
Abstract
Small-conductance Ca2+-activated K+ (SK, KCa2) channels are encoded by KCNN genes, including KCNN1, 2, and 3. The channels play critical roles in the regulation of cardiac excitability and are gated solely by beat-to-beat changes in intracellular Ca2+. The family of SK channels consists of three members with differential sensitivity to apamin. All three isoforms are expressed in human hearts. Studies over the past two decades have provided evidence to substantiate the pivotal roles of SK channels, not only in healthy heart but also with diseases including atrial fibrillation (AF), ventricular arrhythmia, and heart failure (HF). SK channels are prominently expressed in atrial myocytes and pacemaking cells, compared to ventricular cells. However, the channels are significantly upregulated in ventricular myocytes in HF and pulmonary veins in AF models. Interests in cardiac SK channels are further fueled by recent studies suggesting the possible roles of SK channels in human AF. Therefore, SK channel may represent a novel therapeutic target for atrial arrhythmias. Furthermore, SK channel function is significantly altered by human calmodulin (CaM) mutations, linked to life-threatening arrhythmia syndromes. The current review will summarize recent progress in our understanding of cardiac SK channels and the roles of SK channels in the heart in health and disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ca2+ ions are the central player in the regulation of cardiac excitability and contractility. Ca2+ influx through voltage-gated Ca2+ channels is critical not only for initiating cardiac excitation-contraction coupling, but also for activating multiple downstream signaling pathways in response to changes in membrane potentials. Small-conductance Ca2+-activated K+ (SK, KCa2) channels, encoded by KCNN genes, are among the Ca2+-gated ion channels that play critical roles in the regulation of cardiac excitability. The dynamic beat-to-beat changes of intracellular Ca2+ are essential for the function and regulation of SK channels.
Our understandings of cardiac SK channels have greatly expanded in the past 2 decades since the functional and molecular identification of SK channels in the heart [99]. Ca2+-activated K+ currents (IK,Ca) in the heart have previously been considered controversial [26, 69]. However, studies by our group and others have provided evidence to substantiate the pivotal roles of SK channels, not only in healthy heart but also with diseases including atrial fibrillation (AF), ventricular arrhythmia, and heart failure (HF) [10, 27, 28, 34, 59, 71, 99, 107]. SK channels are prominently expressed in atrial myocytes and pacemaking cells, compared to ventricular cells [13, 92, 93, 99, 105]. However, the channels are significantly upregulated in ventricular myocytes in HF and pulmonary veins (PV) in AF models. The potential role of SK channels in human AF has been demonstrated using genome-wide association analysis (GWAS), revealing an association between single-nucleotide polymorphism (SNP) in KCNN2 and KCNN3 genes with lone AF [27, 28]. SK channels may represent novel therapeutic targets against AF [10, 19,20,21, 76, 82, 84, 88, 95]. Furthermore, SK channel function is significantly altered by human calmodulin (CaM) mutations, linked to life-threatening arrhythmia syndromes [38, 81, 104]. The current review will summarize recent progress in our understanding of cardiac SK channels and serve as a discussion platform for the roles of SK channels in the heart in health and disease.
SK channels
The discovery of SK channels dates back to more than 70 years ago [107]. The bee venom toxin apamin, a highly selective blocker of SK channels, enables the molecular identification of SK channels in the mammalian brain [47], which were later observed in various tissues, including smooth muscle, endothelia, epithelia, blood cell, and heart [85, 107]. The family of SK channels consists of three members with differential sensitivity to apamin: SK1 (or KCa2.1 encoded by KCNN1 gene), SK2 (or KCa2.2 encoded by KCNN2 gene), and SK3 (or KCa2.3 encoded by KCNN3 gene) [1, 107]. SK2 is highly sensitive to apamin, with a half-blocking concentration (EC50) of ~40 pM, whereas SK1 channels are the least sensitive to apamin with an EC50 of ~10 nM. SK3 channels show intermediate sensitivity, with an EC50 of ~1 nM [1]. No other class of K+ channels is blocked by apamin. An intermediate-conductance Ca2+-activated K+ channel (IK, SK4, or KCa2.4 encoded by the KCNN4 gene) is structurally and functionally similar to the SK channels and is classified in the same gene family [1, 85]. Functional SK channels assemble to form homomeric or heteromeric tetramers [47, 87, 94]. A recent cryo-electron microscopy (EM) study reveals the structure of human SK4-CaM channels in closed and activated states [48]. It was found that one channel tetramer binds to four CaM molecules. The C-lobe of CaM binds to the channel constitutively, whereas the N-lobe of CaM interacts with the S4-S5 linker in a Ca2+-dependent manner. The S4-S5 linker undergoes conformational changes upon CaM binding to open the channel pore [48]. This structural study lays essential foundations in revealing the physiological and pharmacological properties of SK channels in the heart.
Functional expression of SK channels in the heart
All three isoforms of SK channels are expressed in mouse and human hearts [93, 99]. The channels are expressed in different regions of the hearts in multiple species, including rabbit PV [13, 75] and ventricular myocytes [16, 49], human atrial myocytes [83, 103], and rat ventricular myocytes [72]. SK currents have also been recorded in canine PV and left atrial myocytes using an SK-specific blocker, NS8593 [77]. In addition, SK channels play critical roles in pacemaking cells, including mouse atrioventricular nodal cells [105] and rabbit and mouse sinoatrial nodal cells [13, 92]. SK channels are more abundantly expressed in atrial myocytes, pacemaking cells, and cardiac Purkinje fibers, compared to ventricular myocytes [79, 92, 99, 105]. SK2 and SK3 channels are also functionally expressed in mouse embryonic cardiomyocytes and are required for maintaining excitability in developing cardiomyocytes, important in promoting Ca2+-dependent gene expression [44].
Regulation of cardiac SK channels
Cardiac SK channel interactome
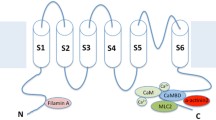
Similar to other membrane proteins, ion channels form multi-protein complexes interacting with the extracellular matrix and cytosolic proteins [2, 9, 63]. SK channels are gated solely by beat-to-beat changes in intracellular Ca2+ via CaM, a ubiquitous Ca2+ sensing protein. CaM binds to the CaM binding domain (CaMBD) in the C termini of SK channels. SK channels are activated upon Ca2+-dependent CaM binding to the S4-S5 linker of the SK channels, resulting in conformational changes leading to channel activation [1, 48, 98]. CaM is essential for Ca2+ sensitivity and critical to the trafficking of SK channels [62]. Specifically, previous studies have demonstrated that Ca2+-independent association between CaM and SK channels is necessary for cell surface expression.
Several cytoskeletal proteins including α-actinin2 [55, 56], filamin A [78], and myosin light chain 2 (MLC2) [57] interact with cardiac SK2 channels. Specifically, α-actinin2 and MLC2 interact via the C-termini, and filamin A interacts via the N-termini of the SK2 channel. Moreover, cardiac SK2 channels couple with L-type Ca2+ channels, Cav1.3 and Cav1.2, through a physical bridge, α-actinin2. Trafficking of SK2 channels is critically dependent on the direct protein-protein interactions of the channels with α-actinin2, MLC2, and filamin A [56, 57, 78]. Knockdown of α-actinin2 or filamin A results in a decrease in SK2 channel expression on the membrane and localization of SK2 channels in the endosome, suggesting a reduction in the recycling of SK2 channels from the endosome. Finally, SK2 channel trafficking is Ca2+-dependent in the presence of α-actinin2. A decrease in intracellular Ca2+ results in a significant reduction of SK2 channel membrane localization [78]. Therefore, an increase in intracellular Ca2+, as evident during rapid AF or atrial tachycardia, is predicted to increase SK2 channel expression, leading to shortening of the atrial APs and maintenance of arrhythmias. One previous study using rapid pacing in isolated rabbit atria demonstrates a significant increase in SK2 immunostaining from a perinuclear pattern to the plasma membrane in the PV after burst pacing, suggesting an increase in forward trafficking of SK2 channels [75]. Indeed, PV is known to be the critical initiation site of human AF [35].
Protein kinase (casein kinase II, CK2) and protein phosphatase 2A (PP2A) are identified as SK2 channel binding proteins [3, 6]. CK2 and PP2A regulate SK channel sensitivity to intracellular Ca2+ by phosphorylating or dephosphorylating CaM [1]. In the heart, CK2 is found to co-localize with SK2 and SK3 in the rat ventricle. The increased expression of PP2A and the decreased interaction of CK2 and SK2 are two mechanisms underlying the upregulation of cardiac SK currents in the rat model of volume-overload HF [100]. A recent study found that junctophilin type 2 (JP2) associates with SK2 channels in cardiomyocytes [29]. JP2 directly interacts with SK2 channels via the membrane occupation and recognition nexus (MORN motifs) in its N-terminus.
In summary, the SK channel interactome in cardiomyocytes consists of homomeric or heteromeric SK channel α subunits, CaM, α-actinin2, filamin A, CK2, and PP2A (Fig. 1). The interactions of the multi-protein SK channel complexes are critical for the expression, function, and trafficking of SK channels in cardiomyocytes. Additional interacting proteins of SK channels are likely to be identified with advanced proteomics approaches.

SK channels interactome. SK channels interactome includes α-actinin2 (Actin2), filamin A (FLNA), myosin light chain 2 (MLC2), CK2, and PP2A. Cardiac SK channels have been shown to couple to L-type Ca2+ channels through a physical bridge, α-actinin2. SK2 channels do not physically interact with the Ca2+ channels, instead the two channels co-localize via their interaction with α-actinin2 along the Z-line in atrial myocytes. An increase in intracellular Ca2+, as evident during rapid AF or atrial tachycardia, is predicted to increase SK2 channel expression leading to shortening of the atrial APs and maintenance of arrhythmias. Schematic representation was generated using BioRender
Coupling of SK channels with L-type Ca2+ channels and ryanodine receptors
The co-localization of SK and Ca2+ channels suggests the possibility that local subsarcolemmal Ca2+, resulting from Ca2+ channel opening, is sufficient to activate SK channels, as was demonstrated for hippocampal neurons [61]. However, two previous studies suggest that sarcoplasmic reticulum (SR) Ca2+ release via ryanodine receptors (RyR2) is required for the activation of cardiac SK channels [66, 89]. Our recent report shows that L-type Ca2+ channels (LTCCs) provide the immediate Ca2+ microdomain for the activation of SK channels in cardiomyocytes, supporting the roles of both Ca2+ entry and Ca2+ release from SR in the activation of SK channels. Further super-resolution imaging quantifies and localizes the three molecules including SK channels, LTCCs, and RyR2 to be within hundreds of nanometers (Fig. 2). The distribution of nearest neighbor distances (NND) between SK2 and RyR2 as well as SK2 and Cav1.2 was bimodal, suggesting a spatial relationship between the channels. Therefore, Ca2+ influx through LTCCs and Ca2+ release via RyR2 provide the immediate and efficient Ca2+ microdomain for the activation of SK channels [108].

Spatial distribution of SK2, RyR2, and Cav1.2 [108]. a Spatial coupling of SK2, RyR2, and Cav1.2 channels. Stimulated emission depletion (STED) microscopy of SK2, RyR2, and Cav1.2 expression in rabbit ventricular myocytes at three Z planes (scale bar 2 μm). b Visualization of the spatial distribution of SK2, RyR2, and Cav1.2. (AU, arbitrary units; NND, nearest neighbor distance)
Phosphorylation of cardiac SK channels
Even though SK channels are expressed at a relatively low level in normal ventricles, SK channels have been shown to be phosphorylated by both Ca2+/CaM-dependent protein kinase II (CaMKII) and protein kinase A (PKA) in different models of cardiac hypertrophy, leading to the upregulation of SK currents in ventricular myocytes [37, 64]. Cardiac SK2 channels form multiprotein complexes with phosphorylated CaMKII at threonine-286 in a spontaneously hypertensive rat model. The modulation of the channels by CaMKII may contribute to ventricular arrhythmias during hypoxia in hypertrophied hearts [88]. Additionally, β-adrenergic stimulation results in enhanced SK channel activation via increased CaMKII activities [43]. In contrast, PKA phosphorylation at serine-465 of SK2 channel attenuates rectification of SK currents by reducing the voltage-dependent inhibition by intracellular Ca2+, leading to the upregulation of SK currents in ventricular myocytes from hypertrophic hearts [37]. In human AF, autophosphorylated CaMKII at Thr287 is significantly increased, leading to the upregulated SK currents in atria with increased Ca2+ sensitivity but decreased expression of SK1, SK2, and SK3 channels [30].
MiRNA regulation of cardiac SK channels
MicroRNAs (miRNAs) are endogenously expressed small non-coding RNAs. MiRNAs in the heart have been extensively studied in the past decade [17, 31, 39, 73, 91, 109]. Cardiac-specific miRNAs play critical roles in heart development, myocardial regeneration, and cardiac remodeling. Recent studies have provided strong evidence for the critical roles of miRNAs in the regulation of cardiac ion channels and transporters [5, 7, 22, 25, 65, 90]. We have investigated the relationship between cardiac-specific miRNA-1/133a, cAMP signaling, and electrical remodeling in a clinically relevant mouse myocardial infarction (MI) model [68]. Chronic over-expression of inducible cAMP early repressor (ICER) from excessive β-adrenergic signaling in MI results in suppression of miRNA-1 and miRNA-133a expression leading to the reduction of transient outward K+ currents (Ito) and prolonged action potential (AP) duration (APD) [68]. MiRNA 499 (MiR-499) is upregulated in the atria of patients with AF, leading to the downregulation of SK3 channels by binding to the 3' untranslated region of KCNN3 gene [53]. The role of miRNAs in cardiac SK channels regulation requires further studies, especially in diseased hearts.
Sex-specific regulation of cardiac SK channels
Expressions of SK2 channel and SK current density are higher in ventricular myocytes of female rabbits compared to male rabbits [14]. Furthermore, action potential triangulation, a known proarrhythmic predictor, is observed in female rabbit hearts upon isoproterenol stimulation, which could be reversed by apamin, suggesting the possible roles of SK channels in ventricular fibrillation in female animals during sympathetic stimulation. This report of sex-specific expression and function of cardiac SK channels highlights the critical need for a deeper understanding of sex-specific therapeutics in cardiac arrhythmia, an area that is currently understudied. Further investigations are necessary to determine the underlying mechanisms of the sex-specific expression of SK channels [23].
Physiological significance of cardiac SK channels
Critical roles in cardiac repolarization
Repolarization of cardiac AP is critically dependent on the orchestrated activity of multiple K+ channels and transporters. The initial study found that inhibition of SK currents by apamin prolonged APD in mouse and human atrial myocytes. However, the effects were less prominent in ventricular myocytes suggesting the unique role of the channels in atrial repolarization [99]. These findings are supported by subsequent studies in global SK2 knockout mice, demonstrating that ablation of KCNN2 results in a significant prolongation of APD, prominently in the late phase of the repolarization in atrial myocytes, and increased susceptibility to AF [51]. In contrast, there is no significant alteration in the APD in ventricular myocytes, and no ventricular arrhythmias are induced in the null mutant animals [51]. Conversely, gain-of-function of SK3 channels results in a significant shortening of APD in atrial myocytes [106].
Consistently, optical mapping in isolated canine left atria demonstrates that inhibition of SK channels by either apamin or UCL1684 [80] prolongs APD [41]. SK channel inhibitors, NS8593, and ICAGEN [32] prolong APD in isolated human atrial myocytes [83]. The above studies support the critical role of SK channels in the repolarization not only in mouse and canine but also in human atrial myocytes [71, 99]. In atrioventricular nodal cells, SK2 channel overexpression results in shortening of spontaneous APs and an increase in the firing frequency, while ablation of SK2 channels results in the opposite effects [105]. These findings are further supported by experimental data in rabbit heart [13], where sinoatrial node (SAN) cells exhibit larger SK currents than pulmonary vein cardiomyocytes. Furthermore, apamin decreases the firing rate and prolongs APD in SAN cells and pulmonary vein cardiomyocytes.
Intracellular signaling and mitochondrial function
K+ channels are critical to normal mitochondrial function by regulating the mitochondrial membrane potentials, plasticity, matrix volume, reactive oxygen species (ROS) production, and respiratory chain activities [12, 97]. The activation of the mitochondrial K+ channels results in cardiac protective effects against ischemia-reperfusion (I/R) injury [54, 74]. Studies demonstrate that SK channels are expressed in neuronal and cardiac inner mitochondrial membrane [24, 86, 101]. An SK channel opener, DCEBIO, is found to protect the heart from the I/R injury, while an SK channel inhibitor, NS8593, antagonizes the protection by DCEBIO [86]. This is further supported by a study identifying the expression and functional effects of SK3 variants in the inner mitochondrial membrane of guinea pig, rat, and human ventricular myocytes [101], and KCNN3 gene silencing with siRNA enhances cell death. The function of mitochondrial SK channels in Ca2+-dependent ventricular arrhythmia was further investigated in a rat model of cardiac hypertrophy [45]. A membrane-permeable SK enhancer, NS309, improves aberrant Ca2+ homeostasis, reduces the oxidation of ryanodine receptors, and abolishes ventricular tachycardia and fibrillation induced by β-adrenergic stimulation. Ca2+ signaling in mitochondria is critical to cardiac energetics, excitation-contraction (E-C) coupling, and cytosolic Ca2+ signaling [18]. Mitochondrial SK channels sense the mitochondrial Ca2+ signaling to regulate the mitochondrial function. Further studies are necessary to understand the molecular and cellular mechanisms of SK channels in the regulation of mitochondrial function in physiological and pathological conditions.
Remodeling of cardiac SK channels in disease conditions
Cardiac SK channel expression and function are significantly altered in disease conditions, as summarized in Fig. 3.

Functional roles of SK channels in normal and diseased hearts. Distinct roles of SK channels in atria and ventricles are depicted together with remodeling in AF, HF, and calmodulinopathy. EAD, early afterdepolarization; PV, pulmonary veins; AF, atrial fibrillation; VT/VF, ventricular tachycardia and fibrillation; LQTS, long QT syndromes; CPVT, polymorphic ventricular tachycardia; IVF, familial idiopathic ventricular fibrillation
Atrial arrhythmia
Emerging evidence from our group and others have shown that SK channels significantly contribute to AF. Ozgen et al. elegantly show that atrial burst pacing in rabbits, a model that mimicked ectopic foci, upregulates SK2 channel proteins in the cytosolic and membrane regions and shortened APD in PV [75]. Similarly, SK current is more prominently expressed in canine PV than left atrium (LA) and is upregulated by atrial tachypacing [77]. Mechanistically, this may result from Ca2+-dependent trafficking of SK2 channels to the membrane during rapid pacing [78].
The importance of SK channels in AF is further highlighted in a genome-wide association study that identified genetic variants in KCNN2 and KCNN3 genes that encode SK2 and SK3 channels be linked to AF susceptibility [27]. Overexpression of SK3 channels in mice results in increased sudden death due to AV block and severe sinus bradyarrhythmias [60]. In addition, atrial cardiomyocytes isolated from SK3 overexpressed mice exhibit significantly shortened APD with abbreviated atrial effective refractory period and increased AF inducibility [106]. In contrast, ablation of KCNN2 results in a significant prolongation of APD and increased AF susceptibility [51]. These studies highlight that both gain- and loss-of-function of SK channels can increase AF susceptibility, likely from distinct underlying mechanisms. K+ channel gain-of-function is predicted to result in decreased atrial refractoriness and reentry mechanisms. On the other hand, K+ channel loss-of-function results in delayed repolarization and possibly triggered arrhythmias.
Downregulation of SK channel expression and current has also been documented in AF patients. Specifically, SK1, SK2, and SK3 channel expression and apamin-sensitive currents are reduced in chronic AF patients [83, 103]. Although the above studies highlight the crucial role for SK channel remodeling in AF, the exact timing for the observed changes in expression and function of SK channels in AF remains incompletely understood.
Heart failure
Abnormal Ca2+ handling [58], complex alterations in gene expression [4], and posttranslational modification [50] have been documented in HF. SK channels are significantly upregulated in HF even though most other K+ channels are downregulated [10]. In a tachycardia-induced rabbit HF model, shortening of APD in ventricular myocytes and recurrent spontaneous ventricular tachyarrhythmias can be reversed by apamin due to an increase in apamin-sensitive K+ current (IKAS) [15]. Moreover, the apamin-sensitive K+ currents are heterogeneously upregulated, with higher density in the epicardial than mid myocardial and endomyocardial myocytes. The upregulation of IKAS is due, in part, to increased Ca2+ sensitivity of the SK channels in HF. Importantly, IKAS is also upregulated in failing human ventricles, attributed to an upregulation in SK2 expression and increased Ca2+ sensitivity [8]. Similar findings are reported in a canine right ventricular tachypacing model [8]. Indeed, SK channels’ upregulation in ventricular myocytes in HF may play a protective role by increasing repolarization reserve [40]. Moreover, SK channels are upregulated in atrial tissues from human and canine HF, which may underlie the increased incidence of AF in HF patients [8]. Although there is a consensus that SK channels are upregulated in HF ventricles, additional mechanistic understandings into the upregulation and heterogeneity of SK channel expression [11] will provide important insights into the clinical implications of SK channel remodeling in HF.
Diabetes
Diabetes is a strong risk factor for both AF and HF [33, 102]. A recent study found that streptozotocin-induced diabetes in a mouse model downregulates SK2 and SK3 channels in atrial tissues by 85% and 92%, respectively, while SK1 remains the same as control [102]. Indeed, in vitro findings with HL-1 cells treated with high glucose demonstrate similar changes in SK channel expression. SK channel-associated electrical remodeling may therefore contribute to pathogenesis or increase susceptibility to AF. The underlying mechanisms for the remodeling require further investigations.
Human calmodulinopathy
Since SK channel activation and membrane localization are critically dependent on CaM binding to the CaMBD in the C terminus of the channel, CaM mutations, associated with human arrhythmia syndrome, significantly alter SK channel function [38, 81, 104]. Five human CaM mutations (N54I, N98S, D96V, D130G, and F90L) significantly reduces SK2 current relative to wild-type CaM without a change in the membrane or intracellular protein expression [104]. Similarly, long QT syndrome (LQTS)-associated CaM variants (D96V, D130G, F142L) and catecholaminergic polymorphic ventricular tachycardia (CPVT)-associated variant (N54I) reduces SK3 current [81]. To address the mechanistic underpinnings of the CaM mutations on SK channel function, we took advantage of human induced pluripotent stem cell-derived cardiomyocyte-like cells (hiPSC-CMs) as well as structural modeling and molecular dynamics simulation [38]. We demonstrate that human calmodulinopathy-associated CaM mutations disrupt cardiac SK channel function via distinct mechanisms. CaMD96V and CaMD130G mutants reduce SK currents through a dominant-negative fashion. By contrast, specific mutations replacing phenylalanine with leucine result in conformational changes that affect helix packing in the C- lobe, which disengage the interactions between apo-CaM and the CaM binding domain of SK channels (Fig. 4). Even though SK currents play a relatively minor role in normal ventricles, the channels are expressed in cardiac Purkinje cells [79], the probable site of origin of cardiac arrhythmias, including in patients with heritable arrhythmia syndrome [36, 96]. Additionally, SK channels are expressed and play important roles in pacemaking cells, including sinoatrial and atrioventricular nodes [92, 105]. Distinct mutant CaMs may significantly reduce the activation of the SK channels, decreasing the key Ca2+-dependent repolarization currents these channels mediate, exacerbating the effects of CaM mutations in human arrhythmia syndrome.

Structural modeling of the interactions between CaM mutants and SK2 CaMBD [38]. a Schematic of the 4 α-helices within the C-lobe of CaM. Location of mutations are shown by labels and colored markers. b–d Comparisons of CaMWT (green) and CaMF90L (red, b), CaMF93L (yellow, c), and CaMF142L (purple, d). Side chains of key amino acid residues are shown in stick representation using color scheme shown in panel a. Conformational changes due to CaM mutation are indicated by black arrows in each panel. e C-lobe of apo-CaMWT (colored in green) bound to the C-terminus of hSK2 channel (colored in light brown). Side chains of key amino acids are shown using space-filling representation. f Panel E rotated 90° to the left around the Y-axis. Molecular modeling was performed in Ca2+ free conditions
SK channels in stem cell cardiomyogenesis
Activation of SK channels and maintaining the activated state by small molecule, 1-ethyl-2- benzimidazolinone (EBIO), has been reported to drive cardiomyogenesis in both the murine and the human pluripotent stem cells (PSCs) [46, 52, 67]. In the murine PSCs, SK channel activation on day 5 of the differentiated embryoid bodies improves the cardiomyogenesis efficiency, as assessed by flow cytometry with a ~4-fold increase in the fraction of troponin-positive cells [46]. Additionally, EBIO induced an upregulation of the pacemaking-specific genes (Hcn4, Tbx3, Shox2, Cx30.2, and Cx45), a downregulation of the ventricular-specific genes (Myl2v, Cx43), and an increase in the number of cardiomyocytes exhibiting pacemaker-like action potentials, suggesting that the pacemaking-like cardiomyocyte subtype was promoted over that of the ventricular-like cells. The promotion of pacemaking-like cardiomyocytes is mediated through the activation of the ERK1/2 signaling pathway [46]. EBIO-induction of cardiomyogenesis with a preferential yield for the pacemaking-like subtype was similarly demonstrated in human PSCs; however, the pacemaking-like electrophysiology was not demonstrated in the study [67]. The overexpression of the dominant SK channel isoform present in the murine PSCs, the SK4 isoform, promotes cardiomyogenesis in an inducible SK4 murine PSC line but without an apparent bias for a particular cardiomyocyte subtype [52]. Since SK4 is not the dominant SK channel isoform in human PSCs [67], SK4 is not likely the key player in EBIO-induced cardiomyogenesis in human cells like the murine PSCs.
Interestingly, a recent publication on cardiomyogenesis reported that the observed EBIO-induced increase in the cardiomyocyte yield and concurrent increase in the fraction of pacemaking-like cardiomyocytes from the human hPSCs were not attributed to directed differentiation but rather an enrichment due to a higher tolerance or survivability of cardiomyocytes—specifically those that have APs with a short APD—to EBIO-induced toxicity than the non-cardiomyocytes [42]. Indeed, the overall cell count in the EBIO-treated hPSCs was 2.5 times fewer than the control cells. The mechanism of action by EBIO was also determined to be SK4-independent since the SK4-specific activator, NS309, did not induce the same cardiomyocyte yield as that of EBIO. Only a SK2/3-selective activator, CyPPA, was able to induce similar EBIO-mediated cardiomyocyte enrichment. This is in agreement with SK2 being the dominant SK isoform in human PSCs [67]. Nevertheless, considering that none of the SK blockers tested (clotrimazole and apamin) were able to negate the EBIO-mediated cardiomyocyte enrichment, the exact enrichment mechanism by EBIO remains to be elucidated.
SK channels as therapeutic targets
The differential expression of cardiac SK channels in autorhythmic and contractile myocytes and their heterogeneous distribution within each tissue type [15] create a remarkable and exciting opportunity for cardiac regional-specific therapy. Indeed, a major limitation of current antiarrhythmic drugs is the risk for proarrhythmia, which can be life-threatening [70]. SK channels may serve as a new therapeutic target for atrial arrhythmias. Moreover, SK channel trafficking is tightly regulated by intracellular Ca2+ and may serve as one future therapeutic avenue for SK channel targeting and cardiomyocyte excitability. However, SK channels are known to be upregulated in HF and may serve to increase cardiac repolarization reserve by counterbalancing the downregulation of other K+ channels. Therefore, elucidating SK channels’ intricate mechanisms in pathological conditions will have profound clinical implications.
Conclusions and perspectives
The identification of cardiac SK channels opens a new avenue of research and provides therapeutic opportunities in cardiac arrhythmias. The significance of cardiac SK channels is supported by studies from multiple investigators over the past two decades. The cumulative findings so far have greatly enhanced our understanding of SK channels in the regulation of cardiac excitability and function. However, there exist many challenges and knowledge gaps. The role of SK channels in AF and ventricular arrhythmias is not entirely understood. Functional roles of SK channels in pacemaking cells and cardiac conduction systems are only beginning to be realized. The study of SK channels in intracellular organelles, including cardiac mitochondria, is in its early stage. Additionally, the local control and feedback of Ca2+ and SK channels within the microdomains are not well understood. On the translational level, the use of SK channel blockers in the treatment of cardiac arrhythmia and the possible pro-arrhythmic effects of SK channel blockers require further consideration and investigation.
References
Adelman JP, Maylie J, Sah P (2012) Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol 74:245–269. https://doi.org/10.1146/annurev-physiol-020911-153336
Adsit GS, Vaidyanathan R, Galler CM, Kyle JW, Makielski JC (2013) Channelopathies from mutations in the cardiac sodium channel protein complex. J Mol Cell Cardiol 61:34–43. https://doi.org/10.1016/j.yjmcc.2013.03.017
Allen D, Fakler B, Maylie J, Adelman JP (2007) Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci 27:2369–2376. https://doi.org/10.1523/JNEUROSCI.3565-06.2007
Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM (2016) Cardiac Remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol 106:62–69. https://doi.org/10.5935/abc.20160005
Benz A, Kossack M, Auth D, Seyler C, Zitron E, Juergensen L, Katus HA, Hassel D (2016) miR-19b Regulates ventricular action potential duration in zebrafish. Sci. Rep 6:36033. https://doi.org/10.1038/srep36033
Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B (2004) Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron 43:847–858. https://doi.org/10.1016/j.neuron.2004.08.033
Binas S, Knyrim M, Hupfeld J, Kloeckner U, Rabe S, Mildenberger S, Quarch K, Stratz N, Misiak D, Gekle M, Grossmann C, Schreier B (2020) miR-221 and -222 target CACNA1C and KCNJ5 leading to altered cardiac ion channel expression and current density. Cell Mol Life Sci 77:903–918. https://doi.org/10.1007/s00018-019-03217-y
Bonilla IM, Long VP III, Vargas-Pinto P, Wright P, Belevych A, Lou Q, Mowrey K, Yoo J, Binkley PF, Fedorov VV, Györke S, Janssen PML, Kilic A, Mohler PJ, Carnes CA (2014) Calcium-activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS One 9:e108824. https://doi.org/10.1371/journal.pone.0108824
Catterall WA (2010) Signaling complexes of voltage-gated sodium and calcium channels. Neurosc lett 486:107–116. https://doi.org/10.1016/j.neulet.2010.08.085
Chang PC, Chen PS (2015) SK channels and ventricular arrhythmias in heart failure. Trends Cardiovasc Med 25:508–514. https://doi.org/10.1016/j.tcm.2015.01.010
Chang PC, Turker I, Lopshire JC, Masroor S, Nguyen BL, Tao W, Rubart M, Chen PS, Chen Z, Ai T (2013) Heterogeneous upregulation of apamin-sensitive potassium currents in failing human ventricles. J Am Heart Assoc 2:e004713. https://doi.org/10.1161/jaha.112.004713
Checchetto V, Azzolini M, Peruzzo R, Capitanio P, Leanza L (2018) Mitochondrial potassium channels in cell death. Biochem Biophys Res Commun 500:51–58. https://doi.org/10.1016/j.bbrc.2017.06.095
Chen WT, Chen YC, Lu YY, Kao YH, Huang JH, Lin YK, Chen SA, Chen YJ (2013) Apamin modulates electrophysiological characteristics of the pulmonary vein and the Sinoatrial Node. Eur J Clin Invest 43:957–963. https://doi.org/10.1111/eci.12125
Chen M, Yin D, Guo S, Xu DZ, Wang Z, Chen Z, Rubart-von der Lohe M, Lin SF, Everett Iv TH, Weiss JN, Chen PS (2018) Sex-specific activation of SK current by isoproterenol facilitates action potential triangulation and arrhythmogenesis in rabbit ventricles. J Physiol 596:4299–4322. https://doi.org/10.1113/JP275681
Chua S-K, Chang P-C, Maruyama M, Turker I, Shinohara T, Shen Mark J, Chen Z, Shen C, Rubart-von der Lohe M, Lopshire John C, Ogawa M, Weiss James N, Lin S-F, Ai T, Chen P-S (2011) Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Cir Res 108:971–979. https://doi.org/10.1161/CIRCRESAHA.110.238386
Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart-von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T, Chen PS (2011) Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res 108:971–979. https://doi.org/10.1161/CIRCRESAHA.110.238386
Colpaert RMW, Calore M (2019) MicroRNAs in cardiac diseases. Cells 8. https://doi.org/10.3390/cells8070737
Dedkova EN, Blatter LA (2013) Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol 58:125–133. https://doi.org/10.1016/j.yjmcc.2012.12.021
Diness JG, Sorensen US, Nissen JD, Al-Shahib B, Jespersen T, Grunnet M, Hansen RS (2010) Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol 3:380–390. https://doi.org/10.1161/CIRCEP.110.957407
Diness JG, Skibsbye L, Jespersen T, Bartels ED, Sorensen US, Hansen RS, Grunnet M (2011) Effects on atrial fibrillation in aged hypertensive rats by Ca(2+)-activated K(+) channel inhibition. Hypertension 57:1129–1135. https://doi.org/10.1161/HYPERTENSIONAHA.111.170613
Diness JG, Skibsbye L, Simo-Vicens R, Santos JL, Lundegaard P, Citerni C, Sauter DRP, Bomholtz SH, Svendsen JH, Olesen SP, Sorensen US, Jespersen T, Grunnet M, Bentzen BH (2017) Termination of Vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca(2+)-activated K(+) channels in pigs. Circ Arrhythm Electrophysiol 10. https://doi.org/10.1161/CIRCEP.117.005125
Divakaran V, Mann DL (2008) The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res 103:1072–U1047. https://doi.org/10.1161/Circresaha.108.183087
Dobrev D (2018) Sex-specific density of ventricular SK currents: is this a general feature of SK channel function? J Physiol 596:4287–4288. https://doi.org/10.1113/JP276663
Dolga AM, Netter MF, Perocchi F, Doti N, Meissner L, Tobaben S, Grohm J, Zischka H, Plesnila N, Decher N, Culmsee C (2013) Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J Biol Chem 288:10792–10804. https://doi.org/10.1074/jbc.M113.453522
Duong E, Xiao J, Qi XY, Nattel S (2017) MicroRNA-135a regulates sodium-calcium exchanger gene expression and cardiac electrical activity. Heart Rhythm 14:739–748. https://doi.org/10.1016/j.hrthm.2017.01.045
Eisner DA, Vaughan-Jones RD (1983) Do calcium-activated potassium channels exist in the heart? Cell Calcium 4:371–386
Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, Lubitz SA, Fox E, Darbar D, Smith NL, Smith JD, Schnabel RB, Soliman EZ, Rice KM, Van Wagoner DR, Beckmann BM, van Noord C, Wang K, Ehret GB, Rotter JI, Hazen SL, Steinbeck G, Smith AV, Launer LJ, Harris TB, Makino S, Nelis M, Milan DJ, Perz S, Esko T, Kottgen A, Moebus S, Newton-Cheh C, Li M, Mohlenkamp S, Wang TJ, Kao WH, Vasan RS, Nothen MM, MacRae CA, Stricker BH, Hofman A, Uitterlinden AG, Levy D, Boerwinkle E, Metspalu A, Topol EJ, Chakravarti A, Gudnason V, Psaty BM, Roden DM, Meitinger T, Wichmann HE, Witteman JC, Barnard J, Arking DE, Benjamin EJ, Heckbert SR, Kaab S (2010) Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42:240–244. https://doi.org/10.1038/ng.537
Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Muller-Nurasyid M, Krijthe BP, Lubitz SA, Bis JC, Chung MK, Dorr M, Ozaki K, Roberts JD, Smith JG, Pfeufer A, Sinner MF, Lohman K, Ding J, Smith NL, Smith JD, Rienstra M, Rice KM, Van Wagoner DR, Magnani JW, Wakili R, Clauss S, Rotter JI, Steinbeck G, Launer LJ, Davies RW, Borkovich M, Harris TB, Lin H, Volker U, Volzke H, Milan DJ, Hofman A, Boerwinkle E, Chen LY, Soliman EZ, Voight BF, Li G, Chakravarti A, Kubo M, Tedrow UB, Rose LM, Ridker PM, Conen D, Tsunoda T, Furukawa T, Sotoodehnia N, Xu S, Kamatani N, Levy D, Nakamura Y, Parvez B, Mahida S, Furie KL, Rosand J, Muhammad R, Psaty BM, Meitinger T, Perz S, Wichmann HE, Witteman JC, Kao WH, Kathiresan S, Roden DM, Uitterlinden AG, Rivadeneira F, McKnight B, Sjogren M, Newman AB, Liu Y, Gollob MH, Melander O, Tanaka T, Stricker BH, Felix SB, Alonso A, Darbar D, Barnard J, Chasman DI, Heckbert SR, Benjamin EJ, Gudnason V, Kaab S (2012) Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 44:670–675. https://doi.org/10.1038/ng.2261
Fan HK, Luo TX, Zhao WD, Mu YH, Yang Y, Guo WJ, Tu HY, Zhang Q (2018) Functional interaction of Junctophilin 2 with small- conductance Ca2+ -activated potassium channel subtype 2(SK2) in mouse cardiac myocytes. Acta Physiol (Oxf) 222. https://doi.org/10.1111/apha.12986
Fan X, Yu Y, Lan H, Ou X, Yang L, Li T, Cao J, Zeng X, Li M (2018) Ca2+/calmodulin-dependent protein kinase II (CaMKII) increases small-conductance Ca2+-activated K+ current in patients with chronic atrial fibrillation. Med Sci Monit 24:3011–3023. https://doi.org/10.12659/MSM.909684
Fung EC, Butt AN, Eastwood J, Swaminathan R, Sodi R (2019) Circulating microRNA in cardiovascular disease. Adv Clin Chem 91:99–122. https://doi.org/10.1016/bs.acc.2019.03.003
Gentles RG, Grant-Young K, Hu S, Huang Y, Poss MA, Andres C, Fiedler T, Knox R, Lodge N, Weaver CD, Harden DG (2008) Initial SAR studies on apamin-displacing 2-aminothiazole blockers of calcium-activated small conductance potassium channels. Bioorg Med Chem Lett 18:5316–5319. https://doi.org/10.1016/j.bmcl.2008.08.023
Grundy Scott M, Benjamin Ivor J, Burke Gregory L, Chait A, Eckel Robert H, Howard Barbara V, Mitch W, Smith Sidney C, Sowers James R (1999) Diabetes and cardiovascular disease. Circulation 100:1134–1146. https://doi.org/10.1161/01.CIR.100.10.1134
Gu M, Zhu Y, Yin X, Zhang DM (2018) Small-conductance Ca(2+)-activated K(+) channels: insights into their roles in cardiovascular disease. Exp Mol Med 50:23. https://doi.org/10.1038/s12276-018-0043-z
Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J (1998) Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 339:659–666. https://doi.org/10.1056/NEJM199809033391003
Haissaguerre M, Shah DC, Jais P, Shoda M, Kautzner J, Arentz T, Kalushe D, Kadish A, Griffith M, Gaita F, Yamane T, Garrigue S, Hocini M, Clementy J (2002) Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet 359:677–678. https://doi.org/10.1016/S0140-6736(02)07807-8
Hamilton S, Polina I, Terentyeva R, Bronk P, Kim TY, Roder K, Clements RT, Koren G, Choi BR, Terentyev D (2020) PKA phosphorylation underlies functional recruitment of sarcolemmal SK2 channels in ventricular myocytes from hypertrophic hearts. J Physiol 598:2847–2873. https://doi.org/10.1113/JP277618
Hannah Ledford SP, Muir D, Woltz R, Lu R, Nguyen P, Sirish P, Wang W, Sihn C-R, George A Jr, Knollmann B, Yamoah E, Yarov-Yarovoy V, Zhang X-D, Chiamvimonvat N (2020) Different arrhythmia-associated calmodulin mutations have distinct effects on cardiac SK channel regulation. J Gen Physiol 152. https://doi.org/10.1085/jgp.202012667
Hata A (2013) Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol 75:69–93. https://doi.org/10.1146/annurev-physiol-030212-183737
Hsieh YC, Chang PC, Hsueh CH, Lee YS, Shen C, Weiss JN, Chen Z, Ai T, Lin SF, Chen PS (2013) Apamin-sensitive potassium current modulates action potential duration restitution and arrhythmogenesis of failing rabbit ventricles. Circ Arrhythm Electrophysiol 6:410–418. https://doi.org/10.1161/CIRCEP.111.000152
Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS, Lin SF (2013) Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm 10:891–898. https://doi.org/10.1016/j.hrthm.2013.01.033
Jara-Avaca M, Kempf H, Ruckert M, Robles-Diaz D, Franke A, de la Roche J, Fischer M, Malan D, Sasse P, Solodenko W, Drager G, Kirschning A, Martin U, Zweigerdt R (2017) EBIO does not induce cardiomyogenesis in human pluripotent stem cells but modulates cardiac subtype enrichment by lineage-selective survival. Stem Cell Reports 8:305–317. https://doi.org/10.1016/j.stemcr.2016.12.012
Kamada R, Yokoshiki H, Mitsuyama H, Watanabe M, Mizukami K, Tenma T, Takahashi M, Takada S, Anzai T (2019) Arrhythmogenic beta-adrenergic signaling in cardiac hypertrophy: The role of small-conductance calcium-activated potassium channels via activation of CaMKII. Eur J Pharmacol 844:110–117. https://doi.org/10.1016/j.ejphar.2018.12.011
Karppinen S, Rapila R, Naumenko N, Tuomainen T, Koivumaki JT, Hanninen SL, Korhonen T, Tavi P (2016) Ca(2+) -activated K(+) current is essential for maintaining excitability and gene transcription in early embryonic cardiomyocytes. Acta Physiol (Oxf) 216:101–111. https://doi.org/10.1111/apha.12540
Kim TY, Terentyeva R, Roder KH, Li W, Liu M, Greener I, Hamilton S, Polina I, Murphy KR, Clements RT, Dudley SC Jr, Koren G, Choi BR, Terentyev D (2017) SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc Res 113:343–353. https://doi.org/10.1093/cvr/cvx005
Kleger A, Seufferlein T, Malan D, Tischendorf M, Storch A, Wolheim A, Latz S, Protze S, Porzner M, Proepper C, Brunner C, Katz SF, Varma Pusapati G, Bullinger L, Franz WM, Koehntop R, Giehl K, Spyrantis A, Wittekindt O, Lin Q, Zenke M, Fleischmann BK, Wartenberg M, Wobus AM, Boeckers TM, Liebau S (2010) Modulation of calcium-activated potassium channels induces cardiogenesis of pluripotent stem cells and enrichment of pacemaker-like cells. Circulation 122:1823–1836
Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP (1996) Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273:1709–1714
Lee CH, MacKinnon R (2018) Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 360:508–513. https://doi.org/10.1126/science.aas9466
Lee YS, Chang PC, Hsueh CH, Maruyama M, Park HW, Rhee KS, Hsieh YC, Shen C, Weiss JN, Chen Z, Lin SF, Chen PS (2013) Apamin-sensitive calcium-activated potassium currents in rabbit ventricles with chronic myocardial infarction. J Cardiovasc Electrophysiol 24:1144–1153. https://doi.org/10.1111/jce.12176
Lee A, Oh JG, Gorski PA, Hajjar RJ, Kho C (2016) Post-translational modifications in heart failure: small changes, big impact. Heart Lung Circ 25:319–324. https://doi.org/10.1016/j.hlc.2015.11.008
Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, Bond CT, Periasamy M, Adelman J, Chiamvimonvat N (2009) Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol 587:1087–1100. https://doi.org/10.1113/jphysiol.2008.167718
Liebau S, Tischendorf M, Ansorge D, Linta L, Stockmann M, Weidgang C, Iacovino M, Boeckers T, von Wichert G, Kyba M, Kleger A (2011) An inducible expression system of the calcium-activated potassium channel 4 to study the differential impact on embryonic stem cells. Stem Cells Int 2011:456815–456812. https://doi.org/10.4061/2011/456815
Ling TY, Wang XL, Chai Q, Lau TW, Koestler CM, Park SJ, Daly RC, Greason KL, Jen J, Wu LQ, Shen WF, Shen WK, Cha YM, Lee HC (2013) Regulation of the SK3 channel by microRNA-499--potential role in atrial fibrillation. Heart Rhythm 10:1001–1009. https://doi.org/10.1016/j.hrthm.2013.03.005
Liu Y, Sato T, O'Rourke B, Marban E (1998) Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation 97:2463–2469. https://doi.org/10.1161/01.cir.97.24.2463
Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N (2007) Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res 100:112–120. https://doi.org/10.1161/01.RES.0000253095.44186.72
Lu L, Timofeyev V, Li N, Rafizadeh S, Singapuri A, Harris TR, Chiamvimonvat N (2009) Alpha-actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+-activated K+ channel (SK2 channel). Proc Natl Acad Sci U S A 106:18402–18407. https://doi.org/10.1073/pnas0908207106
Lu L, Sirish P, Zhang Z, Woltz RL, Li N, Timofeyev V, Knowlton AA, Zhang X, Yamoah EN, Chiamvimonvat N (2014) Regulation of gene transcription by voltage-gated L-type calcium channel, Cav1.3. J Biol Chem. https://doi.org/10.1074/jbc.M114.586883
Luo M, Anderson Mark E (2013) Mechanisms of altered Ca2+ handling in heart failure. Circ Res 113:690–708. https://doi.org/10.1161/CIRCRESAHA.113.301651
Mahida S (2014) Expanding role of SK channels in cardiac electrophysiology. Heart Rhythm 11:1233–1238. https://doi.org/10.1016/j.hrthm.2014.03.045
Mahida S, Mills RW, Tucker NR, Simonson B, Macri V, Lemoine MD, Das S, Milan DJ, Ellinor PT (2014) Overexpression of KCNN3 results in sudden cardiac death. Cardiovasc Res 101:326–334. https://doi.org/10.1093/cvr/cvt269
Marrion NV, Tavalin SJ (1998) Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395:900–905. https://doi.org/10.1038/27674
Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP (2004) Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol 554:255–261. https://doi.org/10.1113/jphysiol.2003.049072
Meadows LS, Isom LL (2005) Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res 67:448–458. https://doi.org/10.1016/j.cardiores.2005.04.003
Mizukami K, Yokoshiki H, Mitsuyama H, Watanabe M, Tenma T, Takada S, Tsutsui H (2015) Small-conductance Ca2+-activated K+ current is upregulated via the phosphorylation of CaMKII in cardiac hypertrophy from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 309:H1066–H1074. https://doi.org/10.1152/ajpheart.00825.2014
Morishima M, Iwata E, Nakada C, Tsukamoto Y, Takanari H, Miyamoto S, Moriyama M, Ono K (2016) Atrial fibrillation-mediated upregulation of miR-30d regulates myocardial electrical remodeling of the G-protein-gated K(+) channel, IK.ACh. Circ J 80:1346–1355. https://doi.org/10.1253/circj.CJ-15-1276
Mu YH, Zhao WC, Duan P, Chen Y, Zhao WD, Wang Q, Tu HY, Zhang Q (2014) RyR2 modulates a Ca2+-activated K+ current in mouse cardiac myocytes. PLoS One 9:e94905. https://doi.org/10.1371/journal.pone.0094905
Muller M, Stockmann M, Malan D, Wolheim A, Tischendorf M, Linta L, Katz SF, Lin Q, Latz S, Brunner C, Wobus AM, Zenke M, Wartenberg M, Boeckers TM, von Wichert G, Fleischmann BK, Liebau S, Kleger A (2012) Ca2+ activated K channels-new tools to induce cardiac commitment from pluripotent stem cells in mice and men. Stem Cell Rev 8:720–740. https://doi.org/10.1007/s12015-011-9324-9
Myers R, Timofeyev V, Li N, Kim C, Ledford HA, Sirish P, Lau V, Zhang Y, Fayyaz K, Singapuri A, Lopez JE, Knowlton AA, Zhang XD, Chiamvimonvat N (2015) Feedback mechanisms for cardiac-specific microRNAs and cAMP signaling in electrical remodeling. Circ Arrhythm Electrophysiol 8:942–950. https://doi.org/10.1161/CIRCEP.114.002162
Nagy N, Szuts V, Horvath Z, Seprenyi G, Farkas AS, Acsai K, Prorok J, Bitay M, Kun A, Pataricza J, Papp JG, Nanasi PP, Varro A, Toth A (2009) Does small-conductance calcium-activated potassium channel contribute to cardiac repolarization? J Mol Cell Cardiol 47:656–663. https://doi.org/10.1016/j.yjmcc.2009.07.019
Nattel S (1998) Experimental evidence for proarrhythmic mechanisms of antiarrhythmic drugs. Cardiovasc Res 37:567–577. https://doi.org/10.1016/S0008-6363(97)00293-9
Nattel S, Qi XY (2014) Calcium-dependent potassium channels in the heart: clarity and confusion. Cardiovasc Res 101:185–186. https://doi.org/10.1093/cvr/cvt340
Ni Y, Wang T, Zhuo X, Song B, Zhang J, Wei F, Bai H, Wang X, Yang D, Gao L, Ma A (2013) Bisoprolol reversed small conductance calcium-activated potassium channel (SK) remodeling in a volume-overload rat model. Mol Cell Biochem 384:95–103. https://doi.org/10.1007/s11010-013-1785-5
Orenes-Pinero E, Montoro-Garcia S, Patel JV, Valdes M, Marin F, Lip GYH (2013) Role of microRNAs in cardiac remodelling: new insights and future perspectives. International Journal of Cardiology 167:1651–1659. https://doi.org/10.1016/j.ijcard.2012.09.120
O'Rourke B (2004) Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res 94:420–432. https://doi.org/10.1161/01.RES.0000117583.66950.43
Ozgen N, Dun W, Sosunov EA, Anyukhovsky EP, Hirose M, Duffy HS, Boyden PA, Rosen MR (2007) Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res 75:758–769. https://doi.org/10.1016/j.cardiores.2007.05.008
Peyronnet R, Ravens U (2019) Atria-selective antiarrhythmic drugs in need of alliance partners. Pharmacol Res 145:104262. https://doi.org/10.1016/j.phrs.2019.104262
Qi XY, Diness JG, Brundel BJ, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M, Nattel S (2014) Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129:430–440. https://doi.org/10.1161/CIRCULATIONAHA.113.003019
Rafizadeh S, Zhang Z, Woltz RL, Kim HJ, Myers RE, Lu L, Tuteja D, Singapuri A, Bigdeli AA, Harchache SB, Knowlton AA, Yarov-Yarovoy V, Yamoah EN, Chiamvimonvat N (2014) Functional interaction with filamin A and intracellular Ca2+ enhance the surface membrane expression of a small-conductance Ca2+-activated K+ (SK2) channel. Proc Natl Acad Sci U S A 111:9989–9994. https://doi.org/10.1073/pnas.1323541111
Reher TA, Wang Z, Hsueh CH, Chang PC, Pan Z, Kumar M, Patel J, Tan J, Shen C, Chen Z, Fishbein MC, Rubart M, Boyden P, Chen PS (2017) Small-conductance calcium-activated potassium current in normal rabbit cardiac purkinje cells. J Am Heart Assoc 6. https://doi.org/10.1161/JAHA.117.005471
Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH (1998) Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2+)-activated K+ channel. J Med Chem 41:2–5. https://doi.org/10.1021/jm970571a
Saljic A, Muthukumarasamy KM, la Cour JM, Boddum K, Grunnet M, Berchtold MW, Jespersen T (2019) Impact of arrhythmogenic calmodulin variants on small conductance Ca2+ -activated K+ (SK3) channels. Physiol Rep 7:e14210. https://doi.org/10.14814/phy2.14210
Skibsbye L, Diness JG, Sorensen US, Hansen RS, Grunnet M (2011) The duration of pacing-induced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca2+-activated K+ channels. J Cardiovasc Pharmacol 57:672–681. https://doi.org/10.1097/FJC.0b013e318217943d
Skibsbye L, Poulet C, Diness JG, Bentzen BH, Yuan L, Kappert U, Matschke K, Wettwer E, Ravens U, Grunnet M, Christ T, Jespersen T (2014) Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res 103:156–167. https://doi.org/10.1093/cvr/cvu121
Skibsbye L, Bengaard AK, Uldum-Nielsen AM, Boddum K, Christ T, Jespersen T (2018) Inhibition of small conductance calcium-activated potassium (SK) channels prevents arrhythmias in rat atria during beta-adrenergic and muscarinic receptor activation. Front Physiol 9:510. https://doi.org/10.3389/fphys.2018.00510
Stocker M (2004) Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurol 5:758–770. https://doi.org/10.1038/nrn1516
Stowe DF, Gadicherla AK, Zhou Y, Aldakkak M, Cheng Q, Kwok WM, Jiang MT, Heisner JS, Yang M, Camara AK (2013) Protection against cardiac injury by small Ca2+-sensitive K+ channels identified in guinea pig cardiac inner mitochondrial membrane. Biochim Biophys Acta 1828:427–442. https://doi.org/10.1016/j.bbamem.2012.08.031
Strassmaier T, Bond CT, Sailer CA, Knaus HG, Maylie J, Adelman JP (2005) A novel isoform of SK2 assembles with other SK subunits in mouse brain. J Biol Chem 280:21231–21236. https://doi.org/10.1074/jbc.M413125200
Tenma T, Mitsuyama H, Watanabe M, Kakutani N, Otsuka Y, Mizukami K, Kamada R, Takahashi M, Takada S, Sabe H, Tsutsui H, Yokoshiki H (2018) Small-conductance Ca2+-activated K+ channel activation deteriorates hypoxic ventricular arrhythmias via CaMKII in cardiac hypertrophy. Am J Physiol Heart Circ Physiol 315:H262–H272. https://doi.org/10.1152/ajpheart.00636.2017
Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G, Li W (2014) Sarcoplasmic reticulum Ca2+ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am J Physiol Heart Circ Physiol 306:H738–H746. https://doi.org/10.1152/ajpheart.00621.2013
Topkara VK, Mann DL (2010) Clinical applications of miRNAs in cardiac remodeling and heart failure. Personalized medicine 7:531–548. https://doi.org/10.2217/pme.10.44
Topkara VK, Mann DL (2011) Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc Drug Ther 25:171–182. https://doi.org/10.1007/s10557-011-6289-5
Torrente AG, Zhang R, Wang H, Zaini A, Kim B, Yue X, Philipson KD, Goldhaber JI (2017) Contribution of small conductance K+ channels to sinoatrial node pacemaker activity: insights from atrial-specific Na+/Ca2+ exchange knockout mice. J Physiol 595:3847–3865. https://doi.org/10.1113/JP274249
Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA, Chiamvimonvat N (2005) Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289:H2714–H2723. https://doi.org/10.1152/ajpheart.00534.2005
Tuteja D, Rafizadeh S, Timofeyev V, Wang S, Zhang Z, Li N, Mateo RK, Singapuri A, Young JN, Knowlton AA, Chiamvimonvat N (2010) Cardiac small conductance Ca2+-activated K+ channel subunits form heteromultimers via the coiled-coil domains in the C termini of the channels. Circ Res 107:851–859. https://doi.org/10.1161/CIRCRESAHA.109.215269
Voigt N, Dobrev D (2016) Atrial-selective potassium channel blockers. Card Electrophysiol Clin 8:411–421. https://doi.org/10.1016/j.ccep.2016.02.005
Wilde AAM, Garan H, Boyden PA (2019) Role of the Purkinje system in heritable arrhythmias. Heart Rhythm 16:1121–1126. https://doi.org/10.1016/j.hrthm.2019.01.034
Wrzosek A, Augustynek B, Zochowska M, Szewczyk A (2020) Mitochondrial potassium channels as druggable targets. Biomolecules 10. https://doi.org/10.3390/biom10081200
Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP (1998) Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395:503–507. https://doi.org/10.1038/26758
Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN, Chiamvimonvat N (2003) Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J Biol Chem 278:49085–49094. https://doi.org/10.1074/jbc.M307508200
Yang D, Wang T, Ni Y, Song B, Ning F, Hu P, Luo L, Wang Y, Ma A (2015) Apamin-sensitive K+ current upregulation in volume-overload heart failure is associated with the decreased interaction of CK2 with SK2. J Membr Biol 248:1181–1189. https://doi.org/10.1007/s00232-015-9839-0
Yang M, Camara AKS, Aldakkak M, Kwok WM, Stowe DF (2017) Identity and function of a cardiac mitochondrial small conductance Ca2+-activated K+ channel splice variant. Biochim Biophys Acta Bioenerg 1858:442–458. https://doi.org/10.1016/j.bbabio.2017.03.005
Yi F, Ling T-Y, Lu T, Wang X-L, Li J, Claycomb WC, Shen W-K, Lee H-C (2015) Down-regulation of the small conductance calcium-activated potassium channels in diabetic mouse atria. J Biol Chem 290:7016–7026. https://doi.org/10.1074/jbc.M114.607952
Yu T, Deng C, Wu R, Guo H, Zheng S, Yu X, Shan Z, Kuang S, Lin Q (2012) Decreased expression of small-conductance Ca2+-activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci 90:219–227. https://doi.org/10.1016/j.lfs.2011.11.008
Yu CC, Ko JS, Ai T, Tsai WC, Chen Z, Rubart M, Vatta M, Everett TH, George AL Jr, Chen PS (2016) Arrhythmogenic calmodulin mutations impede activation of small-conductance calcium-activated potassium current. Heart Rhythm 13:1716–1723. https://doi.org/10.1016/j.hrthm.2016.05.009
Zhang Q, Timofeyev V, Lu L, Li N, Singapuri A, Long MK, Bond CT, Adelman JP, Chiamvimonvat N (2008) Functional roles of a Ca2+-activated K+ channel in atrioventricular nodes. Circ Res 102:465–471. https://doi.org/10.1161/CIRCRESAHA.107.161778
Zhang X-D, Timofeyev V, Li N, Myers RE, Zhang D-M, Singapuri A, Lau VC, Bond CT, Adelman J, Lieu DK, Chiamvimonvat N (2014) Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc Res 101:317–325. https://doi.org/10.1093/cvr/cvt262
Zhang XD, Lieu DK, Chiamvimonvat N (2015) Small-conductance Ca2+-activated K+ channels and cardiac arrhythmias. Heart Rhythm 12:1845–1851. https://doi.org/10.1016/j.hrthm.2015.04.046
Zhang XD, Coulibaly ZA, Chen WC, Ledford HA, Lee JH, Sirish P, Dai G, Jian Z, Chuang F, Brust-Mascher I, Yamoah EN, Chen-Izu Y, Izu LT, Chiamvimonvat N (2018) Coupling of SK channels, L-type Ca2+ channels, and ryanodine receptors in cardiomyocytes. Sci rep 8:4670. https://doi.org/10.1038/s41598-018-22843-3
Zhao X, Wang Y, Sun X (2020) The functions of microRNA-208 in the heart. Diabetes Res Clin Pract 160:108004. https://doi.org/10.1016/j.diabres.2020.108004
Acknowledgements
The authors would like to acknowledge Ms. K Perkins for her help in manuscript editing.
Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) R01 HL085727, R01 HL085844, and R01 HL137228 (N.C.), and VA Merit Review Grant I01 BX000576 and I01 CX001490 (N.C.), NIH R56 HL138392 (X.D.Z), Postdoctoral Fellowship from NIH/NHLBI Institutional Training Grant in Basic and Translational Cardiovascular Science (NIH T32 HL086350) and NIH F32 HL149288 (P.N.T.), and California Institute of Regenerative Medicine (CIRM) Basic Biology Grant DISC2-10120 (D.K.L.). NC is the holder of the Roger Tatarian Endowed Professorship in Cardiovascular Medicine and a part-time staff physician at VA Northern California Health Care System, Mather, CA.
Author information
Authors and Affiliations
Contributions
The manuscript was first prepared by Xiao-Dong Zhang, Phung N. Thai, and Deborah K. Lieu. The manuscript was then revised by all authors. Figures were prepared and revised by Xiao-Dong Zhang, Phung N. Thai, and Nipavan Chiamvimonvat. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Calcium Signal Dynamics in Cardiac Myocytes and Fibroblasts: Mechanisms in Pflügers Archiv—European Journal of Physiology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, XD., Thai, P.N., Lieu, D.K. et al. Cardiac small-conductance calcium-activated potassium channels in health and disease. Pflugers Arch - Eur J Physiol 473, 477–489 (2021). https://doi.org/10.1007/s00424-021-02535-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02535-0