Kir4.1/Kir5.1 K+ channel allows recycling of K+ to drive Na+-K+-ATPase activity. Uncoupling of this “pump-leak mechanism” will result in depolarization of basolateral membrane activity and reduced NCC activity by the same mechanisms as described above [97]. The importance of the Kir4.1/Kir5.1 channel was further established by the identification of KCNJ10 and KCNJ16 mutations in patients with hypokalemia and hypomagnesemia, mimicking Gitelman syndrome [13, 68, 98]. Nevertheless, hypokalemia and metabolic alkalosis are only present in a subset of patients with HNF1β defects, which is in line with the phenotype of patients with FXYD2 or ATP1A1 mutations [23, 67]. One might hypothesize that this phenotypic variability is explained by the degree of Na+-K+-ATPase dysfunction and the presence of compensatory effects.
The concomitant HNF1β-dependent regulation of basolateral Na+ and K+ transport by FXYD2 and KCNJ16 demonstrates that transcription factors generally regulate gene networks rather than single genes. Similarly, HNF1β determines a gene network controlling the urine concentrating ability of the kidney [2]. A collecting duct-specific Hnf1b KO mouse model showed a reduced urine osmolality [2]. RNA sequencing and ChIP sequencing identified 27 osmosensitive genes that are dependent on HNF1β binding [2]. Among the HNF1β targets is the farnesoid X receptor (FXR), which is essential for urine concentration by regulating aquaporin 2 (AQP2) expression (Fig. 1) [2, 88]. Indeed, apical plasma membrane expression of AQP2 is reduced in collecting duct cells expressing an Hnf1b mutant [2]. Interestingly, FXR directly activates the expression of Mg2+ channel Trpm6 in mouse intestines [40]. Hence, HNF1β might indirectly regulate Trpm6 expression in the intestines and kidneys through FXR, contributing to disturbed Mg2+ homeostasis in HNF1β patients.
Although HNF1β is also expressed in the thick ascending limb of Henle’s loop (TAL) and this segment transports substantial amounts of Na+, K+, Ca2+, and Mg2+, the role of HNF1β in electrolyte transport in this segment remains elusive. In the TAL, HNF1β was demonstrated to regulate the expression of SLC12A1, encoding the Na+-K+-Cl− co-transporter 2 (NKCC2) (Fig. 1) [36]. As NKCC2 facilitates monovalent ion transport and provides the driving force for paracellular divalent cation transport, one would expect that downregulation of NKCC2 would cause major defects. Particularly, because the downstream DCT segment is affected as well and the compensatory capacity is therefore low. Nevertheless, features of TAL dysfunction such as polyuria, RAAS activation, hypercalciuria, and nephrocalcinosis are generally absent in ADTKD-HNF1β patients.
Several studies have demonstrated that HNF1β activates the expression of uromodulin (UMOD) and the calcium-sensing receptor (CASR) (Fig. 1) [32, 42]. As UMOD mutations are known to cause medullary cysts, this regulatory pathway may contribute to the cystic phenotype of patients with HNF1β defects. Reduced UMOD expression in ADTKD-HNF1β patients may also have implications for renal electrolyte handling since UMOD has been demonstrated to activate NKCC2, NCC, transient receptor potential melastatin type 6 (TRPM6), and TRP vanilloid type 5 (TRPV5) activity [54, 56, 75, 83]. However, as the CaSR is an important negative regulator of UMOD, HNF1β defects may simultaneously inhibit UMOD expression and release the inhibition by the CaSR [76]. Consequently, the reduced UMOD expression may be dampened.
The regulation of CaSR may be of particular importance in the parathyroid gland. CaSR activation in the parathyroid gland inhibits PTH release. The PTH promoter is repressed by HNF1β binding [30]. Hence, HNF1β defects directly increase PTH secretion. On top of that, reduced CaSR expression may also activate PTH secretion [42]. Indeed, ADTKD-HNF1β patients suffer from hyperparathyroidism [30, 55]. However, it should be noted that the in vitro experiments demonstrating the regulation of the CaSR promoter by HNF1β have been performed only in kidney cell lines and should be repeated in parathyroid models. Additionally, both increased PTH secretion and decreased renal CaSR expression are expected to raise calcium levels in the blood. Nonetheless, hypocalcemia is not consistently observed in ADTKD-HNF1β patients.
HNF1β is expressed in all tubule segments of the nephron [20]. Consequently, transcriptional targets of HNF1β have also been identified in the proximal tubule (PT). The expression of organic anion transporters (OAT1, OAT3, OAT4), the Na+-phosphate transporter 1 (NPT1), and the renal urate transporter (URAT1) is regulated by HNF1β (Fig. 1) [37,38,39, 66, 99]. Nevertheless, only a few individual cases were presenting with Fanconi syndrome, suggesting relatively mild PT dysfunction [28]. The absence of a PT phenotype in most patients can potentially be explained by the action of HNF1α, which may compensate for the loss of HNF1β in this segment. As HNF1α is within the kidney exclusively expressed in the PT, other nephron segments do not benefit from this compensatory action [100]. Altogether, systematic studying of HNF1β binding sites in the kidney has resulted in the identification of many genes that are transcriptionally regulated by HNF1β [1, 2, 16, 41, 42]. To date, most studies have investigated HNF1β function by measuring the promoter activity of isolated genes using promoter-luciferase assays. Although these artificial overexpression systems have been instrumental to detect the most prominent regulatory pathways, gene transcription also largely depends on chromatin modifications, the presence of co-activators/co-repressors, or post-translational modifications that are not captured by promoter assays. The recent advances in single-cell genomics and proteomics will allow us to further decipher transcriptional regulation by HNF1β beyond individual genes, by analyzing gene networks and combining -omics approaches.
The role of HNF1β in ureteric bud branching and nephron patterning during kidney development
HNF1β has an essential role during kidney development [20, 32, 90]. The developmental defects may contribute to electrolyte disturbances observed in patients with ADTKD-HNF1β. In Gitelman syndrome, impaired DCT development has been postulated as one of the main causes of Mg2+ wasting [97]. Consequently, defects in kidney tubule patterning should be considered when studying the molecular pathogenesis of ADTKD-HNF1β. Various kidney-specific or inducible mice models have been generated over the past years to determine the role of HNF1β in kidney development (Table 1).
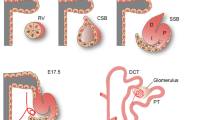
Mice with heterozygous Hnf1b null mutations have no phenotype, while complete deletion of Hnf1b in a mouse model is embryonically lethal due to its crucial role in embryonic visceral endoderm formation [21, 90]. Around E10.5, the development of the kidney starts with the outgrowth of the ureteric bud (UB) from the Wolffian duct (WD) into the metanephric mesenchyme (MM) (Fig. 2). The UB undergoes branching morphogenesis to form the collecting duct system and ureter, after which MM cells surrounding the tips of the ureteric branches form cap mesenchyme. Triggered by signals from the UB tips, these cap mesenchymal cells will polarize into primitive epithelial spheres (pretubular aggregates) to form the renal vesicles. Renal vesicles differentiate into comma- and S-shaped bodies; eventually, part of the S-shaped body will associate with capillaries to form the glomerulus, and other parts will form the nephron tubule that will connect to the collecting duct system. This tightly regulated process called nephrogenesis determines the development and segmentation of the kidney tubule. Although kidney development in humans and mice is very similar at a macroscopic level, organization (e.g., numbers of nephron progenitors and UB tips in human kidneys are increased compared to mice kidneys), timing, and gene expression patterns differ [44]. Therefore, extrapolating data obtained from mice to humans should be done with caution.

HNF1β is required for UB branching and nephron segmentation. Schematic representation of different stages of mouse metanephric nephron development. At E10.5, kidney development starts with the outgrowth of the UB into the MM. HNF1β is essential for normal branching of the UB that eventually will form the collecting duct system. Around E12.5, cells of the cap mesenchyme polarize into pretubular aggregates that will form renal vesicles which require MET. Whether HNF1β is involved in this early stage of nephrogenesis is not yet conclusive. Subsequently, renal vesicles differentiate into comma and S-shaped bodies. Hnf1b KO mice develop S-shaped bodies that lack the epithelial bulge that will give rise to the proximal and Henle’s loop tubule in the WT situation. Eventually at E17.5, part of the S-shaped body will associate with capillaries to form the glomerulus and other parts will form the nephron tubule. WD Wolffian duct, UB ureteric bud, MM metanephric mesenchyme, MET mesenchymal-epithelial transition
In early kidney development, Hnf1b is expressed in the WD and UB [46]. Whereas it is expressed during all nephrogenesis steps including the renal vesicle and comma- and S-shaped body, it is not expressed in the cap mesenchyme [46, 50]. Inactivation of Hnf1b in the mouse UB led to a massively mispatterned ureteric tree network along with defective collecting duct differentiation and polarization (Fig. 2) [25]. Moreover, using constitutive inactivation of Hnf1b in the epiblast by tetraploid aggregation, researchers show that HNF1β is required for UB branching and timing of outgrowth as well as WD maintenance [46]. Although most kidney development studies have been conducted in mouse models, recently heterozygous HNF1β KO (HNF1β+/−) ureteric bud organoids derived from human-induced pluripotent stem cells (iPSCs) were developed [101]. Wild-type (WT) ureteric bud organoids were polarized, had clear tubular lumen, and showed repeated branching morphogenesis [101]. Similar to Hnf1b KO mouse models, human HNF1β+/− organoids showed loss of apical-basolateral polarity and had reduced numbers of budding regions [101].
In addition, several studies uncovered an important role for HNF1β in early nephron segmentation, more specifically in the development of the PT and TAL. HNF1β is required for the formation of a specific mid-limb subcompartment of the S-shaped body, the so-called epithelial bulge, that gives rise to the TAL and the PT (Fig. 2) [35, 50]. In mice, the absence of Hnf1b in the MM resulted in S-shaped bodies without the epithelial bulge and led to the development of nephrons characterized by dilated glomeruli directly connected to collecting ducts via short, primitive tubules displaying early distal markers [50]. Likewise, conditional inactivation of Hnf1b in nephron progenitors results in a reduction of tubular structures with a drastic decrease in PT clusters, medullar Henle’s loop tubules, and DCTs in kidneys from newly born mice (P0) [35]. Expression levels of Notch signaling molecules were strongly decreased in these mice, which may explain the lack of proximal-intermediate nephron segment fate acquisition [35, 50]. In line with these findings, expression of early PT (Hnf4a, Cubn, and Lrp2), mature PT (LTA), TAL (Slc12a1), and DCT (Pvalb) markers was drastically decreased in kidneys of mutant pups at P0 [35, 50]. Mutant S-shaped bodies may express early distal markers, but fail to differentiate into mature distal tubules [35]. Although HNF1β is important for early nephrogenesis, it is still unclear if it also plays a role during the initiation stage that requires mesenchymal-epithelial transition of the MM. In particular, inactivation of Hnf1b in the MM or in nephron precursors resulted in correctly polarized renal vesicles, indicating that HNF1β is not required to initiate nephrogenesis [35, 50]. In contrast, decreased numbers of pretubular aggerates were observed in Hnf1b-deficient mouse kidneys potentially caused by decreased levels of Wnt9b required for mesenchymal-to-epithelial transition underlying the initiation of nephrogenesis (Fig. 2) [46].
Comparable to the mice models, human iPSC-derived organoids with HNF1β KO formed podocytes and GATA3 + distal nephron segments but lacked cells expressing of PT (LRP2, HNF4α) and TAL markers (UMOD, SLC12A1) [64]. These findings are concomitant with a statistical overrepresentation of HNF1β-binding sites in the promoters of PT-specific genes [14, 102]. Altogether, these findings suggest that HNF1β is essential for UB branching and nephrogenesis and particularly affects the PT and TAL segments.
As KO mice models may not represent the effects of human mutations, Niborski et al. generated a mouse model introducing a human splice site mutation (< IVS2nt + 1G > T) [103]. Their mouse model displayed delayed PT differentiation, hydronephrosis, and cysts. Consistent with other mice models, PT markers were decreased from E14.5 to E17.5; however, S-shaped bodies appeared normal and PT maker expression was restored at P0 [103]. Interestingly, at 6 but not 12 months of age, Hnf1b mutant mice exhibited a reduced ability to concentrate urine associated with hypercalciuria but no hypomagnesemia or hyperkalemia was observed [103]. These findings suggest that HNF1β dysfunction in development may be compensated for at a later age.
How do these developmental defects translate to the electrolyte defects in the adult kidney? Remarkably, PT defects are rare in ADTKD-HNF1β, which is difficult to match with maldevelopment of the PT [28]. However, it should be noted that kidney development has been mostly studied in mice. In addition, PT defects could be compensated for by HNF1α transcriptional activity in postnatal life, as evidenced by partial restoration of several PT markers in adult kidneys of mice with a heterozygous splice site mutation in Hnf1b [103]. The impact of heterozygous mutations on kidney development in humans is largely unknown. Histological analysis of a limited number of cystic kidneys from human fetuses carrying HNF1β mutations showed defective or delayed nephrogenesis characterized by a decrease in nephron structures labeled by either LTA, NKCC2, or UMOD [11, 34, 47]. How and to what extent, developmental abnormalities in mice and humans, in particular the rudimentary nephrons lacking mature PT, TAL, and DCT observed in mice models, influence ion transport in adults is unknown. In recent years, an impressive number of human kidney organoids models have been generated and successfully employed to improve our understanding of kidney diseases (reviewed in [104]). Hence, organoid models may provide a valuable tool to better understand the role of HNF1β in human kidney development and electrolyte transport using relevant genetic models instead of full KOs.
The role of HNF1β in apical-basolateral polarity, tight junction integrity, and primary cilia
Apical-basolateral polarity and tight junctions are key regulators of controlled water and ion movement in the kidney epithelium [24, 73]. Moreover, the primary cilium influences renal electrolyte transport in response to changes in tubular flow [52, 63, 72, 81]. In the following part of this review, we will discuss the proposed role of HNF1β in apical-basolateral polarity, tight junction function, and primary cilia development.
Apical-basolateral polarity
Apical-basolateral polarity allows the distribution of channels and transporters to distinct membrane domains and is critical for directional transport of ions and water from the pro-urine to the blood and vice versa [73]. Several polarity markers show aberrant localization or expression during kidney development in HNF1β mutant mice models [25, 103]. For instance, removal of Hnf1b from the UB in mice results in reduced expression of polarity markers Cdh16 and Pkhd1 in UB epithelium [25]. Moreover, in mice with a heterozygous splice site mutation in Hnf1b, decreased levels of HNF1β appear to disturb basal membrane organization without affecting apical cell polarity markers [103]. Interestingly, NKCC2 expression in TAL cells, normally apically expressed, was normal in non-cystic tubules, but the expression was downregulated in cystic tissue [103]. Studies performed by our group using an immortalized mouse collecting duct cell line with disrupted HNF1β function demonstrated a decrease in cell height compared to cells expressing WT HNF1β (unpublished data). Apical-basal growth is a characteristic of polarizing epithelia; likewise, studies using different types of epithelial cells have shown that a loss of cell integrity is associated with a decrease in cell height [59, 71]. In addition, HNF1β+/− ureteric bud organoids derived from human iPSCs display loss of apical-basolateral polarity shown by reduced mRNA expression of apical markers, villin-2 (EZRIN) and protein kinase C zeta type (PRKCζ) [101]. Consistent with this putative role for HNF1β in establishing cell polarity, HNF1β-binding site motifs are enriched in ATAC-sequencing peaks and promoters of upregulated genes during in vitro 3D spheroid formation [105]. Together, this suggests that gene activation by HNF1β is important for cells to establish cell polarization.
Tight junction integrity
Tight junctions establish a border between the functionally different apical and basolateral membrane and act as a barrier for paracellular transport of water and ions [24, 89]. These structures contain a wide variety of proteins (occluding, claudins, junctional adhesion molecules) that define the permeability characteristics of epithelia [24, 58]. Structurally, Desgrange et al. showed that tight junctions appeared well-organized in the UB tips of developing Hnf1b mutant kidneys; however, lateral cell–cell junctions were irregular and the space between cells was larger [25]. Both disruptions in Ca2+ and Mg2+ homeostasis are frequently observed in ADTKD-HNF1β patients. Our unpublished data in immortalized cells showed a significant decrease in transepithelial resistance (TEER) values, a measure of paracellular pathway resistance involving tight junction integrity, in cells with disrupted HNF1β function compared to cells expressing WT Hnf1b.
Primary cilia development
HNF1β regulates an impressive number of genes that localize to the primary cilium including PKHD1, PKD1, PKD2, IFT88, KIF12, CYS1, and PDE4C (reviewed in [70]). Consequently, ciliary defects have been widely considered as the main cause of cyst formation in ADTKD-HNF1β patients [32, 70]. Nevertheless, it is unclear whether HNF1β is directly involved in primary cilium formation, despite the direct transcriptional activation of cilia genes. Two independent studies observed a decrease (25% and not quantified, respectively) of cilia in the cystic epithelium of developing mutant mice compared to WT mice [25, 103]. However, a different study observed normal cilia in cystic tubular cells compared to WT cells of mice with kidney-specific inactivation of Hnf1b (not quantified) [32]. Furthermore, humans and mice with HNF1β deficiency do display an absence of normal primary cilia in the bile duct.
The role of HNF1β in cilia function may also be relevant for electrolyte transport. The cilium acts as an antenna to sense tubular flow and converts changes in tubular pressure into signals that affect electrolyte transport along the nephron [52, 63, 72, 81]. Evidence for the involvement of cilia in flow sensing is based on the fact that flow-sensitive proteins polycystin 1 and transient receptor potential cation channel vanilloid-type 4 (TRPV4) localize to the primary cilium [43, 84, 87]. Furthermore, several examples demonstrate the putative importance of cilia in flow-mediated electrolyte transport. For instance, mice without ciliated TAL cells have diminished Na+ excretion in response to increased water intake causing differences in tubular pressure [72]. In addition, the removal of cilia in immortalized mouse DCT cells reduced transepithelial Ca2+ transport [52]. Additional quantitative studies and the use of high-resolution microscopy techniques to visualize key ciliary proteins should clarify whether HNF1β is involved in cilia function in the kidney.
The importance of cell polarity and tight junction integrity in ion homeostasis has been recognized for decades. Even though the analyzed studies demonstrate that HNF1β defects disturb apical-basolateral cell polarity and tight junction integrity, these mechanisms have never been considered in the pathogenesis of electrolyte disturbances observed in ADTKD-HNF1β patients [25, 103, 105]. Although many Hnf1b animal models have been developed, electrolyte disturbances and polarity defects are often not measured (Table 1). Systematic analysis of apical-basolateral polarity markers and intracellular signaling pathways may help further elucidate the role of cell polarity in electrolyte homeostasis.
Additional pathways
Our literature review has demonstrated that several mechanisms contribute to electrolyte disturbances in patients with HNF1β defects. Nevertheless, it cannot be excluded that additional factors influence ion transport in these patients.
Firstly, the presence of cysts in the kidneys of ADTKD-HNF1β patients can lead to electrolyte disturbances, as observed in patients with autosomal dominant polycystic kidney disease (ADPKD) [60, 62]. Interestingly, the deletion of a transcriptional target of HNF1β and frequently mutated gene in ADPKD patients, called Pkd1, caused aberrant Mg2+, Ca2+, and phosphate (Pi) handling in a precystic mice model [80]. Given the precystic stage of the mice, these changes could not be caused by dilated and cystic tubular structures but were instead attributed to the downregulation of key regulators in Mg2+ and Ca2+ reabsorption in the TAL (Cldn16, Kcnj1, Slc12a1), DCT (Trpm6, Slc12a3), and connecting tubule (Calb1, Slc8a1, Atp2b4). Several of these genes are also downregulated in (developing) kidney tissue of Hnf1b mutant mice [25, 50, 103]. The presence of cysts in glomerular and tubular nephron structures of ADPKD patients can dramatically impair electrolyte and water homeostasis. However, no association has been described to date between the presence of cysts and hypomagnesemia or other electrolyte phenotypes in ADTKD-HNF1β patients.
Secondly, in vitro and in vivo experiments have shown that HNF1β controls mitochondrial respiration in the PT [15, 61]. Inhibition or KO of HNF1β in a human PT cell line resulted in either downregulation of Ppargc1a (important for mitochondrial biogenesis) and altered mitochondrial morphology or ATP reduction and increased glycolysis, respectively [15, 61]. The kidney requires large quantities of ATP to maintain electrochemical gradients across membranes which are particularly important for transcellular ion transport [9]. Given the high energetic demand of the kidneys, the energy deficiency triggered by HNF1β defects might influence transport processes in the PT, and potentially TAL and DCT-mediated transport of Mg2+, Ca2+, and K+. Indeed, mutations in the mitochondrial DNA were recently demonstrated to cause a Gitelman-like phenotype of hypomagnesemia and hypokalemia [82].
Finally, over the past years, HNF1β has been implicated in a broad spectrum of pathways ranging from WNT signaling to planar cell polarity and cholesterol synthesis [1, 17, 31]. The role of these pathways in electrolyte transport has never been examined.
Conclusions and perspectives
Hypomagnesemia, hyperuricemia, and hypocalciuria are common in patients with ADTKD-HNF1β. In subgroups of patients, these electrolyte disturbances are associated with hyperparathyroidism, hypokalemia, and metabolic alkalosis. These clinical findings suggest that the electrolyte disturbances in patients with HNF1β defects have a distal tubular origin. Indeed, our literature review demonstrated that HNF1β regulates the expression of genes involved in distal tubule electrolyte transport, including FXYD2, KCNJ16, CASR, and FXR. In this review, we propose additional mechanisms that may further contribute to electrolyte disorders. HNF1β defects have been demonstrated to impair kidney development, apical-basolateral polarity, tight junction integrity, and cilia development.
The function of HNF1β in kidney physiology has mainly been studied in a wide range of mouse models. Our systematic comparison of all published mouse models identified large differences in phenotypes depending on the genetic defect and strain (Table 1). Complete HNF1β KO may result in different molecular consequences than heterozygous deletions and missense mutations. Consequently, the pathophysiological mechanism of ADTKD-HNF1β may not be captured by most available mouse studies. Moreover, phenotyping of the electrolyte disturbances in HNF1β patients and mouse models is limited, resulting in a knowledge gap in the literature. A more systematic approach is required to associate specific polarity, cilia, or tight junction defects with electrolyte disturbances.
A promising development is the generation of organoid models from patient-derived iPSCs. Recently, kidney organoids were successfully generated from urinary iPSCs of HNF1β patients [53]. Although the current generation kidney organoids are still immature compared with fetal and adult human kidney, these models provide the first patient-derived model to study HNF1β defects in kidney development and function [85].
In conclusion, the causes of electrolyte disturbances in ADTKD-HNF1β may partially be beyond direct transcriptional regulation of specific channels and transporters. Further studies should determine which additional pathways contribute to the molecular mechanisms of electrolyte disturbances observed in ADTKD-HNF1β patients. More systematic phenotyping and the development of patient-specific organoid models are essential next steps in HNF1β research.
Change history
24 May 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00424-022-02706-7
References
Aboudehen K, Kim MS, Mitsche M, Garland K, Anderson N, Noureddine L, Pontoglio M, Patel V, Xie Y, DeBose-Boyd R, Igarashi P (2016) Transcription factor hepatocyte nuclear factor-1 regulates renal cholesterol metabolism. J Am Soc Nephrol 27:2408–2421. https://doi.org/10.1681/ASN.2015060607
Aboudehen K, Noureddine L, Cobo-stark P, Avdulov S, Igarashi P (2017) Hepatocyte nuclear factor–1b regulates urinary concentration and response to hypertonicity. J Am Soc Nephrol 28:2887–2900
Aboudehen K, Noureddine L, Cobo-Stark P, Avdulov S, Farahani S, Gearhart MD, Bichet DG, Pontoglio M, Patel V, Igarashi P (2017) Hepatocyte nuclear factor–1 β regulates urinary concentration and response to hypertonicity. J Am Soc Nephrol 28:2887–2900. https://doi.org/10.1681/ASN.2016101095
Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, Van HW, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU (2009) HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20:1123–1131
Adalat S, Woolf AS, Johnstone KA, Wirsing A (2009) HNF1B mutations associate with hypomagnesemia and renal magnesium. J Am Soc Nephrol 20:1123–1131
Adalat S, Hayes WN, Bryant WA, Booth J, Woolf AS, Kleta R, Subtil S, Clissold R, Colclough K, Ellard S, Bockenhauer D (2019) HNF1B mutations are associated with a gitelman-like tubulopathy that develops during childhood. Kidney International Reports 4:1304–1311. https://doi.org/10.1016/j.ekir.2019.05.019
Ashton EJ, Legrand A, Benoit V, Roncelin I, Venisse A, Zennaro MC, Jeunemaitre X, Iancu D, van’t Hoff WG, Walsh SB, Godefroid N, Rotthier A, del Favero J, Devuyst O, Schaefer F, Jenkins LA, Kleta R, Dahan K, Vargas-Poussou R, Bockenhauer D (2018) Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int 93:961–967
Bech AP, Wetzels JF, Bongers EMHF, Nijenhuis T (2016) Thiazide responsiveness testing in patients with renal magnesium wasting and correlation with genetic analysis: a diagnostic test study. Am J Kidney Dis 68:168–170
Bhargava P, Schnelmann RG (2017) Mitochondrial energetics in the kidney. Nat Rev Nephrol 13:629–646
Bingham C, Hattersley AT (2004) Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1β. Nephrol Dial Transplant 19:2703–2708
Bingham C, Ellard S, Allen L, Bulman M, Shepherd M, Frayling T, Berry PJ, Clark PM, Lindner T, Bell GI, Ryffel GU, Nicholls AJ, Hattersley AT (2000) Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1β. Kidney Int 57:898–907
Bockenhauer D, Jaureguiberry G (2016) HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol 31:707–714. https://doi.org/10.1007/s00467-015-3142-2
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–1970
Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, Kaimal V, Jegga AG, Grimmond S, McMahon AP, Patterson LT, Little MH, Potter SS (2008) Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell 15:781–791. https://doi.org/10.1016/j.devcel.2008.09.007
Casemayou A, Fournel A, Bagattin A, Schanstra J, Belliere J, Decramer S, Marsal D, Gillet M, Chassaing N, Huart A, Pontoglio M, Knauf C, Bascands J-L, Chauveau D, Faguer S (2017) Hepatocyte nuclear factor-1 β controls mitochondrial respiration in renal tubular cells. J Am Soc Nephrol 28:3205–3217
Chan SC, Zhang Y, Shao A, Avdulov S, Herrera J, Aboudehen K, Pontoglio M, Igarashi P (2018) Mechanism of fibrosis in HNF1B-related autosomal dominant tubulointerstitial kidney disease. J Am Soc Nephrol 29:2493–2509. https://doi.org/10.1681/ASN.2018040437
Chan SC, Zhang Y, Pontoglio M, Igarashi P (2019) Hepatocyte nuclear factor-1β regulates Wnt signaling through genome-wide competition with β-catenin/ lymphoid enhancer binding factor. Proc Natl Acad Sci USA 116:24133–24142. https://doi.org/10.1073/pnas.1909452116
Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C (2015) HNF1B-associated renal and extra-renal disease—an expanding clinical spectrum. Nat Publ Group 11:102–112
Clissold RL, Shaw-smith C, Turnpenny P, Bunce B, Bockenhauer D, Kerecuk L, Waller S, Bowman P, Ford T, Ellard S, Hattersley AT (2016) Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int 90:203–211. https://doi.org/10.1016/j.kint.2016.03.027
Coffinier C, Barra J, Babinet C, Yaniv M (1999) Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev 89:211–213
Coffinier C, Barra J, Babinet C, Yaniv M (1999) Expression of the vHNF1/HNF1b homeoprotein gene during mouse organogenesis. Mech Dev 89:211–213
Coffinier C, Gresh L, Fiette L, Tronche F, Schütz G, Babinet C, Pontoglio M, Yaniv M, Barra J (2002) Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1β. Dev 129:1829–1838
de Baaij JHF, Dorresteijn EM, Hennekam EAM, Kamsteeg EJ, Meijer R, Dahan K, Muller M, van den Dorpel MA, Bindels RJM, Hoenderop JGJ, Devuyst O, Knoers NVAM (2015) Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant 30:952–957. https://doi.org/10.1093/ndt/gfv014
Denker BM, Sabath E (2011) The biology of epithelial cell tight junctions in the kidney. J Am Soc Nephrol 22:622–625
Desgrange A, Heliot C, Skovorodkin I, Akram SU, Heikkilä J, Ronkainen V-P, Miinalainen I, Vainio SJ, Cereghini S (2017) HNF1B controls epithelial organization and cell polarity during ureteric bud branching and collecting duct morphogenesis. Dev 144:4704–4719
Dubois-Laforgue D, Cornu E, Saint-Martin C, Coste J, Bellanné-Chantelot C, Timsit J (2017) Diabetes, associated clinical spectrum, long-term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care 40:1436–1443
Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O (2015) Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management - A KDIGO consensus report. Kidney Int 88:676–683
Faguer S, Decramer S, Chassaing N, Bellanné-Chantelot C, Calvas P, Beaufils S, Bessenay L, Lengelé JP, Dahan K, Ronco P, Devuyst O, Chauveau D (2011) Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 80:768–776. https://doi.org/10.1038/ki.2011.225
Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, Casemayou A, Huart A, Schanstra JP, Calvas P (2014) The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 86:1007–1015
Ferrè S, Bongers EMHF, Sonneveld R, Cornelissen EAM, van der Vlag J, van Boekel GAJ, Wetzels JFM, Hoenderop JGJ, Bindels RJM, Nijenhuis T (2013) Early development of hyperparathyroidism due to loss of PTH transcriptional repression in patients with HNF1/β mutations? J Clin Endocrinol Metab 98:4089–4096
Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M (2006) Defective planar cell polarity in polycystic kidney disease. Nat Genet 38:21–23. https://doi.org/10.1038/ng1701
Gresh L, Fischer E, Tanguy M, Shao X, Hiesberger T, Fiette L, Igarashi P, Pontoglio M (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668
Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S (2005) Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. PNAS 102:1490–1495
Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide AL, Cereghini S (2006) Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1β/MODY5 mutations. Hum Mol Genet 15:2363–2375
Heliot C, Desgrange A, Buisson I, Prunskaite-Hyyryläinen R, Shan J, Vainio S, Umbhauer M, Cereghini S (2013) HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Dev 140:873–885
Igarashi P, Whyte DA, Li K, Nagami GT (1996) Cloning and kidney cell-specific activity of the promoter of the murine renal Na-K-Cl cotransporter gene. J Biol Chem 271:9666–9674. https://doi.org/10.1074/jbc.271.16.9666
Jin L, Kikuchi R, Saji T, Kusuhara H, Sugyiama Y (2012) Regulation of tissue-specific expression of renal organic anion transporters by hepatocyte nuclear factor 1 α/β and DNA methylation. J Pharmacol Exp Ther 29:648–655
Kikuchi R, Kusuhara H, Hattori N, Shiota K, Kim I, Gonzalez FJ, Sugiyama Y (2006) Regulation of the expression of human organic anion transporter 3 by hepatocyte nuclear factor 1α/β and DNA methylation. Mol Pharmacol 70:887–896. https://doi.org/10.1124/mol.106.025494
Kikuchi R, Kusuhara H, Hattori N, Kim I, Shiota K, Gonzalez FJ, Sugiyama Y (2007) Regulation of tissue-specific expression of the human and mouse urate transporter 1 gene by hepatocyte nuclear factor 1 α/β and DNA methylation. Mol Pharmacol 72:1619–1625
Kim EY, Lee JM (2022) Transcriptional control of Trpm6 by the nuclear receptor FXR. Int J Mol Sci 23:1980
Kompatscher A, de Baaij JHF, Aboudehen K, Hoefnagels APWM, Igarashi P, Bindels RJM, Veenstra GJC, Hoenderop JGJ (2017) Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1β drives autosomal dominant tubulointerstitial kidney disease. Kidney Int 92:1145–1156
Kompatscher A, de Baaij JHF, Aboudehen K, Farahani S, van Son LHJ, Milatz S, Himmerkus N, Veenstra GC, Hoenderop JGJ (2018) Transcription factor HNF1B regulates expression of the calcium-sensing receptor in the thick ascending limb of the kidney. Am J Renal Physiol 315:F27–F35
Kunnen SJ, Malas TB, Formica C, Leonhard WN, ’t Hoen PAC, Peters DJM (2018) Comparative transcriptomics of shear stress treated Pkd1 −/− cells and pre-cystic kidneys reveals pathways involved in early polycystic kidney disease. Biomed Pharmacother 108:1123–1134. https://doi.org/10.1016/j.biopha.2018.07.178
Lindström NO, Mcmahon JA, Guo J, Tran T, Guo Q, Rutledge E, Parvez RK, Saribekyan G, Schuler RE, Liao C, Kim AD, Abdelhalim A, Ruffins SW, Thornton ME, Basking L, Grubbs B, Kesselman C, Mcmahon AP (2018) Conserved and divergent features of human and mouse kidney organogenesis significance statement. J Am Soc Nephrol 29:785–805
Lokmane L, Haumaitre C, Garcia-Villalba P, Anselme I, Schneider-Maunoury S, Cereghini S (2008) Crucial role of vHNF1 in vertebrate hepatic specification. Dev 135:2777–2786
Lokmane L, Heliot C, Garcia-villalba P, Fabre M, Cereghini S (2010) vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Dev 137:347–357
Madariaga L, Morinière V, Jeanpierre C, Bouvier R, Loget P, Martinovic J, Dechelotte P, Leporrier N, Thauvin-Robinet C, Jensen UB, Gaillard D, Mathieu M, Turlin B, Attie-Bitach T, Salomon R, Gübler MC, Antignac C, Heidet L (2013) Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol 8:1179–1187
Madariaga L, García-Castaño A, Ariceta G, Martínez-Salazar R, Aguayo A, Castaño L (2019) Variable phenotype in HNF1B mutations: extrarenal manifestations distinguish affected individuals from the population with congenital anomalies of the kidney and urinary tract. Clin Kidney J 12:373–379. https://doi.org/10.1093/ckj/sfy102
Martínez V, Trasancos C, Ramos F, Alcázar C, Cabezuelo JB, García M (2016) Poliquistosis renal autosómica recesiva diagnosticada en mujer de 39 años con fallo renal y calambres. Nefrologia 36:318–320. https://doi.org/10.1016/j.nefro.2016.02.002
Massa F, Garbay S, Bouvier R, Sugitani Y, Noda T, Gubler M-C, Heidet L, Pontoglio M, Fischer E (2013) Hepatocyte nuclear factor 1 controls nephron tubular development. Dev 140:886–896
Meij IC, Koenderink JB, de Jong JC, de Pont JJHHM, Monnens LAH, van den Heuvel LPWJ, Knoers NVAM (2003) Dominant isolated renal magnesium loss is caused by misrouting of the Na+, K+-ATPase γ-subunit. Ann N Y Acad Sci 986:437–443. https://doi.org/10.1111/j.1749-6632.2003.tb07226.x
Mohammed SG, Arjona FJ, Latta F, Bindels RJM, Roepman R, Hoenderop JGJ (2017) Fluid shear stress increases transepithelial transport of Ca2+ in ciliated distal convoluted and connecting tubule cells. FASEB J 31:1796–1806
Mulder J, Sharmin S, Chow T, Rodrigues DC, Hildebrandt MR, D’Cruz R, Rogers I, Ellis J, Rosenblum ND (2020) Generation of infant- and pediatric-derived urinary induced pluripotent stem cells competent to form kidney organoids. Pediatr Res 87:647–655. https://doi.org/10.1038/s41390-019-0618-y
Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castañeda-Bueno M, Gamba G, Bachmann S (2011) Activation of the bumetanide-sensitive Na+K+2Cl -Cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem 286:30200–30210
Nagano C, Morisada N, Nozu K, Kamei K, Tanaka R, Kanda S, Shiona S, Araki Y, Ohara S, Matsumura C, Kasahara K, Mori Y, Seo A, Miura K, Washiyama M, Sugimoto K, Harada R, Tazoe S, Kourakata H, Enseki M, Aotani D, Yamada T, Sakakibara N, Yamamura T, Minamikawa S, Ishikura K, Ito S, Hattori M, Iijima K (2019) Clinical characteristics of HNF1B-related disorders in a Japanese population. Clin Exp Nephrol 23:1119–1129. https://doi.org/10.1007/s10157-019-01747-0
Nie M, Bal MS, Liu J, Yang Z, Rivera C, Wu XR, Hoenderop JGJ, Bindels RJM, Marciano DK, Wolf MTF (2018) Uromodulin regulates renal magnesium homeostasis through the ion channel transient receptor potential melastatin 6 (TRPM6). J Biol Chem 293:16488–16502
Okorn C, Goertz A, Vester U, Beck BB, Bergmann C, Habbig S, König J, Konrad M, Müller D, Oh J, Ortiz-brüchle N, Patzer L, Schild R, Seeman T, Staude H, Thumfart J, Tönshoff B, Walden U, Weber L, Weber S (2019) HNF1B nephropathy has a slow-progressive phenotype in childhood—with the exception of very early onset cases: results of the German Multicenter HNF1B Childhood Registry. Pediatr Nephrol 34:1065–1075
Otani T, Furuse M (2020) Tight junction structure and function revisited. Trends Cell Biol 30:805–817
Paniagua AE, Segurado A, Dolón JF, Esteve-Rudd J, Velasco A, Williams DS, Lillo C (2021) Key role for CRB2 in the maintenance of apicobasal polarity in retinal pigment epithelial cells. Front Cell Dev Biol 9:1–15. https://doi.org/10.3389/fcell.2021.701853
Pavik I, Jaeger P, Kistler AD, Poster D, Krauer F, Cavelti-Weder C, Rentsch KM, Wüthrich RP, Serra AL (2011) Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int 79:234–240. https://doi.org/10.1038/ki.2010.375
Piedrafita A, Balayssac S, Casemayou A, Saulnier-Blache JS, Lucas A, Iacovoni JS, Breuil B, Chauveau D, Decramer S, Malet-Martino M, Schanstra JP, Faguer S (2021) Hepatocyte nuclear factor-1β shapes the energetic homeostasis of kidney tubule cells. FASEB J 35:1–16
Pietrzak-Nowacka M, Safranow K, Bober J, Olszewska M, Birkenfeld B, Nowosiad M, Ciechanowski K (2013) Calcium-phosphate metabolism parameters and erythrocyte Ca2+ concentration in autosomal dominant polycystic kidney disease patients with normal renal function. Arch Med Sci 9:837–842. https://doi.org/10.5114/aoms.2012.30834
Praetorius HA, Spring KR (2003) The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12:517–520
Przepiorski A, Sander V, Tran T, Hollywood JA, Sorrenson B, Shih JH, Wolvetang EJ, McMahon AP, Holm TM, Davidson AJ (2018) A simple bioreactor-based method to generate kidney organoids from pluripotent stem cells. Stem Cell Reports 11:470–484. https://doi.org/10.1016/j.stemcr.2018.06.018
Raaijmakers A, Corveleyn A, Devriendt K, van Tienoven TP, Allegaert K, van Dyck M, van den Heuvel L, Kuypers D, Claes K, Mekahli D, Levtchenko E (2015) Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol Dial Transplant 30:835–842
Saji T, Kikuchi R, Kusuhara H, Kim I, Gonzalez FJ (2008) Transcriptional regulation of human and mouse organic anion transporter 1 by hepatocyte nuclear factor 1 α/β. J Pharmacol Exp Ther 324:784–790
Schlingmann KP, Bandulik S, Mammen C, Tarailo-Graovac M, Holm R, Baumann M, König J, Lee JJY, Drögemöller B, Imminger K, Beck BB, Altmüller J, Thiele H, Waldegger S, van’t Hoff W, Kleta R, Warth R, van Karnebeek CDM, Vilsen B, Bockenhauer D, Konrad M (2018) Germline de novo mutations in ATP1A1 cause renal hypomagnesemia, refractory seizures, and intellectual disability. Am J Hum Genet 103:808–816
Schlingmann KP, Renigunta A, Hoorn EJ, Forst AL, Renigunta V, Atanasov V, Mahendran S, Barakat TS, Gillion V, Godefroid N, Brooks AS, Lugtenberg D, Lake J, Debaix H, Rudin C, Knebelmann B, Tellier S, Rousset-Rouvière C, Viering D, de Baaij JHF, Weber S, Palygin O, Staruschenko A, Kleta R, Houillier P, Bockenhauer D, Devuyst O, Vargas-Poussou R, Warth R, Zdebik AA, Konrad M (2021) Defects in KCNJ16 cause a novel tubulopathy with hypokalemia, salt wasting, disturbed acid-base homeostasis, and sensorineural deafness. J Am Soc Nephrol 32:1498–1512
Seeman T, Weigel F, Blahova K, Fencl F, Pruhova S, Hermes K, Klaus R, Lange-Sperandio B, Grote V, John-Kroegel U (2021) Blood pressure in children with renal cysts and diabetes syndrome. Eur J Pediatr 180:3599–3603
Shao A, Chan SC, Igarashi P (2020) Role of transcription factor hepatocyte nuclear factor-1β in polycystic kidney disease. Cell Signal 71:1–24. https://doi.org/10.1016/j.cellsig.2020.109568
Shen L, Weber CR, Raleigh DR, Yu D, Turner JR (2011) Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 73:283–309
Song J, Wang L, Fan F, Wei J, Zhang J, Lu Y, Fu Y, Wang S, Juncos LA, Liu R (2017) Role of the primary cilia on the macula densa and thick ascending limbs in regulation of sodium excretion and hemodynamics. Hypertens 70:324–333
Stoops EH, Caplan MJ (2014) Trafficking to the apical and basolateral membranes in polarized epithelial cells. J Am Soc Nephrol 25:1375–1386
Sugimoto R, Watanabe H, Ikegami K, Enoki Y, Imafuku T, Sakaguchi Y, Murata M, Nishida K, Miyamura S, Ishima Y, Tanaka M, Matsushita K, Komaba H, Fukagawa M, Otagiri M, Maruyama T (2017) Down-regulation of ABCG2, a urate exporter, by parathyroid hormone enhances urate accumulation in secondary hyperparathyroidism. Kidney Int 91:658–670
Tokonami N, Takata T, Beyeler J, Ehrbar I, Yoshifuji A, Christensen EI, Loffing J, Devuyst O, Olinger EG (2018) Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int 94:701–715
Tokonami N, Olinger E, Debaix H, Houillier P, Devuyst O (2018) The excretion of uromodulin is modulated by the calcium-sensing receptor. Kidney Int 94:882–886
van der Made CI, Hoorn EJ, de La Faille R, Karaaslan H, Knoers NVAM, Hoenderop JGJ, Vargas Poussou R, de Baaij JHF (2015) Hypomagnesemia as first clinical manifestation of ADTKD-HNF1B: a case series and literature review. Am J Nephrol 42:85–90. https://doi.org/10.1159/000439286
Verdeguer F, Le Corre S, Fischer E, Callens C, Garbay S, Doyen A, Igarashi P, Terzi F, Pontoglio M (2010) A mitotic transcriptional switch in polycystic kidney disease. Nat Med 16:106–110
Verhave JC, Bech AP, Wetzels JFM, Nijenhuis T (2016) Hepatocyte nuclear factor 1—associated kidney disease: more than renal cysts and diabetes. J Am Soc Nephrol 27:345–353
Verschuren EHJ, Mohammed SG, Leonhard WN, Overmars-Bos C, Veraar K, Hoenderop JGJ, Bindels RJM, Peters DJM, Arjona FJ (2018) Polycystin-1 dysfunction impairs electrolyte and water handling in a renal precystic mouse model for ADPKD. Am J Physiol Renal Physiol 315:F537–F546
Verschuren EHJ, Castenmiller C, Peters DJM, Arjona FJ, Bindels RJM, Hoenderop JGJ (2020) Sensing of tubular flow and renal electrolyte transport. Nat Rev Nephrol 16:337–351. https://doi.org/10.1038/s41581-020-0259-8
Viering D, Schlingmann KP, Hureaux M, Nijenhuis T, Mallett A, Chan MMY, van Beek A, van Eerde AM, Coulibaly J-M, Vallet M, Decramer S, Pelletier S, Klaus G, Kömhoff M, Beetz R, Patel C, Shenoy M, Steenbergen EJ, Anderson G, Bongers EMHF, Bergmann C, Panneman D, Rodenburg RJ, Kleta R, Houillier P, Konrad M, Vargas-Poussou R, Knoers NVAM, Bockenhauer D, de Baaij JHF (2022) Gitelman-like syndrome caused by pathogenic variants in mtDNA. J Am Soc Nephrol 33:305–325
Wolf MTF, Wu XR, Huang CL (2013) Uromodulin upregulates TRPV5 by impairing caveolin-mediated endocytosis. Kidney Int 84:130–137
Wu L, Gao X, Brown RC, Heller S, O’Neil RG (2007) Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am J Physiol Renal Physiol 293:1699–1713
Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD (2018) Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23:869-881.e8. https://doi.org/10.1016/j.stem.2018.10.010
Yi-zhi C, Qing GAO, Xue-zhi Z, Ying-zhang C, Bennett CL, Xi-shan X, Chang-lin MEI, Yong-quan SHI, Xiang-mei C (2010) Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J 123:3326–3333
Yoder BK, Hou X, Guay-Woodford LM (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13:2508–2516
Zhang X, Huang S, Gao M, Liu J, Jia X, Han Q, Zheng S, Miao Y, Li S, Weng H, Xia X, Du S, Wu W, Gustafsson JA, Guan Y (2014) Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc Natl Acad Sci USA 111:2277–2282
Zihni C, Mills C, Matter K, Balda MS (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17:564–580. https://doi.org/10.1038/nrm.2016.80
Barbacci E, Reber M, Ott M, Breillat C, Huetz F, Cereghini S (1999) Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. 4805:4795–4805
Owen K (2014) HNF1B-related autosomal dominant tubulointerstitial kidney disease. In: Orphanet. Accessed 02-20-2022 https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=93111
Kołbuc M, Bieniaś B, Habbig S, Kołek M, Szczepanska M, Kiliś-Pstrusińska K, Wasilewska A, Adamczyk P, Motyka R, Tkaczyk M, Sikora P, Beck BB, Zaniew M (2021) Hyperuricemia is relatively common in children with HNF1B mutation, but its utility as a clinically useful marker for predicting the mutation is limited. Nephrol Dial Transplant 36. https://doi.org/10.1093/ndt/gfab080.0015
Knoers NVAM, Levtchenko EN (2008) Gitelman syndrome. Orphanet J Rare Dis 3
Simon DB, Nelson-Williams C, Johnson Bia M, Ellison D, Karet FE, Morey Molina A, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitelman HJ, LiftonL RP (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter
Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB (2001) Gitelman’s syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int 59:710–717. https://doi.org/10.1046/j.1523-1755.2001.059002710.x
Ferrè S, Igarashi P (2018) New insights into the role of HNF-1β in kidney (patho)physiology. Pediatric Nephrol 1–11. https://doi.org/10.1007/s00467-018-3990-7
Franken GAC, Adella A, Bindels RJM, de Baaij JHF (2021) Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney. Acta Physiologica 231
Scholl UI, Choi M, Liu T, Ramaekers VT, Hä Usler C MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10
Cheret C, Doyen A, Yaniv M, Pontoglio M (2002) Hepatocyte nuclear factor 1 a controls renal expression of the Npt1-Npt4 anionic transporter locus. 2836:929–941. https://doi.org/10.1016/S0022-2836(02)00816-1
Pontoglio M, Barra J (1996) Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal fanconi syndrome
Mae SI, Ryosaka M, Sakamoto S, Matsuse K, Nozaki A, Igami M, Kabai R, Watanabe A, Osafune K (2020) Expansion of human iPSC-derived ureteric bud organoids with repeated branching potential. Cell Rep 32. https://doi.org/10.1016/j.celrep.2020.107963
Miao Z, Balzer MS, Ma Z, Liu H, Wu J, Shrestha R, Aranyi T, Kwan A, Kondo A, Pontoglio M, Kim J, Li M, Kaestner KH, Susztak K (2021) Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat Comm 12:2277. https://doi.org/10.1038/s41467-021-22266-1
Niborski LL, Paces-Fessy M, Ricci P, Bourgeois A, Magalhaes P, Kuzma-Kuzniarska M, Lesaulnier C, Reczko M, Declercq E, Zurbig P, Doucet A, Umbhauer M, Cereghini S (2021) Hnf1b haploinsufficiency differentially affects developmental target genes in a new renal cysts and diabetes mouse model. Dis Model Mech 14. https://doi.org/10.1242/dmm.047498
Romero-Guevara R, Ioannides A, Xinaris C (2020) Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front Phys 11:563981. https://doi.org/10.3389/fphys.2020.563981
Wang T, Kwon SH, Peng X, Urdy S, Lu Z, Schmitz RJ, Dalton S, Mostov KE, Zhao S (2020) A qualitative change in the transcriptome occurs after the first cell cycle and coincides with lumen establishment during MDCKII cystogenesis. iScience 23:101629. https://doi.org/10.1016/j.isci.2020.101629
Hiesberger T, Bai Y, Shao X, Mcnally BT, Sinclair AM, Tian X, Somlo S, Igarashi P (2004) Mutation of hepatocyte nuclear factor-1β inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest 113
Funding
This work was financially supported by grants from the Dutch Kidney Foundation (17OKG07 and 20OP + 018) and the IMAGEN project which is co-funded by the PPP Allowance made available by Health ~ Holland, Top Sector Life Sciences & Health, to stimulate public–private partnerships (IMplementation of Advancements in GENetic Kidney Disease, LSHM20009).
Author information
Authors and Affiliations
Contributions
L.E.T., J.G.J.H., and J.H.F.d.B. wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Kidney Control of Homeostasis in Pflügers Archiv—European Journal of Physiology
The original online version of this article was revised: Originally, the article has been published online with error in Table 1. See below list.
Na+, K+, and urea concentrations = Decreased Na+, K+, and urea urine concentrations.
Mg2+, Na, K+, and urea concentrations = Increased total Mg2+, Na+ and K+ urine excretiond
In Table 1 footer:
d < 12 months of age
e In the C57BL/6 N background but not in 129/sv background
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tholen, L.E., Hoenderop, J.G.J. & de Baaij, J.H.F. Mechanisms of ion transport regulation by HNF1β in the kidney: beyond transcriptional regulation of channels and transporters. Pflugers Arch - Eur J Physiol 474, 901–916 (2022). https://doi.org/10.1007/s00424-022-02697-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02697-5