
Abstract
Inward rectifier K+ channels are important for maintaining normal electrical function in many cell types. The proper function of these channels requires the presence of membrane phosphoinositide 4,5-bisphosphate (PIP2). Stimulation of the Ca2+-sensing receptor CaR, a pleiotropic G protein-coupled receptor, activates both Gq/11, which decreases PIP2, and phosphatidylinositol 4-kinase (PI-4-K), which, conversely, increases PIP2. How membrane PIP2 levels are regulated by CaR activation and whether these changes modulate inward rectifier K+ are unknown. In this study, we found that activation of CaR by the allosteric agonist, NPSR568, increased inward rectifier K+ current (I K1) in guinea pig ventricular myocytes and currents mediated by Kir2.1 channels exogenously expressed in HEK293T cells with a similar sensitivity. Moreover, using the fluorescent PIP2 reporter tubby-R332H-cYFP to monitor PIP2 levels, we found that CaR activation in HEK293T cells increased membrane PIP2 concentrations. Pharmacological studies showed that both phospholipase C (PLC) and PI-4-K are activated by CaR stimulation with the latter played a dominant role in regulating membrane PIP2 and, thus, Kir currents. These results provide the first direct evidence that CaR activation upregulates currents through inward rectifier K+ channels by accelerating PIP2 synthesis. The regulation of I K1 plays a critical role in the stability of the electrical properties of many excitable cells, including cardiac myocytes and neurons. Further, synthetic allosteric modulators that increase CaR activity have been used to treat hyperparathyroidism, and negative CaR modulators are of potential importance in the treatment of osteoporosis. Thus, our results provide further insight into the roles played by CaR in the cardiovascular system and are potentially valuable for heart disease treatment and drug safety.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kir2.x channels are inward rectifier K+ channels that play an important role in maintaining stable resting membrane potentials, controlling excitability, and shaping the initial depolarization and final repolarization of ventricular myocytes [13, 19, 21, 27]. Gain and loss of function of Kir2.x channels, which mediate cardiac inwardly rectifying currents (I K1), can cause reentry and arrhythmia, respectively [19]. Reentry is facilitated by shortening of the action potential duration (APD), which abbreviates refractoriness. On the other hand, excessive APD prolongation may cause torsades de pointes arrhythmia and sudden cardiac death [25].
We previously demonstrated that extracellular spermine inhibits the outward current through Kir2.1 channels expressed in oocytes and the outward I K1 of myocytes, but the effect was much greater in oocytes than in cardiac myocytes [3]. However, why the effects of extracellular spermine are quantitatively different in oocytes and guinea pig myocytes is unclear [3]. It may be simply attributable to the fact that the effects of spermine on Kir channels in different cell types vary. Alternatively, the actions of extracellular spermine may be more diverse in complex cell types such as cardiac myocytes. For example, extracellular spermine can activate the calcium-sensing receptor, CaR [34]. This receptor was first discovered in the parathyroid gland, where its activation by extracellular Ca2+ was shown to decrease the release of parathyroid hormone [22]. CaR is also highly expressed in the kidney, bone, blood vessels, brain, and heart [34]. CaR is a pleiotropic G protein-coupled receptor and thus couples to more than one type of G protein [4]. Notably, several signaling pathways activated by stimulation of CaR regulate I K1. For example, increases in intracellular Ca2+ concentration ([Ca2+] i ) and activation of protein kinase C (PKC) inhibit this current [5, 6, 15], whereas a rise in phosphoinositide 4,5-bisphosphate (PIP2) levels enhances the current [8]. PIP2, acting as a second messenger, plays an important role in modulating several ion channels and transporters [8, 30]. Stimulation of CaR activates both Gq/11, which decreases PIP2, and phosphatidylinositol 4-kinase (PI-4-K), which increases PIP2. But how membrane PIP2 levels are regulated by CaR activation and whether the resulting changes regulate inward rectifier K+ channels are unknown.
In this study, we monitored PIP2 levels using the fluorescent PIP2 probe tubby-R332H-cYFP [12] and investigated how this regulation affects Kir2.1 channels expressed in HEK293T and I K1 in guinea pig ventricular myocytes.
Materials and methods
Cell culture and transfection procedures
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma Chemical Co., St. Louis, MO, USA) containing 10% (v/v) fetal bovine serum (FBS; Life Technologies, Paisley, Scotland) and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. HEK293T cells plated on poly-l-lysine-coated no. 1 glass coverslips (35 mm) were transiently transfected with 2 μg of the expression constructs Kir2.1-cyan fluorescent protein(eCFP), CaR-green fluorescent protein (GFP) (OriGene Technologies Inc., MD, USA), and/or tubby-R332H-yellow fluorescent protein (cYFP) using Lipofectamine 2000 (Invitrogen Co., Carlsbad, CA, USA). Cells were used 1–2 days after transfection. The Kir2.1-eCFP construct was generated by subcloning Kir2.1 cDNA into an XhoI/HindIII-digested peCFP vector (Clontech Lab Inc., Mountain View, CA, USA).
Isolation of guinea pig cardiac myocytes
Guinea pig (Hartley) ventricular myocytes were isolated using an enzymatic procedure described previously [1]. Briefly, guinea pigs were anesthetized with sodium pentobarbital (50 mg/kg, i.v.) and hearts were isolated and retrogradely perfused, first with a Ca2+-free Tyrode’s solution (5 mM HEPES pH 7.4, 145 mM NaCl, 5 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 5.5 mM glucose) and then with the same solution containing 0.5 mg/ml collagenase, 0.25 mg/ml protease, 1 mg/ml albumin, and 50 μM CaC12. The heart was minced, and cells were dissociated by gentle agitation in the enzyme solution. Isolated cells were stored at room temperature in modified Tyrode’s solution (pH 7.4) containing 100 mg/ml albumin and 10 mM glucose.
Electrophysiological recordings
Currents were recorded at room temperature (21–24 °C) using the patch-clamp technique [7] in the whole-cell configuration and an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). The bath contained Tyrode’s solution (see composition previously mentioned), and the pipette solution (pH 7.2) contained 138 mM K-aspartate, 3 mM MgATP, 5 mM Na2-phosphocreatine, 1 mM MgCl2, 5 mM EGTA, 5 mM HEPES, and 2 mM creatine. For recordings of I K1 in myocytes, 5 μM nifedipine, 1 μM atropine, 10 μM glibenclamide, and 1 mM 4-aminopyridine were added in Tyrode’s solution.
Currents were sampled and filtered at frequencies of 20 and 5 kHz, respectively. Command voltages were controlled by and data were acquired using the pCLAMP10 software (Molecular Devices). Recordings in HEK293T cells and myocytes were corrected for the liquid junction potential (−15 mV).
Measurement of fluorescence signals
Total internal reflection fluorescence (TIRF) fluorescence images of HEK293T cells were obtained at 22 °C using a Nikon Eclipse microscope with a Ti-E TIRF module (PFS) (Nikon Co, Tokyo, Japan) and an iXON Ultra 897 EMCCD camera (Andor Technology Ltd., Belfast, UK). Cells were visualized using a ×100 1.45 oil immersion objective lens. YFP fluorescence was monitored by exciting with a 515-nm laser (Cobolt Laser, Sweden) and collecting the emission at 535/30 nm (Chroma, Rockingham, VT). Averaged fluorescence intensity F of a region of interest was selected to cover the majority of the cell with background subtraction.
Data analysis
Averaged data are presented as means ± SEMs. Student’s t test for independent samples was used to assess the statistical significance of differences. Asterisks *, **, and *** indicate p < 0.05, 0.01, and 0.005, respectively.
Results
Effects of CaR activation on the inhibition of Kir2.1 channels by extracellular spermine in HEK293T cells
To test whether spermine can indirectly modulate Kir2.1 channels through activation of CaR, we compared the effects of spermine in the absence and presence of exogenously expressed CaR. Currents were recorded from HEK293T cells transfected with Kir2.1 channels in the whole-cell configuration. Spermine decreased both inward and outward currents, but the magnitude of this decrease was diminished in the presence of CaR (Fig. 1a, b). The effects of spermine on the voltage dependence of normalized current (I) are shown in Fig. 1c, d. A comparison of the time course of the decrease in current recorded at −115 mV (Fig. 1e) and peak outward current (Fig. 1f) in the absence and presence of CaR expression showed that spermine-induced inhibition of currents was significantly reduced in HEK293T cells cotransfected with Kir2.1 and CaR compared with that in cells transfected with Kir2.1 alone (Fig. 1g, h). It is noted that the time course of effect was slow with and without CaR expression (Fig. 1e, f), suggesting that the direct effect of spermine on the Kir2.1 channel is as slow as the indirect effect (i.e., accumulation of PIP2 against the phospholipase C (PLC)-dependent hydrolysis via CaR activation). Our previous study suggests that extracellular spermine bound to the mouth of the extracellular pore of the Kir2.1 channel may induce an allosteric effect on voltage-dependent decay of outward currents, a process in which a region in the vicinity of the selectivity filter and cytoplasmic pore is involved [3]. The slow time course of effect by spermine may be due to this complicated allosteric regulation.

Effects of extracellular spermine on Kir2.1 currents. a Representative traces showing currents recorded using a voltage step protocol (−115 to −15 mV in 10-mV increments) from a holding potential of −15 mV in the whole-cell mode from a HEK293T cell transfected with Kir2.1 alone. b Currents recorded from a HEK293T cell transfected with Kir2.1 and CaR. c, d Effects of extracellular spermine on normalized I–V relationships in the absence and presence of CaR activation. Currents were normalized to that recorded at −115 mV under control condition. e, f Time course of spermine-inhibited currents, showing currents recorded at −115 mV (e) and peak outward currents (f) in the absence (n = 8) and presence (n = 6) of CaR expression. g, h Quantification of results in e, f, showing the percent decrease in the current recorded at −115 mV (g) and peak outward currents (h) in CaR-expressing cells compared with controls
These results suggest that spermine can enhance Kir2.1 channel activity through activation of CaR in addition to its direct inhibitory effect on the channel.
CaR activation increases currents mediated by Kir2.1 channels expressed in HEK293T cells
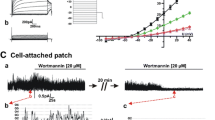
Next, we examined the effects of CaR activation alone on the Kir2.1 channel. Stimulation of CaR with 1 μM NPSR568 resulted in increases in both inward and outward currents at all voltages tested, and the effects were reversible upon washout (Fig. 2a, b). The time courses of the increases in current recorded at −115 mV and of peak outward current are shown in Fig. 2c, d, respectively. Fitting the concentration-response curve of NPSR568 to current recorded at −115 mV (Fig. 2e) and peak outward current (Fig. 2f) yielded K a values of 0.86 μM at −115 mV and 0.71 μM for peak outward current. The maximum increase in current was 68.3% at −115 mV and 111.4% for peak outward current. These results support the conclusion that activation of CaR increases Kir2.1 currents.

CaR stimulation increases Kir2.1 currents. a Effects of NPSR568 on currents recorded from a HEK293T cell transfected with both Kir2.1 and CaR. b Voltage dependence of normalized current. c, d Time courses of NPSR568 (1 μM)-induced averaged changes in currents, showing currents recorded at −115 mV (c) and peak outward currents (d), n = 6. e, f Concentration-response effects of NPSR568 on current (n = 3–6)
CaR activation increases Kir2.1 currents and PIP2 via activation of PI-4-K
CaR activation stimulates both the PLC pathway, which reduces membrane PIP2, and the PI-4-K pathway, which stimulates PIP2 synthesis. The enhancing effects of the CaR agonist, NPSR568, on Kir2.1 channel activity suggest that the predominant pathway is PIP2 synthesis. To test this hypothesis, we monitored PIP2 levels using a YFP-tagged, mutated C-domain of the PIP2-binding protein tubby (tubby-R332H-cYFP) [12, 24]. The application of 3 μM NPSR568 resulted in reversible increases in membrane fluorescence in cells expressing tubby-R332H-cYFP (Fig. 3a). The time courses of fluorescence changes at the membrane are shown in Fig. 3b. In cells expressing both tubby-R332H-cYFP and CaR, application of NPSR568 increased membrane fluorescence by 25.3 ± 2.1%. In cells transfected with tubby-R332H-cYFP alone, fluorescence was not increased upon treatment with NPSR568. The YFP fluorescence of cells transfected with CaR-GFP alone was too low to be detected and thus was not quantified.

Activation of CaR increases membrane PIP2. a Images of tubby-R332H-cYFP fluorescence obtained from cells under control conditions, in the presence of 3 μM NPSR568 and following washout. b Time course of increases in fluorescence intensity at the membrane induced by 3 μM NPSR568, expressed as the percent change in fractional fluorescence (n = 13 for tubby-R332H-cYFP and CaR cotransfection; n = 11 for tubby alone)
The increase in membrane fluorescence induced by CaR activation implies that the PI-4-K pathway predominates over the PLC pathway in regulating Kir2.1 channels. To confirm this, we tested the effects of a potent inhibitor for PI-4-K IIIβ (which was attributed to membrane PIP2 increases upon CaR activation [11]), PIK-93 [2, 16], on CaR-induced increases in Kir2.1 currents and PIP2. With PIK-93 pretreatment (0.15 μM, 10–15 min), NPSR568 decreased both inward and outward currents (Fig. 4a, b), and this inhibitory effect increased over time (Fig. 4c, d). On average, with PIK-93 pretreatment, NPSR568 decreased currents at −115 mV and peak outward currents by 32.5 ± 3.2 and 40.5 ± 4.9%, respectively.

Effects of PIK-93 on NPSR568-induced increases in Kir2.1 currents. a Effects of NPSR568 with 150 nM PIK-93 pretreatment (10–15 min), on currents recorded from a HEK293T cell transfected with both Kir2.1 and CaR. b Voltage dependence of normalized I. c, d Time courses of NPSR568 (1 μM)-induced averaged changes in currents, showing currents recorded at −115 mV (c) and peak outward currents (d) with 150 nM PIK-93 pretreatment. e, f Quantification of results in c, d, showing the percent increase in currents recorded at −115 mV (e) and peak outward currents (f) induced by 1 μM NPSR568 alone (n = 6) and by 1 μM NPSR568 + 150 nM PIK-93 (n = 3)
Next, we examined the effects of PIK-93 on PIP2 levels. PIK-93 decreased membrane fluorescence, and the subsequent activation of CaR with 3 μM NPSR568 further reduced the signal (Fig. 5a). The time courses of fluorescence changes at the membrane are shown in Fig. 5b. PIK-93 treatment decreased the fluorescence by 18.8 ± 2.9%, and subsequently, application of NPSR568 further significantly decreased the signal to −30.1 ± 4.0% (Fig. 5c). PIK-93 treatment alone decreased the fluorescence by 19.8 ± 2.9%.

Effects of PIK-93 on NPSR568-induced increases in tubby-R332H-cYFP fluorescence on membrane. a Images of tubby-R332H-cYFP fluorescence obtained from cells under control (left panel), during PIK-93 pretreatment (middle panel), and after 3-μM NPSR568 application with 300 nM PIK-93 (right panel). b Time course of averaged changes in tubby-R332H-cYFP fluorescence at the membrane induced by treatment with 3 μM NPSR568 + 300 nM PIK-93 and PIK-93 alone. c Quantification of results in b, showing the percent increase in fractional fluorescence of tubby-R332H-cYFP at the membrane induced by treatment with 3 μM NPSR568 alone versus 3 μM NPSR568 + 300 nM PIK-93 (n = 11 for both sets of experiments)
To further support the involvement of PI-4-K in the enhancement of membrane (PIP2) induced by CaR activation, we next examined the recovery of membrane PIP2 level following PLC activation. Applying CCL20 (300 ng/ml) activated PLC via CCR6 [28] in stable transfected HEK293 cells and resulted in decreases of membrane fluorescence (Fig. 6a). Applying NPSR568 facilitated the recovery of tubby-R332H-cYFP fluorescence as compared to no NPSR treatment (Fig. 6b). Stimulation of CaR facilitated fluorescence recovery (τ = 3.7 min) as compared to the washout (τ = 5.8 min). CaR activation increased membrane fluorescence from −31.0 ± 4.0 to 0.1 ± 5.5%, and washout increased the signal to −9.3 ± 3.9% (Fig. 6c). These results support the conclusion that activation of CaR increases Kir2.1 channel-mediated currents by increasing the membrane PIP2 levels through activation of PI-4-K.

Stimulation of CaR promotes tubby-R332H-cYFP fluorescence recovery from PLC activation. a Images of tubby-R332H-cYFP fluorescence obtained from cells with CCR6, tubby, and CaR expression. The cell was first treated with CCL20 (300 ng/ml, middle panel) followed by washout (right panel). b Images of tubby-R332H-cYFP fluorescence obtained from cells first treated with CCR20 (middle panel) followed by application of 3-μM NPSR568 application (right panel). c Averaged percent of changes in fractional fluorescence of tubby-R332H-cYFP at the membrane induced with (n = 14) and without NPSR (n = 12)
CaR signaling through the Gq/11 pathway inhibits Kir2.1 channel activity
In addition to activating PI-4-K, CaR stimulation also activates PLC via the Gq/11 pathway and thus degrades membrane PIP2. To examine whether this pathway is involved in the regulation of Kir2.1 channels by CaR activation, we tested the effects of pretreatment of the PLC inhibitor, U73122, on CaR activation-induced increases in Kir2.1 currents and PIP2. With U73122 pretreatment (10–15 min), NPSR568 increased currents (Fig. 7a, b), and this effect increased over time (Fig. 7c, d). U73122 pretreatment significantly increased the effects of NPSR 568 on inward currents at −115 mV from 34.1 ± 1.6 to 51.1 ± 2.0 and peak outward currents from 57.3 ± 4.9 to 71.6 ± 3.0 (Fig. 7e, f).

Effects of U73122 pretreatment on NPSR568-induced increases in Kir2.1 currents. a Effects of NPSR568 with 5-μM U73122 pretreatment (10–15 min) on currents recorded from a HEK293T cell transfected with both Kir2.1 and CaR. b Voltage dependence of normalized currents. c, d Time courses of NPSR568 (1 μM)-induced changes in currents, showing currents recorded at −115 mV (c) and peak outward currents (d), with 5-μM U73122 pretreatment. e, f Quantification of results in c, d, showing the percent change in currents recorded at −115 mV (e) and peak outward currents (f) induced by 1 μM NPSR568 alone (n = 6) and 1 μM NPSR568 plus 5 μM U73122 (n = 4)
Next, we examined the effects of U73122 pretreatment on PIP2 levels. With pretreatment of U73122, NPSR568 increased membrane tubby-R332H-cYFP fluorescence (Fig. 8a, right panel). The time courses of fluorescence changes at the membrane are shown in Fig. 8b. In cells treated with NPSR568 only, tubby-R332H-cYFP fluorescence was increased by 25.3 ± 2.1%. This effect was significantly enhanced by U73122 pretreatment to 52.7 ± 5.4% (Fig. 8c). The treatment of U73122 alone increased the fluorescence by 7.7 ± 4.8%, significantly lower than the treatment of U73122 + NPSR568 (Fig. 8c).

Effects of U73122 on NPSR568-induced increases in tubby-R332H-cYFP fluorescence on membrane. a Images of tubby-R332H-cYFP fluorescence obtained from cells under control (left panel), during U73122 pretreatment (middle panel), and after (right panel) of 3-μM NPSR568 application with 5 μM U73122 presence all the time. b Time course of averaged changes in tubby-R332H-cYFP fluorescence at the membrane induced by treatment with 3 μM NPSR568, 3 μM NPSR568 + 5 μM U73122, and 5 μM U73122 alone. c Averaged percent increase in fractional fluorescence of tubby-R332H-cYFP at the membrane induced by NPSR568 with and without U73122 pretreatment (n = 12) and by U73122 alone (n = 11)
These results suggest that activation of CaR decreases Kir2.1 channel-mediated currents through activation of PLC.
CaR activation increases I K1 in guinea pig ventricular myocytes
Next, we explored whether activating CaR regulates I K1 in native ventricular myocytes exposed to physiological solutions. Activation of endogenous CaR in ventricular myocytes with 3 μM NPSR568 resulted in increases in both inward and outward currents, and these effects were reversible upon washout (Fig. 9a, b). The time courses of averaged increases in currents recorded at −115 mV and in peak outward current are shown in Fig. 9c, d, respectively. An analysis of the concentration-response effect of NPSR568 on currents recorded at −115 mV (Fig. 9e) and on peak outward current (Fig. 9f) yielded K a values of 1.82 and 3.22 μM, respectively. The maximum increase in current was 96.8% at −115 mV and 81.1% for peak outward current. These results support the conclusion that activation of CaR increases I K1 in guinea pig ventricular myocytes.

CaR stimulation increases I K1 in guinea pig ventricular myocytes. a Effects of NPSR568 on I K1 recorded from a guinea pig ventricular myocyte. b Voltage dependence of normalized I K1. c, d Time courses of averaged changes in I K1 induced by 3 μM NPSR, showing currents recorded at −115 mV (c) and peak outward currents (d), n = 6. e, f Concentration-response relationship, showing I K1 recorded at −115 mV (e) and peak outward I K1 (f) as a function of NPSR568 concentration (n = 3–6)
Figure 4 shows that the enhancing effect of the CaR agonist NPSR568 on Kir2.1 channel activity was attributable to PIP2 synthesis through activation of PI-4-K. To confirm this mechanism in guinea pig ventricular myocytes, we tested the effect of PIK-93 pretreatment on CaR activation-induced increases in I K1. With PIK-93 pretreatment (0.15 μM, 10–15 min), 3 μM NPSR 568 decreased both inward and outward I K1 (Fig. 10a, b), and this inhibitory effect increased over time (Fig. 10c, d). With PIK-93 (0.15 μM) pretreatment, CaR activation decreased currents at −115 mV by 27.4 ± 3.9% and peak outward currents by 26.2 ± 4.0%.

Effects of PIK-93 on NPSR568-induced increases in I K1. a Effects of NPSR568 with 150 nM PIK-93 pretreatment (10–15 min) on I K1 recorded from a guinea pig ventricular myocyte. b Voltage dependence of normalized I K1. c, d Time courses of averaged changes in I K1 induced by 3 μM NPSR, showing currents recorded at −115 mV (c) and peak outward currents (d) with 150 nM PIK-93 pretreatment. e, f Quantification of results in c, d, showing the percent change in currents recorded at −115 mV (e) and peak outward currents (f) induced by 3 μM NPSR568 alone (n = 6) and by 3 μM NPSR568 plus 150 nM PIK-93 pretreatment (n = 4)
Finally, to determine whether activation of PLC via the Gq/11 pathway is involved in the regulation of I K1 by CaR activation in guinea pig ventricular myocytes, we tested the effect of U73122 pretreatment on CaR activation-induced increases in I K1. With U73122 pretreatment (5 μM, 10–15 min), NPSR 568 increased inward and outward I K1 (Fig. 11a–d). U73122 pretreatment significantly enhanced the stimulatory effect of NPSR568 on I K1 from 57.6 ± 2.4 to 102.7 ± 10.4% at −115 mV and from 35.7 ± 3.6 to 82.6 ± 3.5% for peak outward I K1 (Fig. 11e, f).

Effects of U73122 pretreatment on NPSR568-induced increases in I K1. a Effects of NPSR568 on I K1, with 5-μM U73122 pretreatment (10–15 min). b Voltage dependence of normalized I K1. c, d Time course of averaged changes in I K1 induced by 3 μM NPSR, showing currents recorded at −115 mV (c) and peak outward currents (d) with 5-μM U73122 pretreatment. e, f Quantification of results in c, d, showing the percent change in currents recorded at −115 mV (e) and peak outward currents (f) induced by 3 μM NPSR568 alone (n = 6) and by 3 μM NPSR568 plus 5 μM U73122 (n = 5)
In summary, CaR stimulation activates both the PLC and PI-4-K pathways, but the effect on PI-4-K dominates, resulting in increases of Kir2.1 currents recorded from HEK293T cells transfected with Kir2.1 and I K1 in guinea pig ventricular myocytes.
Discussion
CaR activation increases membrane PIP2 levels and Kir currents
The purpose of this study was to examine whether CaR activation regulates inward rectifier K+ channels by modulating the level of membrane PIP2. To achieve this goal, we investigated the effects of activating CaR on currents mediated by Kir2.1 channels expressed in HEK293T cells and on I K1 in guinea pig ventricular myocytes. We found that activation of CaR by an allosteric agonist (NPSR568) increased both endogenous I K1 and currents mediated by exogenously expressed Kir2.1 to a similar extent. Further, monitoring PIP2 levels using the PIP2-binding probe tubby-R332H-cYFP demonstrated that CaR activation increased PIP2 concentration at the plasma membrane in HEK293T cells. These effects were abolished by PIK-93, suggesting the involvement of PI-4-K. CaR activation facilitates the recovery of membrane PIP2 following PLC activation, further supporting this notion. Collectively, these results provide the first direct evidence that CaR activation increases inward rectifier K+ channel currents by accelerating PIP2 synthesis in HEK293T cells and possibly also in guinea pig ventricular myocytes.
PIP2 acts as a second messenger molecule to play an important role in modulating a number of ion channels and transporters [8, 9, 30]. Previous literature reports have enhanced our understanding of this topic, yet significant gaps remain. First, although it has been shown that CaR can regulate ion channels in cells [33], CaR regulation of ion channels through modulation of PIP2 is a mechanism that has not been explored. Second, most previous investigations have focused on single signaling pathways in PIP2 regulation and its association with channel activities. Yet, multiple pathways regulate membrane PIP2, and how they are integrated to regulate ion channels remains elusive. In the current study, we explored the involvement of two signaling pathways engaged by activation of CaR in the regulation of PIP2. Our data revealed that activation of CaR results in increases in membrane PIP2 and Kir currents through PI-4-K activation, although the activation of PLC also makes contribution to the regulation of Kir channels.
Third, most studies have focused on the effect of PIP2 depletion on ion channels expressed in heterologous systems [23, 24, 30]. PIP2 depletion has also been studied in native cells. For example, it has been shown that, in normal isolated Müller cells, activation of Gq/11-coupled metabotropic glutamate receptors (mGluRs) with the mGluR I agonist (S)-3,5-dihydroxyphenylglycine (DHPG) suppresses Kir currents through the intracellular Ca2+-dependent PLC/IP3-ryanodine/PKC signaling pathway [14]. On the other hand, little is known about whether resynthesis of PIP2 regulates ion channels in heterologous expression systems or in native cells. PI-4-K is involved in the constitutive biosynthesis of PIP2 and in PIP2 resynthesis after its breakdown by PLC. Can physiological regulators that target PI-4-K affect PIP2 levels and modulate channel activities? A previous study has shown that resynthesis of PIP2 via PI-4-K mediates adaptation of caffeine responses in taste receptor cells by regulating Kir and KV channels [38]. Our study provides additional evidence that increasing membrane PIP2 by stimulating PI-4-K via CaR activation can regulate ion channel function.
Fourth, it is unclear whether physiological fluctuations in the levels of PIP2, as may occur with the activation of intracellular signaling pathways, are sufficient in and of themselves to regulate ion channels [9]. It has been hypothesized that some channels interact with lipid partners with high affinity such that the lipid-binding site of the channel remains saturated during most physiological changes in lipid levels. By contrast, other channels bind with low affinity; for these channels, variations in lipid concentration may act as physiological signals to regulate the computational properties of neurons and transport properties of secretory cells [23]. Among Kir channels, the constitutively active Kir1.1 and Kir2.1 interact with PIP2 with high affinity [10], whereas Kir3 channels show weaker interactions with PIP2 [26]. Application of exogenous PIP2 dynamically activates these channels in reverse order, namely Kir3.1/3.4 > Kir2.1 > Kir1.1 [10, 26, 36]. Whether Kir2.1 channels are sensitive to changes in membrane PIP2 level under physiological conditions remains unclear. Our study provides the first direct evidence that activation of CaR can increase I K1 via the PI-4-K pathway.
The modulation of Kir2.1 channels by PIP2 via CaR activation appears to be voltage dependent. The underlying mechanisms are unknown. It has been previously shown that polyamines bound to the Kir2.1 channel at positive driving force interact with the gating of Kir2.1 channels by PIP2 [35]. It is possible that the voltage-dependent effect of PIP2 is related to the voltage-dependent polyamine block of the channel. Further, it is shown that PIP2 shifts the voltage dependence of conductance by promoting the opening of the ion conduction gate and by a negative surface charge in Slo1 BK channels [32]. PIP2 may also exert similar effects on Kir, and thus, the effect appears to be different at different voltages.
In addition to regulating PIP2, CaR also modulates several cellular events including increases of [Ca2+] i , PKC, and arachidonic acids as well as a decrease in PKA [20, 33]. Several of these intracellular signaling molecules regulate Kir2.1 and/or I K1 [5, 17, 18, 40]. This study investigated only the effect of PIP2 on Kir2.1 upon CaR activation. How other intracellular signaling is involved in the regulation of Kir2.1/I K1 via CaR activation requires further studies. Further, besides I K1, the electrical properties of the membrane involve other ion channels. The overall physiological and pathological functions require further investigation when all the effects during CaR activation are taken into consideration.
In this study, we used PIK-93 to inhibit PI-4-K IIIβ. Because PIK-93 inhibits PI-3-K and PI-4-K with similar potency [2, 16], the contribution of PI-4-K-induced increases of Kir2.1 currents and I K1 upon CaR activation is underestimated. Our study provides the qualitative analysis of involvement of PI-4-K signaling in CaR activation. For more quantitative analysis, future studies with specific PI-4-K inhibitors are required.
Pathophysiological implications of CaR regulation of I K1
It has been previously shown that putrescine significantly reduces the arrhythmia associated with cardiac ischemia and reperfusion [31]. Furthermore, ischemia/reperfusion results in depletion of the myocardial polyamine pool. Application of exogenous spermine restores the intracellular polyamine pool and reduces cardiac myocyte necrosis, suggesting that the loss of spermine might be involved in the cardiac injury produced by reperfusion [39]. These cardioprotective effects have been attributed to the membrane-stabilizing and antioxidative effects of putrescine [31]. However, because ischemia and reperfusion can lead to upregulation of CaR in the heart during a myocardial infarction [37] and because polyamines are potent agonists of CaR, it is possible that the cardioprotective effects of polyamines are the result of CaR activation. The results from our study suggest that CaR activation may stabilize the electrical properties of the cardiac membrane by increasing I K1. This action may play an important role in the cardioprotective effects of polyamines during ischemia/reperfusion injury, although further studies are required to confirm this hypothesis. CaR functions as an integrator of extracellular stimuli and is probably always activated to some extent [29]. It remains to be determined whether CaR calcilytics are capable of decreasing I K1 and thus affect cardiac electrical properties.
Extracellular Ca2+ is a low-affinity agonist for CaR, and thus, under physiological conditions, CaR is subjected to the tonic stimulation of extracellular Ca2+. Investigations on this tonic activation of CaR may shed light on the physiological functions of CaR.
Conclusions
Our data show that increases of endogenous PIP2 in cardiac myocytes regulate I K1. This novel relationship supports a model in which autonomic stimulation of cardiac CaR may alter I K1-dependent repolarization and excitability and impact cardiac function. Our study explored important questions related to PIP2 modulation by CaR activation and showed that CaR can upregulate inward rectifier K+ channels in both native cells and a heterologous expression system. Given the increasing clinical use of calcimimetics in the treatment of hyperparathyroidism and the potential for using calcilytics in the treatment of osteoporosis, it is essential to understand the role of CaR in cardiac function [29].
References
Arreola J, Dirksen RT, Shieh RC, Williford DJ, Sheu SS (1991) Ca2+ current and Ca2+ transients under action potential clamp in guinea pig ventricular myocytes. Am J Phys 261:C393–C397
Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T (2008) Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol Biol Cell 19:711–721
Chang HK, Shieh RC (2013) Voltage-dependent inhibition of outward Kir2.1 currents by extracellular spermine. BBA-Bioembranes 1828:765–775
Chang HK, Yeh SH, Shieh RC (2005) A ring of negative charges in the intracellular vestibule of Kir2.1 channel modulates K+ permeation. Biophys J 88:243–254
Fakler B, Brandle U, Glowatzki E, Zenner HP, Ruppersberg JP (1994) Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron 13:1413–1420
Fauconnier J, Lacampagne A, Rauzier JM, Vassort G, Richard S (2005) Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc Res 68:204–212
Hamill OP, Marty A, Heher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflug Arch Eur J Phy 391:85–100
Hilgemann DW, Feng S, Nasuhoglu C (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 111:re19
Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC (2015) Phosphoinositides regulate ion channels. BBA-Mol Cell Biol L 1851:844–856
Huang CL, Feng S, Hilgemann DW (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391:803–806
Huang C, Handlogten ME, Miller RT (2002) Parallel activation of phosphatidylinositol 4-kinase and phospholipase C by the extracellular calcium-sensing receptor. J Biol Chem 277:20293–20300
Hughes S, Marsh SJ, Tinker A, Brown DA (2007) PIP(2)-dependent inhibition of M-type (Kv7.2/7.3) potassium channels: direct on-line assessment of PIP(2) depletion by Gq-coupled receptors in single living neurons. Pflug Arch Eur J Phy 455:115–124
Hume JR, Uehara A (1985) Ionic basis of the different action potential configurations of single guinea-pig atrial and ventricular myocytes. J Physiol 368:525–544
Ji M, Miao Y, Dong LD, Chen J, Mo XF, Jiang SX, Sun XH, Yang XL, Wang Z (2012) Group I mGluR-mediated inhibition of Kir channels contributes to retinal Muller cell gliosis in a rat chronic ocular hypertension model. J Neurosci 32:12744–12755
Karle CA, Zitron E, Zhang W, Wendt-Nordahl G, Kathofer S, Thomas D, Gut B, Scholz E, Vahl CF, Katus HA, Kiehn J (2002) Human cardiac inwardly-rectifying K+ channel Kir(2.1b) is inhibited by direct protein kinase C-dependent regulation in human isolated cardiomyocytes and in an expression system. Circulation 106:1493–1499
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM (2006) A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733–747
Koumi S, Wasserstrom JA, Ten Eick RE (1995) Beta-adrenergic and cholinergic modulation of inward rectifier K+ channel function and phosphorylation in guinea-pig ventricle. J Physiol 486:661–678
Liu Y, Liu D, Heath L, Meyers DM, Krafte DS, Wagoner PK, Silvia CP, W Y, Curran ME (2001) Direct activation of an inwardly rectifying potassium channel by arachidonic acid. Mole Pharmacol 59:1061–1068
Lopatin AN, Nichols CG (2001) Inward rectifiers in the heart: an update on IK1. J Mol Cell Cardiol 33:625–638
Magno AL, Ward BK, Ratajczak T (2011) The calcium-sensing receptor: a molecular perspective. Endocr Rev 32:3–30
Matsuoka S, Sarai N, Kuratomi S, Ono K, Noma A (2003) Role of individual ionic current systems in ventricular cells hypothesized by a model study. Jpn J Physiol 53:105–123
Nemeth EF, Scarpa A (1986) Cytosolic Ca2+ and the regulation of secretion in parathyroid cells. FEBS Lett 203:15–19
Quinn KV, Cui Y, Giblin JP, Clapp LH, Tinker A (2003) Do anionic phospholipids serve as cofactors or second messengers for the regulation of activity of cloned ATP-sensitive K+ channels? Circ Res 93:646–655
Quinn KV, Behe P, Tinker A (2008) Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol 586:2855–2871
Ravens U, Cerbai E (2008) Role of potassium currents in cardiac arrhythmias. Europace 10:1133–1137
Rohacs T, Chen J, Prestwich GD, Logothetis DE (1999) Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J Biol Chem 274:36065–36072
Shimoni Y, Clark RB, Giles WR (1992) Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol 448:709–727
Shirakawa AK, Liao F, Zhang HH, Hedrick MN, Singh SP, Wu D, Farber JM (2010) Pathway-selective suppression of chemokine receptor signaling in B cells by LPS through downregulation of PLC-beta2. Cell Mol Immunol 7:428–439
Smajilovic S, Tfelt-Hansen J (2007) Calcium acts as a first messenger through the calcium-sensing receptor in the cardiovascular system. Cardiovasc Res 75:457–467
Suh BC, Hille B (2005) Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol 15:370–378
Tagliavini S, Genedani S, Bertolini A, Bazzani C (1991) Ischemia- and reperfusion-induced arrhythmias are prevented by putrescine. Euro J Pharmacol 194:7–10
Tian Y, Ullrich F, Xu R, Heinemann SH, Hou S, Hoshi T (2015) Two distinct effects of PIP2 underlie auxiliary subunit-dependent modulation of Slo1 BK channels. J Gen Physiol 145:331–343
Ward DT (2004) Calcium receptor-mediated intracellular signalling. Cell Calcium 35:217–228
Weston AH, Geraghty A, Egner I, Edwards G (2011) The vascular extracellular calcium-sensing receptor: an update. Acta Physiol (Oxf) 203:127–137
Xie LH, John SA, Ribalet B, Weiss JN (2005) Long polyamines act as cofactors in PIP2 activation of inward rectifier potassium (Kir2.1) channels. J Gen Physiol 126:541–549
Zhang H, He C, Yan X, Mirshahi T, Logothetis DE (1999) Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nature Cell Biol 1:183–188
Zhang WH, Fu SB, Lu FH, Wu B, Gong DM, Pan ZW, Lv YJ, Zhao YJ, Li QF, Wang R, Yang BF, Xu CQ (2006) Involvement of calcium-sensing receptor in ischemia/reperfusion-induced apoptosis in rat cardiomyocytes. Biochem Bioph Res Co 347:872–881
Zhao FL, Herness S (2009) Resynthesis of phosphatidylinositol 4,5-bisphosphate mediates adaptation of the caffeine response in rat taste receptor cells. J Physiol 587:363–377
Zhao YJ, Xu CQ, Zhang WH, Zhang L, Bian SL, Huang Q, Sun HL, Li QF, Zhang YQ, Tian Y, Wang R, Yang BF, Li WM (2007) Role of polyamines in myocardial ischemia/reperfusion injury and their interactions with nitric oxide. Euro J Pharmacol 562:236–246
Zitron E, Gunth M, Scherer D, Kiesecker C, Kulzer M, Bloehs R, Scholz EP, Thomas D, Weidenhammer C, Kathofer S, Bauer A, Katus HA, Karle CA (2008) Kir2.X inward rectifier potassium channels are differentially regulated by adrenergic alpha1A receptors. J Mole Cell Cardiol 44:84–94
Acknowledgements
We thank Dr. Andrew Tinker for generously providing the tubby-R332H-cYFP construct and Dr. Fang Liao for the stable CCR6 HEK cells. This work was supported by a grant from Academia Sinica and the MOST of Taiwan (102-2320-B-001-004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All applicable international and institutional guidelines for the care and use of animals were followed. All procedures performed in this study involving animals were in accordance with the ethical standards of and approved by Academia Sinica Institutional Animal Care and Utilization Committee.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, CH., Chang, HK., Lee, SP. et al. Activation of the Ca2+-sensing receptors increases currents through inward rectifier K+ channels via activation of phosphatidylinositol 4-kinase. Pflugers Arch - Eur J Physiol 468, 1931–1943 (2016). https://doi.org/10.1007/s00424-016-1901-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1901-y