
Abstract
The steadily increasing obesity epidemic affects currently 30% of western populations and is causative for numerous disorders. It has been demonstrated that immune cells such as macrophages reside in or infiltrate metabolic organs under obese conditions and cause the so-called low-grade inflammation or metaflammation that impairs insulin action thus leading to the development of insulin resistance. Here, we report on data that specifically address macrophage biology/physiology in obesity-induced inflammation and insulin resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The steadily rising prevalence of obesity incorporates a major health issue because it is attended by fatal obesity-associated disorders including not only the development of type 2 diabetes and fatty liver diseases but also the rising incidence for certain cancer entities [7, 54]. In the first instance, obesity alters whole body metabolism that frequently results in insulin resistance [5]. Insulin is produced by the pancreatic beta cells in response to rising blood glucose levels, thereby leading to glucose uptake in insulin responsive organs. Excessive glucose is stored in the white adipose tissue (WAT) as lipids and in the liver as glycogen that can be converted back to glucose during fasting or periods of increased energy demands. All these metabolic processes and many more are controlled by insulin [93]. The actions of insulin are mediated via binding to the insulin receptor (IR) [5]. The IR and the homologous insulin-like growth factor 1 receptor (IGF1R) are receptor tyrosine kinases that use adaptor molecules for their downstream signaling [48]. These molecules belong to the insulin receptor substrate (IRS) family of proteins that upon engagement of the IR or IGF1R are tyrosine phosphorylated further leading to phosphatidylinositide 3-kinase (PI3K) and AKT activation. In diabetes patients, insulin action is impaired. While type 1 diabetes patients exhibit beta cell/insulin loss due to autoimmune reactions against the pancreas, type 2 diabetes develops as a consequence of insulin resistance that is frequent in obese patients. In obesity, the compromised glucose uptake into metabolic organs induces hyperglycemia in turn accelerating insulin production in beta cells. The excessive insulin production can partly compensate for decreased insulin sensitivity but progresses to increased beta cell mass and ultimately to beta cell death. How obesity facilitates the development of insulin resistance has been discovered over the last decade. Obesity has been accepted as low-grade inflammatory state that is also known as metaflammation [38]. Metaflammation is mainly derived from innate immune cells, e.g., macrophages whose derivation, fate, and functional consequences are discussed in this review.
Tissue resident macrophages
Macrophages are cells of the innate immune system that populate every organ. They display great functional plasticity and are required for maintenance of tissue homeostasis, immunity against invading pathogens, and tissue repair. Different organs harbor different specialized tissue resident macrophages, which include red pulp and marginal zone macrophages in the spleen, microglia in the brain, peritoneal macrophages, osteoclasts in the bone, alveolar macrophages in the lung, and the two major metabolic tissue macrophage subsets in the liver and adipose tissue—liver Kupffer cells and adipose tissue macrophages, respectively [73]. Certain tissue macrophage subsets populate their organs during embryogenesis, while in adulthood, tissue resident macrophage subsets are replenished by monocytes that are recruited from the bone marrow [22]. The two major subsets of monocytes are termed inflammatory and patrolling monocytes. They are distinguished by a defined panel of surface markers and chemokine receptors and have distinct chemotactic properties. Inflammatory monocytes are C-C chemokine receptor type 2 (CCR2) and lymphocyte antigen 6 c (Ly6C) positive, whereas patrolling monocytes are CCR2 and Ly6C negative, but express high levels of CX3 chemokine receptor 1 (CX3CR1). Expression of these different chemokine receptors allows them to follow diverging chemokine gradients. Under steady state, patrolling monocytes crawl along the vasculature where they function as immune sentinels. They can enter tissues through expression of CX3CR1 and differentiate into tissue resident cells with dendritic cell- and macrophage-like features [21, 86]. The degree of macrophage turnover under homeostatic conditions varies between organs [22]. Inflammatory monocytes are recruited to tissues in response to infection or tissue damage via chemokine (C-C motif) ligand 2 (CCL-2). In the absence of inflammation, they remain in the blood circulation [21].
Macrophage ontogeny
Precursors of several tissue macrophage subsets emerge during embryogenesis. Hematopoiesis during embryonic development occurs at two sites in two stages—first extra-embryonically in the yolk sac and later on in the fetal liver [22]. Hematopoiesis in the yolk sac was termed “primitive” and thought to solely preserve macrophage pools during embryogenesis, while tissue resident macrophages are derived from adult hematopoietic stem cells (HSC). However, fate-mapping studies revealed that numerous tissue resident macrophage populations are of prenatal origin and emerge from the yolk sac or the fetal liver. Microglia for example were shown to originate from the yolk sac, while Langerhans cells are mainly of fetal liver origin [23]. The use of (Myb-deficient) mice, which lack the bone marrow hematopoietic stem cell compartment, shed more light on the development of tissue resident macrophages [96]. These mice still develop tissue resident macrophage populations including Langerhans cells, Kupffer cells, microglia, lung alveolar, splenic red pulp, and peritoneal macrophages, indicating that yolk sac-derived progenitors give rise to several long-lasting tissue macrophage subsets beyond embryonic development [124]. Notably, these macrophage populations are maintained during adulthood under homeostatic conditions owing to their self-renewal potential.
It is an ongoing debate whether determination to the distinct tissue resident macrophage subsets occurs during embryonic development or is induced by signals from the local environment once macrophage populate their final tissue. On the one hand, some findings support the concept that macrophage precursors could already be committed to give rise to a certain subset by the time they enter the circulation to populate their target organ. Recent studies for example suggested that microglia are derived from a distinct yolk sac precursor, while other tissue resident macrophages derive from embryonic hematopoietic precursors [36, 98]. Other studies in contrast provided evidence that cues from the local tissue environment are crucial for differentiation into the distinct tissue resident macrophage subsets. By comparing different tissue resident macrophage populations, it has been assessed that tissue-specific factors shape the chromatin and enhancer landscape of macrophages thus enabling the transcription of subset-specific genes [27, 59]. Retinoic acid for example drives the differentiation of peritoneal macrophages by inducing the expression of the transcription factor GATA-6, which in turn activates a peritoneal macrophage-specific transcriptional program that is crucial for maintenance and functionality of these cells [81, 90]. The idea of in situ differentiation is supported by findings that tissue resident macrophages originate from a common erythro-myeloid progenitor (EMP) in the yolk sac, which populates the fetal liver before entering the blood stream to give rise to tissue resident macrophages [25]. Using single-cell RNA sequencing, a recent study investigated the chronology of macrophage differentiation during embryogenesis. EMPs give rise to premature macrophages that share a common gene expression signature. Induction of specific expression profiles is initiated during organogenesis when pre-mature macrophages enter the tissue [65].
Polarization states of macrophages
Tissue resident macrophages express a multitude of pattern recognition receptors (PRRs) that enable them to sense a wide range of microbial molecules and danger signals. Upon pathogen encounter, they induce a cascade that signals the quality of infection or danger, induces an inflammatory state, and recruits other immune cells to the site of infection [44]. Together with neutrophils, they produce bactericidal molecules and phagocytose pathogens to terminate the infection. After the infection has been cleared, they resolute inflammation by anti-inflammatory cytokines and lipid mediators and govern tissue repair by phagocytosing debris and promoting regeneration of extracellular matrix [73].
Macrophages exhibit a high degree of functional plasticity, and the nature of an inflammatory trigger as well as the local cytokine milieu will determine the respective macrophage polarization and its functional state. In this regard, early on, the distinction between classically activated and alternatively activated or—in analogy to the Th1/Th2 nomenclature of T cells—M1 and M2 macrophages has been made [64]. In vitro, these subsets can be induced by incubation with interferon gamma (IFNγ) and lipopolysaccharide (LPS) or interleukin-4 (IL-4), respectively. The M1/M2 nomenclature was in particular used to classify macrophages into cells with pro-inflammatory or anti-inflammatory properties or certain effector functions. The following studies revealed that distinct pro- or anti-inflammatory stimuli elicit distinct transcriptomic profiles, which led to the proposal of the spectrum model or more recently the multidimensional model [24]. However, many important findings were made using the aforementioned stimuli. Also, many classical M1 and M2 markers are still used to assess the functional state of macrophages in vivo. We thus will refer to macrophages with pro-inflammatory properties as Mpro and macrophages with anti-inflammatory properties as Manti.
Triggers like the Th1 cytokine IFNγ and/or toll-like receptor (TLR) ligands such as LPS initiate a pro-inflammatory response that equips macrophages to fight bacterial infections [64]. These stimuli activate signaling cascades that result in a global transcriptional reprogramming. In this context, signal transducer and activator of transcription (STAT) 1 and 2 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) are key transcription factors of IFNγ and TLR signaling, respectively [60]. Pro-inflammatory macrophages (Mpro) generally have inflammatory properties and are crucial for fighting bacterial infections and immunity against tumors. However, excessive activation can also result in tissue damage and autoimmunity. They present antigen and produce bactericidal agents such as reactive oxygen species (ROS) and nitric oxide (NO). The latter is synthesized by inducible nitric oxide synthase (iNOS) from arginine. They furthermore secrete a multitude of inflammatory cytokines including tumor necrosis factor alpha (TNFα), IL1-β, IL-6, IL-12, and IL-18 [63].
Three major pro-inflammatory cytokines are TNFα, Il-1β, and IL-6. TNFα is one of the first cytokines secreted by macrophages during infection and crucially involved in septic shock. It activates the vascular endothelium and initiates the acute phase in the liver. IL-6 also activates the acute phase and induces fever. In addition, it acts on lymphocytes and activates cytotoxic cells or stimulates differentiation of plasma cells. Depending on the signaling pathways that are activated upon receptor binding, IL-6 can also have anti-inflammatory properties [68]. IL-1β is a strong pyrogen, but in addition can induce the secretion of prostaglandins in the central nervous system. Notably, IL-1β and TNFα are both potent inducer of IL-6 and thereby amplify the inflammatory cascade [17]. IL-12 induces Th1 differentiation and together with IL-18 induces IFNγ production by Th1 and natural killer (NK) cells which in turn acts on macrophages in a feed forward loop [63].
Alternatively activated macrophages (Manti) were initially described during helminth infections and exhibit an anti-inflammatory phenotype. In vitro, they can be induced by the Th2 cytokines IL-4 and IL-13. Similarly as described for pro-inflammatory stimuli, binding of IL-4 to its receptor will result in a global transcriptional reprogramming. One of the key transcription factors mediating these changes is STAT6. It acts in concert with peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and peroxisome proliferator-activated receptor gamma and delta (PPARγ and PPARδ) which, as will be discussed below, are particularly involved in reprogramming cellular energy metabolism [60]. Manti are important players during helminth infection, response to tissue damage, resolution of inflammation, and wound healing, but can also foster fibrosis and tumor growth [26]. Their most important effector molecules include arginase, lectins, scavenger receptors and the cytokines IL-10 and the IL-1 receptor antagonist (IL1-RA). Arginase converts arginine to ornithine, which in turn is used during tissue repair for polyamine and collagen synthesis. Scavenger receptors and lectins mediate clearance of debris and apoptotic cells during resolution of inflammation, while IL-10 and IL-1RA are potent suppressors of inflammation [26, 63].
Macrophage metabolism
The differential use of arginine was an early indication that pro- and anti-inflammatory triggers induce diverging metabolic changes in macrophages. Over the last years, a series of discoveries have highlighted a tight linkage between cellular metabolism and macrophage effector functions [83]. While distinct metabolic features of many immune cells were described, we are just beginning to understand how nutrient availability can shape immune responses. In the following, we will briefly summarize these findings with a focus on glycolysis, β-oxidation, and amino acid metabolism.
Glycolysis describes the sequential breakdown of glucose to pyruvate, which is either converted to lactate and secreted or imported into mitochondria. When shuttled into mitochondria, pyruvate is converted to acetyl-coenzyme A (CoA), which enters the tricarboxylic acid (TCA) cycle by condensation with oxaloacetate. The TCA cycle generates NADH and FADH2, which are used to generate ATP via oxidative phosphorylation (OXPHOS). Apart from glycolysis, multiple catabolic pathways converge into the TCA cycle such as β-oxidation and glutaminolysis. β-Oxidation of fatty acids generates acetyl-CoA, while glutamine can enter the TCA by sequential conversion to α-ketoglutarate [15]. Apart from its central role in catabolism, the TCA cycle also serves as a metabolic hub that can redirect its intermediates for anabolic reactions when required. Citrate for example can be exported from mitochondria and cleaved to acetyl-CoA and oxaloacetate by ATP-citrate lyase. Acetyl-CoA in turn serves as a precursor for fatty acid or cholesterol biosynthesis and is substrate for acetylation reactions in the cytoplasm and the nucleus. OXPHOS is the most effective way for cells to generate ATP and is used by most quiescent cell types to cover their energetic demands [15]. OXPHOS, however, requires oxygen and has rather slow ATP generation kinetics. Thus, under hypoxic conditions or conditions of increased ATP demand, cells switch from OXPHOS to glycolysis to generate ATP. This phenomenon—glycolytic activity under normoxic conditions—is termed Warburg effect after its discoverer Otto Warburg. Initially discovered in cancer cells, proliferating cells or cells with high anabolic demands such as the developing embryo, epithelial cells, or activated immune cells exhibit Warburg metabolism [108]. Switching to glycolysis does not only allow for fast ATP generation, but also enables cells to diverge glycolytic intermediates into anabolic pathways such as amino acid, lipid, or nucleotide biosynthesis, instead of oxidizing them via TCA and OXPHOS [108].
Increased glycolysis was described early on in LPS-stimulated macrophages [83]. A series of recent studies has underpinned that commitment to Warburg metabolism equips macrophages to fulfill their effector functions such as production of ROS or NO, phagocytosis, and secretion of inflammatory mediators in the context of bacterial infection. Upon activation with pro-inflammatory stimuli like LPS or IFNγ, macrophages undergo metabolic reprogramming and exhibit increased rates of glycolysis and decreased OXPHOS. By diverting ATP generation from OXPHOS to glycolysis, mitochondria are available for ROS production [115]. Furthermore, instead of being used for ATP production, citrate is exported from mitochondria and used for fatty acid biosynthesis [42]. To compensate for decreased conversion of citrate to α-ketoglutarate, glutamine is funneled into the TCA via anaplerosis to α-ketoglutarate [102]. The molecular mechanisms driving these changes are just being uncovered. Some are NF-κB dependent: LPS for example induces PFKFB3, which increases glycolytic flux by generating fructose-2,6-bisphosphate [91]. Moreover, the NF-κB responsive gene hypoxia-inducible factor 1 alpha (HIF1α) was identified as another central metabolic regulator in response to LPS [12]. HIF1α is an essential mediator of the hypoxic response, partly by promoting a shift from OXPHOS to glycolysis. Under normoxic conditions, it is constantly degraded by the proteasome and only stabilized under hypoxic conditions. Ubiquitination and subsequent degradation of HIF1α are initiated by hydroxylation of proline residues by prolyl hydroxylases (PHDs). In certain cancer cells that exhibit Warburg metabolism, inhibition of PHDs can lead to HIF1α stabilization under normoxic conditions [97]. A similar effect was observed in response to LPS stimulation. In this context, ROS, succinate accumulation and iron were shown to contribute to PHD inhibition [76, 99, 102]. While ROS and succinate inhibit PHD activity, PHDs require iron as cofactor for the hydroxylation reaction. Indeed, ferritin decreases iron availability to PHDs after LPS stimulation and thereby downregulates PHD-dependent HIF1α hydroxylation [99]. While HIF1α sustains proliferation in cancer cells [97], it upregulates glycolysis [12], and also specifically boosts IL-1β expression [102] in response to LPS stimulation. As described before, arginine metabolism to citrulline and NO is a key effector function of pro-inflammatory macrophages. Indeed, changes in glutamine metabolism upon stimulation supply arginine and boost NO production by iNOS [47].
While LPS-activated macrophages mainly rely on glycolysis, IL-4-activated macrophages exhibit an oxidative phenotype [47]. Upon IL-4 stimulation, macrophages increase fatty acid uptake, β-oxidation, and OXPHOS [109]. These metabolic rearrangements are initiated by STAT6 and PGC-1β. In concert with PPARγ and PPARδ, they induce mitochondrial biogenesis and expression of genes that are involved in β-oxidation and OXPHOS [79]. These studies also highlighted a role for PPARs in the control of metabolic disease and maintaining insulin sensitivity. Furthermore, recent studies have highlighted the tight connection between macrophage metabolism and its effector functions in helminth infection and identified lysosomal lipolysis as alternative pathway to foster β-oxidation. Notably, the authors of this study showed that blockade of lysosomal liposysis during H. polygyrus infection results in defective clearance of the pathogen and inhibits commitment to OXPHOS by macrophages [41]. IL-4-stimulated macrophages maintain an intact active TCA cycle and generate ATP mainly via OXPHOS. While changes in β-oxidation are the most striking metabolic adaptations in response to IL-4, metabolomic studies also revealed that glycolysis as well as glutaminolysis contribute to TCA activity [47]. Notably, glutamine deprivation inhibits Manti macrophage induction in vitro [47], whereas inhibition of glycolysis only affects a small subset of IL-4 target genes [11]. Thus, while macrophages undergo drastic metabolic changes in response to pro- and anti-inflammatory triggers, like LPS and IL-4, respectively, their diverse effector functions and locations differentially affect whole body metabolism underlining their central role in organismal physiology.
Adipose tissue macrophages and Kupffer cells in homeostasis
Besides their role in sensing infection and tissue damage, tissue resident macrophages have important homeostatic and trophic functions. Limiting inflammation and maintaining tissue homeostasis are extra crucial functions of the liver resident Kupffer cells (KC) and adipose tissue macrophages (ATM). In obesity, insulin resistance develops as a consequence of metaflammation in which elevated circulating levels of pro-inflammatory cytokines such as TNFα and IL-6 negatively affect the insulin signaling cascade [37]. The main source for these inflammatory mediators in obesity is hepatic and WAT macrophages [122]. Macrophages adapt in their residing tissue to local circumstances and exert numerous effector functions such as phagocytosis and cytokine production. In the obese state, macrophages in the WAT and the liver are major players in regulating metaflammation. Macrophages sense factors derived from pathogens or from cells belonging to innate and adaptive systems as well as from specialized cells in the affected tissue. We will refer here on the impact of ATM and liver-derived KC in the development of obesity-associated insulin resistance.
Adipose tissue macrophages
Adipose tissue is one of the major metabolic organs that stores excess nutrients as triacylglycerides and releases fatty acids in the fasted state, which serve as energy source for peripheral tissues. Under homeostatic conditions, adipose tissue is populated with macrophages that exhibit a Manti like phenotype and govern adipocyte lipid metabolism by secreting factors such as IL-10 and catecholamines. IL-10 enhances adipocyte insulin sensitivity and lipogenesis [62], whereas catecholamines trigger lipolysis in adipocytes [75]. Under conditions of excessive lipolysis, they control release of fatty acids into the circulation by serving as buffer [55]. While the ontogeny of other tissue macrophage subsets is well studied, less is known about ATM. Under inflammatory conditions, monocytes enter adipose tissue in a CCR2-dependent manner [62]. The origin of ATMs under homeostatic conditions is a matter of debate. Interestingly, WAT contains a pool of c-Kit+/Lin−/Sca-1+ cells that share features of hematopoietic stem cells [10]. This population fails to populate bone marrow in non-irradiated mice, but is capable of replenishing the innate immune cell pool in adipose tissue [85]. ATMs might thus be regenerated in situ independent of the bone marrow.
A pioneering study from Hotamisligil and Spiegelman identified adipocytes as source of TNFα in the WAT that ultimately impaired insulin signaling in obesity [39]. However, findings by Xu et al. demonstrated that mainly the stromal vascular fraction of the obese WAT expresses inflammatory cytokines [122]. Currently, the view that the majority of other cells than adipocytes in the obese WAT are macrophages is supported, whereas in lean conditions, these cells represent approximately 10% [112]. While in lean WAT, mainly alternative Manti like macrophages express anti-inflammatory molecules, in obese, WAT macrophage polarization is shifted towards a pro-inflammatory Mpro like phenotype. The increased abundance and activation of macrophages in the obese WAT can be accounted by adipose tissue stress that includes elevated amounts of free fatty acids and LPS [28]. LPS, which is presumably microbiome derived, is not only abundant in WAT, but also found in the circulation of obese individuals [8]. LPS and fatty acids such as palmitate activate TLR4 signaling in ATMs that polarizes them towards Mpro macrophages [13, 50]. Subsequently, these stimuli trigger expression of TNFα and IL-6 in ATMs that compromise insulin action not only locally in the WAT, but also systemically since they are released to circulation [82]. Thus, it is tempting to speculate that metaflammation is a consequence of local innate immune response in the WAT that spills over via the blood to other organs due to the blood soluble factors involved. Of note, caloric restriction-induced weight loss including improvements in systemic insulin sensitivity and whole body glucose metabolism ameliorated metaflammation in the liver but not in adipose tissue suggesting that long-lived ATMs maintain WAT inflammation [95].
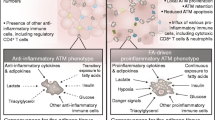
The Manti like ATMs in lean WAT express the cell surface marker CD206 and exhibit anti-inflammatory properties such as IL-10 expression. These Manti like ATMs synergize with regulatory T cells (Treg) and innate type 2 lymphoid cells (ILC2) to maintain the anti-inflammatory WAT environment [18, 70]. Tregs, T cells, ILC2 cells, and even adipocytes provide anti-inflammatory IL-4, IL-13, and IL-33 in the lean WAT to keep ATMs in an Manti like state [49, 117]. In the course of obesity, monocyte recruitment as well as local proliferation gives rise to novel ATMs that polarize towards a pro-inflammatory Mpro like phenotype that express the surface marker CD11c [51, 62, 113]. Abruptly, the WAT environment has changed from anti- to pro-inflammatory conditions indicated by the lack of Treg cells and infiltration of cytotoxic and Th1 T cells as well as NK cells [78, 114]. Besides the already mentioned Mpro polarizing LPS, T cells and NK cells in the obese WAT provide IFNγ, which sustains polarization towards the Mpro phenotype [114]. ATMs accumulate intracellular lipids not only via phagocytosis of dying adipocytes resulting in crown like structures in the WAT, but also via fatty acid transporter-mediated uptake [123]. The metabolism of obese ATMs has changed to glycolysis, which is necessary for the production of nitric oxide by iNos to increase pro-inflammatory macrophage responses [20]. Mpro macrophages take up glucose via glucose transporter 1 that further triggers Mpro polarization [19].
Therefore, it is not surprising that the polarizing environment in the obese WAT in vivo cannot be completely recapitulated in vitro in bone marrow-derived macrophages (BMDM) [123]. For instance, upregulation of CD11c expression is not induced by sole LPS and IFNγ treatment in BMDM, but can be restored by coculture with adipocytes [56]. Treatment with high glucose, insulin, and palmitate induces a Mpro like phenotype in BMDM culture that releases inflammatory cytokines [56]. Consistently, insulin receptor inactivation in macrophages prevents Mpro like polarization [66].
Conversely, Manti macrophages in vitro similarly as present in the lean WAT have not been reported, but Manti can be differentiated in BMDM cultures by several means. Supplementation of culture media with IL-4 or IL-13 creates different CD206 expressing Manti macrophages, than those that require TLR and IL1R agonists [61, 100]. Another Manti population can be differentiated by IL-10 which shares anti-inflammatory properties with the other two BMDM subtypes [72]. Strikingly, reactivity to IL-6 is required to polarize towards all of these Manti type macrophages. In particular, arginase 1 and IL-4Rα expression critically depend on IL-6 signaling [67]. Taken together, reallocation from Mpro like towards Manti type ATMs might be a promising strategy to resume whole body insulin sensitivity that would prevent fatal diseases associated with obesity such as development of metabolic syndrome and the progression to cancer. In line with this evidence, nematode infection or vaccination with nematode antigens reprograms the obese WAT microenvironment towards anti-inflammatory conditions resulting in improved insulin sensitivity and glucose tolerance [4].
Kupffer cells and liver infiltrating macrophages
The liver is essential for life due to its metabolic as well as immunoregulatory functions. On the one hand, under high energy conditions and hyperglycemia, hepatocytes in the liver import excessive glucose that is converted to glycogen. During fasting periods, the liver maintains blood glucose levels via hepatic glucose production that includes degradation of glycogen by glycogenolysis and breakdown of proteins and lipids through gluconeogenesis upon prolonged fasting. On the other, the liver is the first line defense against pathogens via the acute phase response and clears infected as well as exhausted cells. In line with its immune function, the blood stream entering the liver through the portal vein runs through the gut as well as the WAT before. In liver sinusoids, specialized liver resident macrophages, the KC, sense and combat invading commensals from the gut to prevent spreading along circulation [88]. Gut-derived LPS for example can be detected in portal vein but less in circulation [45]. In obesity, impaired storage of excessive lipids in the WAT leads to liver fat accumulation resulting in steatosis and fatty liver diseases [107]. The inappropriate fat storage in the liver results in lipotoxicity which in turn leads to liver damage and inflammation [74, 118]. Thus, in obesity, lipotoxicity and elevated microbial load from the microbiota result in excessive inflammation mediated by KCs and infiltrating macrophages. Interestingly, depletion of phagocytic cells in the liver via clodronate liposomes prevents steatosis, inflammation, and the development of insulin resistance thereby identifying hepatic macrophages as mediators of obesity-associated pathologies [58]. Hepatic macrophages crosstalk to liver non-parenchymal cells and adapt their polarization to states of liver condition. Obesity-induced pro-inflammatory cytokines and LPS polarize KC towards Mpro that in turn induce a vicious cycle of TNFα, IL-6, and IL-1β that further boosts and deteriorates liver functions [46, 53]. The inflammatory boost in obesity does not alter KC numbers but dramatically increases infiltration of CCR2-positive monocytes [52, 71]. Furthermore, inflammatory TNFα released by hepatic macrophages limits systemic insulin action, and IL-6 signaling in hepatocytes instructs downregulation of the inflammatory response in hepatic macrophages [120]. Collectively, inflammatory signaling in the liver differentially affects hepatic cell types and might result in complicating outcomes in whole body metabolism.
Obesity-induced low-grade inflammation and insulin resistance
Obesity contributes to the development of insulin resistance through the so-called obesity-associated low-grade inflammation or metaflammation. Over the course of this process, immune cells infiltrate metabolic organs, mainly WAT and liver, where they secrete pro-inflammatory cytokines that act locally but also systemically after being released into circulation [82].
The cytokine levels in obesity do not reach levels upon infection, but instead are elevated 2–3-fold compared to homeostatic conditions. Moreover, while during infection, pro-inflammatory cytokines increase acutely and stagnate with the elimination of the pathogen, the obesity-associated low-grade inflammation exhibits chronic character suggesting that dynamic modes of action have to be taken into account. The best-studied inflammatory players in obesity are TNFα and IL-6, but also include IL-17, CCL-2, and many others. In this paragraph, we will delineate how TNFα- and IL-6-induced signaling impact on the insulin signaling cascade.
TNFα
In a ground breaking report, Hotamisligil and colleagues discovered that the obese WAT contains high levels of the pro-inflammatory cytokine TNFα [39]. In a follow-up study, they could show in tissue culture experiments that media supplemented with TNFα impaired insulin action [40]. On a molecular level, TNFα compromises activating tyrosine phosphorylations in the insulin signaling cascade mainly of IRS molecules, but also the IR. While at that time, adipocytes were believed to be the source of TNFα in obesity, Xu et al. demonstrated that the stromal vascular fraction of WAT secretes pro-inflammatory cytokines that inhibit insulin signaling [122]. Moreover, bone marrow transplantation experiments revealed that mainly macrophages are the source of TNFα in the obese WAT [14]. TNFα interferes with insulin recpetor signaling at the level of IRS molecules. IRS molecules are phosphorylated on inhibitory serine residues by TNFα-induced kinases such as IκB kinase (IKK), c-Jun N-terminal kinase (JNK), and atypical protein kinase C (aPKC) thereby preventing further downstream signaling [77]. Of note, these kinases have redundant as well as individual functions in IRS phosphorylation and point mutations of IRS1 serine residues to non-phosporylatable counterparts yielded the conflicting result that mutant mice developed insulin resistance [9]. Nevertheless, genetic mouse models provided novel insight into how TNFα-induced signaling interferes with insulin signaling in obesity. On the one hand, TNFα knockout mice exhibit normal glucose tolerance when exposed to normal food, but are protected from the development of obesity-induced insulin resistance in the absence of body weight gain alterations on the other [106]. While this study demonstrated the critical importance of TNFα in the development of insulin resistance, the dissection of further downstream signaling at the TNFα-induced kinase level has revealed surprising results. TNFα induces a dual kinase system that comprises the IKK complex and the JNK kinases [101]. The IKK complex contains the kinases IKK-1 and IKK-2 as well as the NFκB essential modulator NEMO, all of which are essential for mouse viability as revealed by knockout studies. Muscle-specific IKK-2 inactivation showed no effect on diet-induced obesity and alterations in glucose homeostasis [89]. However, while hepatic IKK-2 inactivation conferred insulin sensitivity in this organ, but not in muscle and WAT, myeloid IKK-2 inactivation resulted in systemic improvements of insulin sensitivity upon high-fat diet (HFD) challenge mainly due to reduced inflammatory cytokine release [1, 6]. Otherwise, hepatic NEMO inactivation resulted in global improvements in insulin sensitivity under obese conditions, but in contrast to IKK-2 KO mice, these mice developed liver tumors due to ongoing TNFα-induced cell death and compensatory hyperproliferation [119]. Thus, though activated by the same upstream stimulus, kinases may play redundant and non-redundant roles in impairing the actions of insulin.
In contrast to the IKK complex genes, knockout of one of the three individual JNK kinases (JNK-1, JNK-2, and JNK-3) is well tolerated in mice, whereas double knockout for the most abundant peripheral JNK-1 and JNK-2 is embryonic lethal [57]. It has been shown that JNK-1 but not JNK-2 knockout mice are protected against obesity-induced impairments of glucose homeostasis suggesting an essential role for JNK-1 in serine phosphorylation of IRS molecules [35]. However, conditional mouse models aimed at unraveling the cell type-specific as well as redundant functions of the JNK genes in the development of obesity-associated insulin resistance. Opposite to what was expected, hepatic inactivation of JNK-1 revealed a modestly impaired glucose tolerance and hepatic lipid accumulation suggesting a function of JNK-1 in the prevention of steatosis and liver fat accumulation [110]. Moreover, skeletal muscle-specific JNK-1 deficiency revealed a minor role in glucose metabolism [84], whereas WAT-specific JNK-1 deletion decreased obesity-induced IL-6 levels and thus ameliorated diet-induced insulin resistance [92]. However, neuronal-specific JNK-1 deficiency most closely resembled the phenotype of complete JNK-1 knockout mice indicating that a redundancy between JNK isoforms in peripheral organs exist [3]. In line with these findings, JNK-1/JNK-2 double-deficient macrophages are unable to produce inflammatory cytokines, and thus, mice with macrophage-specific deletion of JNK-1 and 2 are protected against obesity-induced disorders [30, 31]. Collectively, deciphering organ-specific downstream actions of TNFα in obesity-induced insulin resistance revealed redundant as well as non-redundant kinase functions on inhibitory IRS serine phosphorylation.
IL-6
IL-6 is a pleiotropic cytokine that plays crucial roles in metabolic and immune cells. Similar to TNFα, IL-6 is also slightly increased in serum of obese individuals and mice, which is believed to be detrimental for metabolism [2]. Here, a bulk of IL-6 is produced by the stromal vascular fraction of visceral fat depots, which is directly delivered to the liver via the portal vein [92]. In contrast, IL-6 is increased manifold during intense exercise in muscle (regulated by JNK-1) that provides beneficial effects on metabolism [116]. IL-6 exerts its function by binding to the IL-6 receptor α chain (IL-6Rα) and the GP130 signaling chain complex in classical membrane-bound pathway. The IL-6Rα is expressed mainly on hepatocytes and immune cells, but also non-IL-6Rα-expressing cells can be rendered IL-6-responsive by a mechanism called trans-signaling [33, 94]. IL-6 trans-signaling is the process where adam proteases cleave/shed the IL-6Rα from the surface of IL-6Rα-expressing cells that when bound to serum IL-6 generates the soluble IL-6Rα (sIL-6Rα). sIL-6Rα in turn binds ubiquitously expressed GP130 on cells not expressing IL-6Rα to activate the same signaling cascade as the classical membrane IL-6Rα signaling [94]. Both cascades initiate Janus kinase (JAK)2/STAT-3-dependent transcriptional activation of target genes such as SOCS-3 [33]. SOCS-3 is not only a negative regulator of IL-6 signaling but also inhibits insulin signal transduction at the IRS protein level. Here, IL-6-induced SOCS-3 leads to ubiquitination and subsequent proteasomal degradation of IRS1 [104, 105]. Consistently, clinical studies link obesity-induced insulin resistance with increased IL-6 levels. Importantly, weight loss reduces circulating IL-6 and improves insulin sensitivity [2]. On the other hand, however, IL-6 itself provides beneficial effects on metabolic processes such as regulation of hepatic gluconeogenesis indicating that the molecular mechanism of how IL-6 affects metabolism and insulin sensitivity is not completely understood [43, 87]. When IL-6 would exert negative effects on glucose metabolism exclusively, the expectation for IL-6 knockout mice would be the maintenance of insulin sensitivity. However, while Di Gregorio et al. did not observe metabolic alterations in IL-6 knockout mice [16], Wallenius and colleagues demonstrated that IL-6 inactivation favors the development of mature onset obesity and diabetes implicating that IL-6 action on metabolism might be even more complex than hitherto assumed [111].
A potential aspect that may explain these differences might be the chronic/constant presence of IL-6 under obesity conditions. We have demonstrated that in diet-induced obesity, the chronically high IL-6 levels lead to the development of hepatic IL-6 resistance [29]. IL-6 resistance is caused by basal IL-6-activated STAT3 that chronically increases expression of SOCS-3 [121]. SOCS-3 in turn inhibits IL-6 receptor signaling, which can be identified by the inability of liver cells to react with STAT3 phosphorylation upon exogenous IL-6 treatment. Such high hepatic SOCS-3 levels might not only have impact on IL-6 signaling, but also on the insulin receptor signaling cascade by interfering with IRS proteins. Consistently, inactivation of SOCS-3 in hepatocytes improves hepatic insulin action and steatosis in young mice, but at older age, these mice develop obesity and insulin resistance due to the activation of acute phase response and overt inflammation [103]. IL-6 signaling in hepatocytes therefore somehow crosstalks with liver resident KCs that are the source for the inflammatory response. In line with this evidence, inactivation of the IL-6 receptor in hepatocytes fulminates in the development of systemic insulin resistance as a consequence of KC-mediated inflammation. Thus, IL-6 signaling in hepatocytes controls whole body insulin sensitivity by limiting KC-mediated inflammation [120]. Therefore, considering the differential aspects of IL-6 action under lean and obese conditions will contribute to our molecular knowledge how the low-grade metaflammation impacts on insulin signaling to ultimately result in the development of metabolic disorders. Given that IL-6 not only impacts on metabolism but also on the development of cancer and that obesity increases the incidence of cancer entities with an inflammatory microenvironment, the context-specific dissection of signaling cascades will be necessary for the development of novel therapeutic interventions to combat such fatal obesity-associated diseases [68].
Conclusion/outlook
Inflammation triggered by macrophages constitutes a turning point in the development of obesity-related insulin resistance. It is not only that mediators secreted by macrophages trigger insulin resistance, at the same time, also beneficial effects exerted by ATM and KCs under homeostasis are compromised. These include maintenance of a local anti-inflammatory milieu, insulin sensitivity, and control of lipolysis and energy expenditure. Research over the last 15 years has uncovered mechanisms that drive changes in macrophage polarization and how molecules secreted by macrophages affect lipid metabolism and insulin receptor signaling in metabolic organs. The use of mouse models pointed out that macrophage polarization in adipose tissue and the liver is critical for development and progression of metabolic disease. Reprogramming from inflammatory Mpro towards alternative Manti macrophages might represent a promising strategy to recover whole body insulin sensitivity that would prevent fatal diseases associated with obesity. Indeed, thiazolidinediones and omega 3 fatty acids are such exemplary drugs or dietary factors that improve metabolic disease partly by dampening macrophage-mediated inflammation [34, 80]. In this context, targeting immune cell metabolism might also hold great potential. Systemic insulin resistance is also observed during pregnancy or puberty, but most notably also during infection, in particular sepsis. During sepsis, inflammatory cytokines in particular IL-6 and TNFα induce a state of insulin resistance [69]. Moreover, while in type 2 diabetes, lipolysis in adipose tissue and gluconeogenesis in the liver slowly emerge as a result of insulin resistance, inflammatory cytokines actively induce lipolysis in adipose tissue, protein catabolism in the muscle, and gluconeogenesis in the liver and muscle during sepsis. Central mediators include glucagon, epinephrine, and cortisol, which are released as a consequence of the inflammatory cascade [32]. From an evolutionary point of view, this metabolic response serves to support the immune system in fighting the infection and limiting its spread throughout the body in the context of sepsis. As outlined above Mpro macrophages and also other immune cells rely on glycolysis to fuel their inflammatory response to fight infection [83]. In the context of type 2 diabetes and obesity, increased glucose availability to immune cells might initiate a feed forward loop that fosters inflammation and further aggravates disease.
References
Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M (2005) IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11:191–198. doi:10.1038/nm1185
Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B (2000) Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab 85:3338–3342. doi:10.1210/jcem.85.9.6839
Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, Bruning JC (2010) Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci U S A 107:6028–6033. doi:10.1073/pnas.1001796107
Berbudi A, Surendar J, Ajendra J, Gondorf F, Schmidt D, Neumann AL, Wardani AP, Layland LE, Hoffmann LS, Pfeifer A, Hoerauf A, Hubner MP (2016) Filarial infection or antigen administration improves glucose tolerance in diet-induced obese mice. J Innate Immun 8:601–616. doi:10.1159/000448401
Boucher J, Kleinridders A, Kahn CR (2014) Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6. doi:10.1101/cshperspect.a009191
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190. doi:10.1038/nm1166
Calle EE, Kaaks R (2004) Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4:579–591. doi:10.1038/nrc1408
Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NO, Curi R, Carvalheira JB, Saad MJ (2011) Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol 9:e1001212. doi:10.1371/journal.pbio.1001212
Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF (2010) Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab 11:84–92. doi:10.1016/j.cmet.2009.11.003
Corre J, Barreau C, Cousin B, Chavoin JP, Caton D, Fournial G, Penicaud L, Casteilla L, Laharrague P (2006) Human subcutaneous adipose cells support complete differentiation but not self-renewal of hematopoietic progenitors. J Cell Physiol 208:282–288. doi:10.1002/jcp.20655
Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, Zhang Y, Blair IA, Horng T (2016) Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. elife 5. doi:10.7554/eLife.11612
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657
Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME (2008) Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 16:1248–1255. doi:10.1038/oby.2008.210
De Taeye BM, Novitskaya T, McGuinness OP, Gleaves L, Medda M, Covington JW, Vaughan DE (2007) Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab 293:E713–E725. doi:10.1152/ajpendo.00194.2007
DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2:e1600200. doi:10.1126/sciadv.1600200
Di Gregorio GB, Hensley L, Lu T, Ranganathan G, Kern PA (2004) Lipid and carbohydrate metabolism in mice with a targeted mutation in the IL-6 gene: absence of development of age-related obesity. Am J Physiol Endocrinol Metab 287:E182–E187. doi:10.1152/ajpendo.00189.2003
Dinarello CA (2007) Historical insights into cytokines. Eur J Immunol 37(Suppl 1):S34–S45. doi:10.1002/eji.200737772
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D (2009) Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15:930–939. doi:10.1038/nm.2002
Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L (2014) Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289:7884–7896. doi:10.1074/jbc.M113.522037
Galvan-Pena S, O’Neill LA (2014) Metabolic reprograming in macrophage polarization. Front Immunol 5:420. doi:10.3389/fimmu.2014.00420
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82
Ginhoux F, Guilliams M (2016) Tissue-resident macrophage ontogeny and homeostasis. Immunity 44:439–449. doi:10.1016/j.immuni.2016.02.024
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845. doi:10.1126/science.1194637
Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK (2016) New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17:34–40. doi:10.1038/ni.3324
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551. doi:10.1038/nature13989
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35. doi:10.1038/nri978
Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK (2014) Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159:1327–1340. doi:10.1016/j.cell.2014.11.023
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445. doi:10.1146/annurev-immunol-031210-101322
Gruber S, Straub BK, Ackermann PJ, Wunderlich CM, Mauer J, Seeger JM, Buning H, Heukamp L, Kashkar H, Schirmacher P, Bruning JC, Wunderlich FT (2013) Obesity promotes liver carcinogenesis via mcl-1 stabilization independent of IL-6Ralpha signaling. Cell Rep 4:669–680. doi:10.1016/j.celrep.2013.07.023
Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ (2013) JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339:218–222. doi:10.1126/science.1227568
Han MS, Barrett T, Brehm MA, Davis RJ (2016) Inflammation mediated by JNK in myeloid cells promotes the development of hepatitis and hepatocellular carcinoma. Cell Rep 15:19–26. doi:10.1016/j.celrep.2016.03.008
Hartl WH, Jauch KW (2014) Metabolic self-destruction in critically ill patients: origins, mechanisms and therapeutic principles. Nutrition 30:261–267. doi:10.1016/j.nut.2013.07.019
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334(Pt 2):297–314
Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass CK, Ricote M (2007) Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 117:1658–1669. doi:10.1172/JCI31561
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336. doi:10.1038/nature01137
Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, Poidinger M, Zolezzi F, Larbi A, Ng LG, Chan JK, Greter M, Becher B, Samokhvalov IM, Merad M, Ginhoux F (2015) C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42:665–678. doi:10.1016/j.immuni.2015.03.011
Hotamisligil GS (2003) Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 27(Suppl 3):S53–S55. doi:10.1038/sj.ijo.0802502
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867. doi:10.1038/nature05485
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM (1994) Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 91:4854–4858
Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr, Artyomov MN, Pearce EL, Pearce EJ (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15:846–855. doi:10.1038/ni.2956
Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V (2011) The mitochondrial citrate carrier: a new player in inflammation. Biochem J 438:433–436. doi:10.1042/BJ20111275
Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, Teshigawara K, Matsuki Y, Watanabe E, Hiramatsu R, Notohara K, Katayose K, Okamura H, Kahn CR, Noda T, Takeda K, Akira S, Inui A, Kasuga M (2006) Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 3:267–275. doi:10.1016/j.cmet.2006.02.009
Iwasaki A, Medzhitov R (2015) Control of adaptive immunity by the innate immune system. Nat Immunol 16:343–353. doi:10.1038/ni.3123
Jacob AI, Goldberg PK, Bloom N, Degenshein GA, Kozinn PJ (1977) Endotoxin and bacteria in portal blood. Gastroenterology 72:1268–1270
Jager J, Aparicio-Vergara M, Aouadi M (2016) Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med 280:209–220. doi:10.1111/joim.12483
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42:419–430. doi:10.1016/j.immuni.2015.02.005
Kadowaki T, Tobe K, Honda-Yamamoto R, Tamemoto H, Kaburagi Y, Momomura K, Ueki K, Takahashi Y, Yamauchi T, Akanuma Y, Yazaki Y (1996) Signal transduction mechanism of insulin and insulin-like growth factor-1. Endocr J 43(Suppl):S33–S41
Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, Lee CH (2008) Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 7:485–495. doi:10.1016/j.cmet.2008.04.002
Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW (2007) Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 100:1589–1596. doi:10.1161/CIRCRESAHA.106.142851
Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto K, Ginsberg HN, Mukaida N, Kaneko S, Ota T (2012) CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 61:1680–1690. doi:10.2337/db11-1506
Klueh U, Czajkowski C, Ludzinska I, Qiao Y, Frailey J, Kreutzer DL (2016) Impact of CCL2 and CCR2 chemokine/receptor deficiencies on macrophage recruitment and continuous glucose monitoring in vivo. Biosens Bioelectron 86:262–269. doi:10.1016/j.bios.2016.06.026
Kolios G, Valatas V, Kouroumalis E (2006) Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol 12:7413–7420
Kopelman PG (2000) Obesity as a medical problem. Nature 404:635–643. doi:10.1038/35007508
Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW Jr (2010) Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120:3466–3479. doi:10.1172/JCI42845
Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L (2014) Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20:614–625. doi:10.1016/j.cmet.2014.08.010
Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22:667–676
Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA (2010) Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol 298:G107–G116. doi:10.1152/ajpgi.00391.2009
Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I (2014) Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326. doi:10.1016/j.cell.2014.11.018
Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11:750–761. doi:10.1038/nri3088
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, Senior RM, Elias JA (2001) Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194:809–821
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184. doi:10.1172/JCI29881
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. doi:10.1016/j.it.2004.09.015
Mantovani A, Sica A, Locati M (2005) Macrophage polarization comes of age. Immunity 23:344–346. doi:10.1016/j.immuni.2005.10.001
Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, Jacome-Galarza CE, Handler K, Klughammer J, Kobayashi Y, Gomez-Perdiguero E, Schultze JL, Beyer M, Bock C, Geissmann F (2016) Specification of tissue-resident macrophages during organogenesis. Science 353. doi:10.1126/science.aaf4238
Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, Kolanus W, Kahn CR, Bruning JC (2010) Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet 6:e1000938. doi:10.1371/journal.pgen.1000938
Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Bronneke HS, Estevez E, Allen TL, Mesaros A, Partridge L, Febbraio MA, Chawla A, Wunderlich FT, Bruning JC (2014) Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15:423–430. doi:10.1038/ni.2865
Mauer J, Denson JL, Bruning JC (2015) Versatile functions for IL-6 in metabolism and cancer. Trends Immunol 36:92–101. doi:10.1016/j.it.2014.12.008
Mizock BA (2001) Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab 15:533–551. doi:10.1053/beem.2001.0168
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM (2013) Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210:535–549. doi:10.1084/jem.20121964
Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ, Chung H, Bogner-Strauss JG, Evans RM, Olefsky JM, Oh DY (2015) Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64:1120–1130. doi:10.2337/db14-1238
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969. doi:10.1038/nri2448
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. doi:10.1038/nri3073
Neuschwander-Tetri BA (2010) Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52:774–788. doi:10.1002/hep.23719
Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A (2011) Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480:104–108. doi:10.1038/nature10653
Nicholas SA, Sumbayev VV (2010) The role of redox-dependent mechanisms in the downregulation of ligand-induced toll-like receptors 7, 8 and 4-mediated HIF-1 alpha prolyl hydroxylation. Immunol Cell Biol 88:180–186. doi:10.1038/icb.2009.76
Nieto-Vazquez I, Fernandez-Veledo S, Kramer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M (2008) Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem 114:183–194. doi:10.1080/13813450802181047
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R (2009) CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15:914–920. doi:10.1038/nm.1964
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A (2007) Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447:1116–1120. doi:10.1038/nature05894
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142:687–698. doi:10.1016/j.cell.2010.07.041
Okabe Y, Medzhitov R (2014) Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157:832–844. doi:10.1016/j.cell.2014.04.016
Olefsky JM, Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72:219–246. doi:10.1146/annurev-physiol-021909-135846
O’Neill LA, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16:553–565. doi:10.1038/nri.2016.70
Pal M, Wunderlich CM, Spohn G, Bronneke HS, Schmidt-Supprian M, Wunderlich FT (2013) Alteration of JNK-1 signaling in skeletal muscle fails to affect glucose homeostasis and obesity-associated insulin resistance in mice. PLoS One 8:e54247. doi:10.1371/journal.pone.0054247
Poglio S, De Toni F, Lewandowski D, Minot A, Arnaud E, Barroca V, Laharrague P, Casteilla L, Cousin B (2012) In situ production of innate immune cells in murine white adipose tissue. Blood 120:4952–4962. doi:10.1182/blood-2012-01-406959
Poltorak MP, Schraml BU (2015) Fate mapping of dendritic cells. Front Immunol 6:199. doi:10.3389/fimmu.2015.00199
Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B, Peng X, Krause DM, Korytnaya E, Neidert A, Liddle C, Yu RT, Lumeng CN, Oral EA, Downes M, Evans RM, Saltiel AR (2015) A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat Commun 6:6047. doi:10.1038/ncomms7047
Robinson MW, Harmon C, O’Farrelly C (2016) Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol 13:267–276. doi:10.1038/cmi.2016.3
Rohl M, Pasparakis M, Baudler S, Baumgartl J, Gautam D, Huth M, De Lorenzi R, Krone W, Rajewsky K, Bruning JC (2004) Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest 113:474–481. doi:10.1172/JCI18712
Rosas M, Davies LC, Giles PJ, Liao CT, Kharfan B, Stone TC, O’Donnell VB, Fraser DJ, Jones SA, Taylor PR (2014) The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 344:645–648. doi:10.1126/science.1251414
Ruiz-Garcia A, Monsalve E, Novellasdemunt L, Navarro-Sabate A, Manzano A, Rivero S, Castrillo A, Casado M, Laborda J, Bartrons R, Diaz-Guerra MJ (2011) Cooperation of adenosine with macrophage toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene. J Biol Chem 286:19247–19258. doi:10.1074/jbc.M110.190298
Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ (2008) A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322:1539–1543. doi:10.1126/science.1160794
Saltiel AR (2016) Insulin signaling in the control of glucose and lipid homeostasis. Handb Exp Pharmacol 233:51–71. doi:10.1007/164_2015_14
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813:878–888. doi:10.1016/j.bbamcr.2011.01.034
Schmitz J, Evers N, Awazawa M, Nicholls HT, Bronneke HS, Dietrich A, Mauer J, Bluher M, Bruning JC (2016) Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol Metab 5:328–339. doi:10.1016/j.molmet.2015.12.001
Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90. doi:10.1126/science.1219179
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85. doi:10.1016/j.ccr.2004.11.022
Sheng J, Ruedl C, Karjalainen K (2015) Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43:382–393. doi:10.1016/j.immuni.2015.07.016
Siegert I, Schodel J, Nairz M, Schatz V, Dettmer K, Dick C, Kalucka J, Franke K, Ehrenschwender M, Schley G, Beneke A, Sutter J, Moll M, Hellerbrand C, Wielockx B, Katschinski DM, Lang R, Galy B, Hentze MW, Koivunen P, Oefner PJ, Bogdan C, Weiss G, Willam C, Jantsch J (2015) Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1alpha upon LPS activation in the presence of ample oxygen. Cell Rep 13:2048–2055. doi:10.1016/j.celrep.2015.11.005
Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A (2010) Heme oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 215:788–795. doi:10.1016/j.imbio.2010.05.020
Tang F, Tang G, Xiang J, Dai Q, Rosner MR, Lin A (2002) The absence of NF-kappaB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol 22:8571–8579
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496:238–242. doi:10.1038/nature11986
Torisu T, Sato N, Yoshiga D, Kobayashi T, Yoshioka T, Mori H, Iida M, Yoshimura A (2007) The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 12:143–154. doi:10.1111/j.1365-2443.2007.01044.x
Ueki K, Kondo T, Kahn CR (2004a) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 24:5434–5446. doi:10.1128/MCB.24.12.5434-5446.2004
Ueki K, Kondo T, Tseng YH, Kahn CR (2004b) Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci U S A 101:10422–10427. doi:10.1073/pnas.0402511101
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389:610–614. doi:10.1038/39335
van Herpen NA, Schrauwen-Hinderling VB (2008) Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav 94:231–241. doi:10.1016/j.physbeh.2007.11.049
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. doi:10.1126/science.1160809
Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4:13–24. doi:10.1016/j.cmet.2006.05.011
Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, Kim JK, Xu J, Shulha HP, Garber M, Gao G, Davis RJ (2014) The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab 20:512–525. doi:10.1016/j.cmet.2014.06.010
Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, Ohlsson C, Jansson JO (2002) Interleukin-6-deficient mice develop mature-onset obesity. Nat Med 8:75–79. doi:10.1038/nm0102-75
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808. doi:10.1172/JCI19246
Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr (2006) CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116:115–124. doi:10.1172/JCI24335
Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, Bruning JC, Mandelboim O, Polic B (2015) NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16:376–385. doi:10.1038/ni.3120
West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–480. doi:10.1038/nature09973
Whitham M, Chan MH, Pal M, Matthews VB, Prelovsek O, Lunke S, El-Osta A, Broenneke H, Alber J, Bruning JC, Wunderlich FT, Lancaster GI, Febbraio MA (2012) Contraction-induced interleukin-6 gene transcription in skeletal muscle is regulated by c-Jun terminal kinase/activator protein-1. J Biol Chem 287:10771–10779. doi:10.1074/jbc.M111.310581
Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, Maezawa Y, Drucker DJ, Engleman E, Winer D, Dosch HM (2009) Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15:921–929. doi:10.1038/nm.2001
Wree A, Kahraman A, Gerken G, Canbay A (2011) Obesity affects the liver - the link between adipocytes and hepatocytes. Digestion 83:124–133. doi:10.1159/000318741
Wunderlich FT, Luedde T, Singer S, Schmidt-Supprian M, Baumgartl J, Schirmacher P, Pasparakis M, Bruning JC (2008) Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc Natl Acad Sci U S A 105:1297–1302. doi:10.1073/pnas.0707849104
Wunderlich FT, Strohle P, Konner AC, Gruber S, Tovar S, Bronneke HS, Juntti-Berggren L, Li LS, van Rooijen N, Libert C, Berggren PO, Bruning JC (2010) Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12:237–249. doi:10.1016/j.cmet.2010.06.011
Wunderlich CM, Hovelmeyer N, Wunderlich FT (2013) Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAKSTAT 2:e23878. doi:10.4161/jkst.23878
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830. doi:10.1172/JCI19451
Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr (2013) Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18:816–830. doi:10.1016/j.cmet.2013.11.001
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. doi:10.1016/j.immuni.2012.12.001
Acknowledgements
Open access funding provided by Max Planck Society. This work was supported by funds of the Deutsche Forschungsgemeinschaft (SFB1123, MARL, SFB670, FTW, TR134, FTW, KFO286, FTW, CECAD, FTW) and the Deutsche Krebshilfe (FTW).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on macrophages in tissue homeostasis in Pflügers Archiv – European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lauterbach, M.A.R., Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflugers Arch - Eur J Physiol 469, 385–396 (2017). https://doi.org/10.1007/s00424-017-1955-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-1955-5