
Abstract
P2Y receptor activation causes the release of inflammatory cytokines in the bronchial epithelium, whereas G protein-coupled estrogen receptor (GPER), a novel estrogen (E2) receptor, may play an anti-inflammatory role in this process. We investigated the cellular mechanisms underlying the inhibitory effect of GPER activation on the P2Y receptor-mediated Ca2+ signaling pathway and cytokine production in airway epithelia. Expression of GPER in primary human bronchial epithelial (HBE) or 16HBE14o- cells was confirmed on both the mRNA and protein levels. Stimulation of HBE or 16HBE14o- cells with E2 or G1, a specific agonist of GPER, attenuated the nucleotide-evoked increases in [Ca2+]i, whereas this effect was reversed by G15, a GPER-specific antagonist. G1 inhibited the secretion of two proinflammatory cytokines, interleukin (IL)-6 and IL-8, in cells stimulated by adenosine 5′-(γ-thio)triphosphate (ATPγS). G1 stimulated a real-time increase in cAMP levels in 16HBE14o- cells, which could be inhibited by adenylyl cyclase inhibitors. The inhibitory effects of E2 or G1 on P2Y receptor-induced increases in Ca2+ were reversed by treating the cells with a protein kinase A (PKA) inhibitor. These results demonstrated that the inhibitory effects of G1 or E2 on P2Y receptor-mediated Ca2+ mobilization and cytokine secretion were due to GPER-mediated activation of a cAMP-dependent PKA pathway. This study has reported, for the first time, the expression and function of GPER as an anti-inflammatory component in human bronchial epithelia, which may mediate through its opposing effects on the pro‐inflammatory pathway activated by the P2Y receptors in inflamed airway epithelia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Estrogen (E2) is an important hormone that protects the lungs from inflammatory damage. Clinical observations suggested that reduced E2 levels were associated with greater risks of lung pathologies in menopausal women [19, 36]. However, the detailed anti-inflammatory role played by E2 and its pathophysiological mechanism are still unknown. In addition to the classical nuclear hormone receptors, ERα and ERβ, a novel E2 receptor and a G protein-coupled estrogen receptor (GPER), were recently identified [35, 43]. Despite the accumulating body of evidence indicating that the rapid, nongenomic actions of E2 observed in the epithelia are mediated via GPER, few studies have investigated the specific role of GPER in inflammatory airway diseases [31, 32].
Extracellular nucleotide release and the subsequent activation of P2Y receptors have been implicated in the pathogenesis of several inflammatory lung disorders, such as asthma [28]. During airway inflammation, damage to the surface epithelium is due to the secretion of eosinophil-derived, highly toxic, cationic proteins, such as major basic protein (MBP). Our recent study demonstrated that when human bronchial surface epithelia are chemically damaged by poly-l-arginine as a surrogate of MBP [10], nucleotides, such as ATP and UDP, are released into the extracellular medium. The extracellular nucleotides then activate cell surface P2Y receptors to release two proinflammatory cytokines, interleukin (IL)-6 and IL-8, via a Ca2+-dependent process [20].
To the best of our knowledge, no reports have determined whether GPER is expressed in airway epithelia or whether GPER plays a role in the regulation of P2Y receptor-mediated Ca2+ signaling and cytokine secretion in airway epithelia. Therefore, we examined the expression of GPER and its subcellular localization in human bronchial epithelia. We also characterized the cross talk between the GPER and P2Y receptor signaling pathways and its implications on the anti-inflammatory role of GPER.
Materials and methods
Solutions and chemicals
Krebs-Henseleit (KH) solution and the nominally Ca2+-free solution were prepared as previously described [24]. Membrane permeant acetoxymethyl (AM) ester forms of Fura-2 and Pluronic F127 were obtained from Invitrogen (Carlsbad, USA). Uridine 5′-triphosphate (UTP), uridine 5′-diphosphate (UDP), adenosine 5′-(γ-thio)triphosphate (ATPγS), forskolin, poly-l-arginine hydrochloride, SQ 22536, U73122, E2, and G1 were obtained from Sigma-Aldrich (St. Louis, USA). G15 was obtained from Tocris (Bristol, UK). H89 dihydrochloride and MDL 12330A were obtained from Calbiochem (La Jolla, USA). All other general laboratory reagents were obtained from Sigma-Aldrich, and all cell culture reagents were obtained from Invitrogen.
Cell culture
The 16HBE14o- cell line was maintained in minimum essential medium (MEM) supplemented with 10 % fetal bovine serum, 1 % penicillin/streptomycin, and 1 % glutamine (Invitrogen, Carlsbad, CA) and cultured as described previously [47]. In some experiments, MEM with no phenol red (Invitrogen) was applied. Primary HBE cells were obtained from ScienCell Research Laboratories (Carlsbad, USA) and cultured using Bronchial Epithelial Cell Medium (ScienCell Research Laboratories) following the commercial protocol described previously [20].
RNA extraction, reverse transcription PCR, and real-time PCR
Total RNA was extracted with TRIzol Reagent (Invitrogen) and reverse transcribed to cDNA using iScript™ Reverse Transcription Supermix (Bio-Rad Laboratories, Hercules, USA). Reverse transcription PCR (RT-PCR) was performed with TaKaRa Taq™ DNA polymerase. Real-time PCR was performed with an Applied Biosystems Power SYBR Green PCR Master Mix (Invitrogen) on a ViiA™ 7 real-time PCR system. GPER primer sequences were as follows: forward primer, 5′-TCTACACCATCTTCCTCTTCC-3′; and reverse primer, 5′-GTAGCGGTCGAAGCTCATCC-3′. The RT-PCR products were characterized using 2 % agarose gel electrophoresis. Relative expression of GPER was normalized to GAPDH and determined with the Pfaffl method [30]. Each run of PCR included a nontemplate control and a sample without reverse transcriptase.
Western blotting
Western blotting was performed as described previously [10]. Cells grown in culture dishes were lysed on ice in Cytobuster™ Protein Extraction Reagent (Merck Millipore, Billerica, USA), supplemented with a protease inhibitor cocktail (no. 78429, Thermofisher Scientific, Waltham, USA) and a phosphatase inhibitor cocktail (Merck Millipore). Protein samples (20 μg per lane) were transferred to polyvinylidene fluoride (PVDF) membranes (Immobilon-P, Merck Millipore) and immunoblotted with GPER rabbit polyclonal antibody (N-15)-R (1:500; Santa Cruz Biotechnology, Santa Cruz, USA). Blocking peptide (sc-38525 P, Santa Cruz Biotechnology) was used for GPER antibody preabsorption, and mouse monoclonal antibody to GAPDH was used as a loading control. All blots were developed using an enhanced chemiluminescence detection system (Merck Millipore). The apparent molecular masses were calculated using prestained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) midrange protein markers (no. HM0671, Hou-Bio Life Technologies, Hong Kong).
Immunofluorescence microscopy
16HBE14o- or primary HBE cells grown on coverslips in four-well plates were rinsed with phosphate-buffered saline (PBS) and fixed in 4 % paraformaldehyde for 10 min at room temperature. Cells were blocked using PBS with 10 % normal horse serum and 0.1 % Triton X-100 for 1 h and incubated with GPER (N-15)-R rabbit polyclonal antibody (1:60; sc-48525-R, Santa Cruz Biotechnology) overnight at 4 °C [13]. After washing, Alexa Fluor® 488 donkey anti-rabbit IgG (H+L) was added (1:300, Thermofisher Scientific, Waltham, USA). The coverslips were mounted using mounting medium with 1.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a FluoView™-FV1000 confocal microscope (Olympus, Center Valley, USA). In some experiments, cells were co-incubated with purified mouse anti-E-cadherin (1:200; no. 610181, BD Biosciences, Heidelberg, Germany), purified mouse anti-GM130 (1:200; no. 610823, BD Biosciences), or KDEL antibody (1:500; NBP1-97469, Novus Biologicals, Littleton, USA). Alexa Fluor® 555 donkey anti-rabbit IgG was used for visualization (1:400, Thermofisher Scientific). For the negative control group, GPER antibodies were preabsorbed with specific blocking peptides (sc-48525 P, Santa Cruz Biotechnology).
Small interfering RNA lentivirus packaging and transduction
Lentiviral transfer vectors containing small interfering RNA (siRNA)-targeting GPER were purchased from Applied Biological Materials Inc. (Canada). A lentiviral vector with a scramble siRNA sequence was used as the negative control. The VSV-G-pseudotyped lentiviruses were produced by co-transfecting 293T cells with the transfer vectors and three packaging vectors, pMDLg/pRRE, pRSV-REV, and pCMV-VSVG, by calcium phosphate transfection. At 72 h post-transfection, the cell culture supernatant was collected and filtered through a 0.4-μm filter. The lentivirus was concentrated with centrifugation at 20,000 rpm and resuspended in 1× Tris-buffered saline. For lentiviral transduction, 5 × 103 cells were seeded in 24-well plates, and lentivirus was added to the cells in the presence of 8 μg/ml hexadimethrine bromide (Sigma-Aldrich, St. Louis, USA) overnight. After puromycin selection, the knockdown efficiency of GPER expression was determined by real-time PCR and Western blot analysis.
Measurement of intracellular calcium concentrations
Calcium signals in cells grown on glass coverslips were measured as previously described [47, 48]. Fura-2 ratios were used to represent changes in [Ca2+]i using Felix software (Photon Technology International, Edison, USA ). In Ca2+ imaging experiments, the perfusion chamber was mounted on an inverted microscope (Olympus IX70, USA) equipped with a scientific CMOS camera (pco.edge 5.5; PCO AG, Kelheim, Germany). Images were digitized and analyzed using MetaFluor Imaging Software (v7.5, Molecular Devices, USA). The data were also shown quantitatively as a change in Fura-2 ratios.
Manganese quenching
The manganese quench technique was used to estimate calcium influx [15, 45]. 16HBE14o- cells were loaded with Fura-2 as previously described. Since Mn2+ has a similar permeability as Ca2+ through most plasma membrane Ca2+ channels and quenches Fura-2 fluorescence at all excitation wavelengths, Ca2+ influx can be estimated by the Mn2+ quench of Fura-2 fluorescence at the Ca2+-insensitive 360-nm excitation wavelength. During the measurement, cells were treated with 10-μM UTP in the absence (nucleotide alone) or presence of E2 (100 nM) or G1 (10 nM) for 10 min. Then 1-mM MnCl2 was added in perfusion solution to observe the extent of Mn2+ entry. The rate of Mn2+ quenching was assessed by measuring the change of slope of Fura-2 fluorescence decrease before and after the addition of Mn2+ application (using Originlab 8 software, Northampton, USA), as well as the percentage decrease of Fura-2 fluorescence 120 s after Mn2+ application [6, 29, 41].
Monitoring STIM1 oligomerization via FRET microscopy
FRET microscopy was used to monitor the dynamic oligomerization of stromal interaction molecule 1 (STIM1) in 16HBE14o- cells. Plasmids expressing N-terminally tagged cyan fluorescent protein (CFP)- and yellow fluorescent protein (YFP)-STIM1, pEX-SP-CFP-STIM1, and pEX-SP-YFP-STIM1 were obtained from Addgene (Cambridge, USA). Cells were incubated in Ca2+-free Hanks’ Balanced Salt Solution (HBSS) with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) during imaging experiments. E2, G1, or dimethyl sulfoxide (DMSO) vehicle in HEPES-HBSS modified was perfused into the imaging chamber using a perfusion pencil (AutoMate Scientific, Berkeley, USA) from 0 to 15 min, followed by the addition of ATP to the perfusion solution to deplete Ca2+ in the ER. Images were captured using a Nikon Eclipse Ti microscope with a SPOTS RT3 camera (SPOT Imaging Solutions, Sterling Heights, USA). An ND8 filter was used during the experiment to reduce photobleaching. Each set of images (CFP, YFP, and FRET channels) was taken at 0.2 Hz using MetaFluor 7.8 software. The captured images were analyzed with ImageJ software (NIH, Bethesda, USA) using the pixel-to-pixel comparison method [16]. Sensitized emission was employed for the bleed-through correction. These specific bleed-throughs were obtained on a pixel-to-pixel basis from cells transfected with CFP-STIM1 or YFP-STIM1 alone. In our experimental settings, specific bleed-throughs for CFP and YFP were 0.696 and 0.064, respectively. To reduce photobleaching errors, N FRET measurements were adopted as described [49].
Quantitative measurement of STIM1 puncta formation
The formation STIM1 puncta after ER Ca2+ depletion by ATP stimulation was measured as described [18]. In brief, 16HBE14o- cells were transfected with YFP-STIM1 (Addgene, Cambridge, USA). Transfected cells were stimulated with ATP (10 μM) in the absence of extracellular Ca2+ in HEPES-HBSS solution. Single and isolated cells were selected for analyses. Z-stacks of images were acquired in a 0.225-μm separation. A Z-slice image near the close proximity to the attachment surface was selected for puncta analysis (~225 nm thickness) by ImageJ (NIH). YFP puncta were determined by particle analysis plugin, and fluorescent intensity greater than the background by three standard deviations was measured. Those with size less than 0.2 μm2 and greater than 2.5 μm2 were excluded for the analyses. In some experiments, transfected cells were pretreated with G1 (10 nM) or E2 (100 nM) before stimulation with ATP.
Real-time measurement of cAMP levels
CFP-Epac-YFP, an Epac-based polypeptide FRET reporter [46], was used to monitor real-time cyclic adenosine monophosphate (cAMP) changes in 16HBE14o- cells. The experiments were performed using the MetaFluor Imaging system (with the FRET module). Cells were transfected with the Epac-based cAMP sensor and excited at 436-nm wavelengths. CFP and YFP images were simultaneously recorded by the imaging setup equipped with the photometrics DV2 emission splitting system (Photometrics, Tucson, USA) including two emission filters (470/30 nm for CFP; 535/30 nm for FRET). Acquired fluorescence images were background subtracted, and real-time cAMP levels were represented by normalizing the CFP/FRET emission ratios as described previously [20, 24]. Images were digitized and analyzed using MetaFluor imaging software.
Inositol-1-phosphate measurements
16HBE14o- cells were plated 24 h before the experiment into 24-well culture plates at a concentration of 5 × 104 cells/well. Agonist-induced inositol-1-phosphate (IP1) accumulation in 16HBE14o- cells was quantified using the Cisbio IP-One kit (Cisbio Bioassays, Codolet, France) according to the manufacturer’s instructions [51].
Quantification of IL-6 and IL-8 secretion
Quantification of IL-6 and IL-8 secretion was performed using an enzyme-linked immunosorbent assay (ELISA) [10]. Cells were grown in 24-well culture plates. Cell-free supernatants were collected from control and treated cells and analyzed using a commercially available ELISA kit specific for IL-6 (eBioscience, San Diego, USA) and IL-8 (BD Biosciences, San Diego, USA) according to the manufacturers’ protocols. All experiments were performed in duplicate.
Statistical analysis
Data were expressed as the mean ± the standard error of the mean (SEM), and values of n referred to the number of independent experiments for each group. Statistical comparisons between original data were performed using the Student’s t test and analysis of variance (ANOVA) where appropriate. P < 0.05 was considered to be statistically significant.
Results
Expression and subcellular localization of GPER in human bronchial epithelial cells
RT-PCR was performed to assess gper messenger RNA (mRNA) expression in 16HBE14o- (Fig. 1a) and primary human bronchial epithelial (HBE; Fig. 1b) cells. The PCR product of gper mRNA was expressed in both cell types (lane 1), and GPER protein expression was examined by Western blotting. GPER was detected as a single protein band of approximately 40 kDa in both primary HBE and 16HBE14o- cells (Fig. 1c). MCF-7 cells were used as a GPER-positive cell line [3]. Detection of these protein bands was specific, as they were abolished by prior preabsorption of the antibodies with a control antigen (Fig. 1d).

GPER expression in HBE cells. a, b Agarose gel electrophoresis of the gper RT-PCR product obtained from 16HBE14o- (a) and primary HBE (b) cells, respectively. Lane 1, gper PCR product (278 bp); lane 2, RT negative control product; lane 3, Gapdh housekeeping PCR product (236 bp); M, DNA markers (no. 3422A, Takara Bio Inc., Tokyo, Japan) (n = 3–4). c GPER protein bands (40 kDa) in primary HBE cells, 16HBE14o- cells, and MCF-7 cells (positive control). d GPER antibody was blocked by peptide preabsorption. GAPDH (36 kDa) was used as an internal control (n = 3)
Additionally, GPER expression was detected by immunofluorescence in 16HBE14o- cells (Fig. 2a). GPER was labeled with an Alexa Fluor 488-conjugated anti-GPER antibody, and nuclei were visualized by DAPI. There was no overlap between the GPER and nuclear signals. Moreover, no immunoreactivity was observed in sections incubated with GPER antibody preabsorbed with the blocking peptide, indicating that the GPER staining was specific. Similar immunostaining patterns were observed in primary HBE cells (Fig. 2b).

GPER localization in HBE cells. a 16HBE14o- cells immunostained with GPER antibody (sc-48525 N-15; green) and DAPI (nuclei; blue) (upper row). Bright-field photomicrograph shows the same field and cells. 16HBE14o- cells immunostained with GPER antibody preabsorbed with blocking peptide (lower row). Scale bar, 10 μm. b Similar GPER immunostaining results using primary HBE cells. Scale bar, 20 μm. c 16HBE14o- cells immunostained with GPER antibody and the ER marker, KDEL (upper row), the Golgi apparatus marker, GM130 (middle row), or the plasma membrane marker, E-cadherin (lower row). Blue, DAPI, nuclei. Scale bar, 10 μm. Images are representative of three to five independent experiments. (Color figure online)
To further characterize the subcellular localization of GPER, double immunofluorescence labeling was used to determine possible colocalization between GPER and subcellular fractions, such as the endoplasmic reticulum (ER), Golgi apparatus, and plasma membrane, in 16HBE14o- cells (Fig. 2c). The results showed that GPER did not colocalize with Golgi (anti-GM130), while there was a very small amount of overlap between the GPER and the ER (anti-KDEL). In contrast, partial colocalization was observed between GPER and the plasma membrane (anti-E-cadherin) in 16HBE14o- cells.
Inhibitory effects of E2 or the GPER agonist, G1, on nucleotide-induced Ca2+ signaling in HBE cells
Our previous study showed that P2Y receptors were expressed in airway epithelia and could be stimulated by nucleotides, such as ATP, UTP, and UDP, resulting in an increase in [Ca2+]i [47]. Because it has been suggested that E2 inhibits the P2Y receptor-dependent Ca2+ signaling pathway [11], we examined whether activation of GPER had a similar inhibitory effect on P2Y receptor-mediated increases in Ca2+. In this study, both 16HBE14o- (Fig. 3a–d) and primary HBE (Fig. 3e–h) cells were treated with 100-nM E2 or 10-nM G1 for 10 min before being activated by 10-μM UTP (P2Y2 and P2Y4 agonist), 100-μM UDP (P2Y6 agonist), or 10-μM ATPγS (P2Y2 and P2Y11 agonist) [1]. The nucleotide-evoked increases in Ca2+ were inhibited by E2 and G1 with the percentages of inhibition, varying from 37.0 to 62.4 %. Both E2 and G1 were dissolved in DMSO, with a stock concentration 1000-fold higher than the final concentration. DMSO alone was used as vehicle control in all relevant experiments and did not produce any significant effect. Similar findings were obtained in primary HBE cells. The degree of G1-mediated inhibition of UTP-, UDP-, and ATPγS-induced increases in Ca2+ were 46.4, 53.4, and 54.6 %, respectively.

Inhibitory effects of E2 or G1 on nucleotide-induced increases in Ca2+ in HBE cells. 16HBE14o- (n = 4–6) were stimulated with 10-μM UTP (a), 100-μM UDP (b), or 10-μM ATPγS (c) in the absence (nucleotide alone) or presence of E2 (100 nM) or G1 (10 nM) for 10 min, and the maximal increase in the Fura-2 fluorescence ratio was quantified. d Statistical results for a to c. Similarly, primary HBE cells (n = 4–7) were stimulated with 10-μM UTP (e), 100-μM UDP (f), or 10-μM ATPγS (g) in the absence (nucleotide alone) or presence of G1 (10 nM) for 10 min, and h shows the summarized data. i, j UTP-induced Ca2+ increases in 16HBE14o- cells were inhibited in a concentration-dependent manner by E2 (i) or G1 (h) (n = 3–8). Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to nucleotide alone
Various concentrations of E2 and G1 were used to examine their inhibitory effects on P2Y receptor-mediated Ca2+ signaling in 16HBE14o- cells. The 16HBE14o- cells were pretreated with E2 (1–300 nM) or G1 (0.01–100 nM) for 10 min and then stimulated by 10-μM UTP in the presence of E2 or G1. Both E2 and G1 inhibited the UTP-induced increases in Ca2+ in a concentration-dependent manner (Fig. 3i, j). The half maximal inhibitory concentration (IC50) values of E2 and G1 were 12.42 and 0.58 nM, respectively.
To confirm the specificity of the GPER-mediated inhibitory effect of G1, a newly developed GPER antagonist, G15 [14], was used. The 16HBE14o- cells were treated with 10-nM G1 in the presence or absence of 1-μM G15 for 10 min. The cells were then stimulated with 10-μM UTP. In the presence of G15, the inhibitory effect of G1 on the nucleotide-induced increase in Ca2+ was reversed in both 16HBE14o- (Fig. 4a) and primary HBE (Fig. 4b) cells. Furthermore, the inhibitory effect of E2 (100 nM) on the increase in Ca2+ was also reversed in the presence of G15 (Fig. 4c), indicating that E2 could inhibit nucleotide-induced Ca2+ signaling via GPER.

Reversal of E2- and G1-inhibited increases in Ca2+ via the GPER antagonist, G15, or GPER siRNA knockdown in HBE cells. a G15 (1 μM) reversed the inhibitory action of G1 (10 nM) on UTP (10 μM)-, UDP (100 μM)-, and ATPγS (10 μM)-induced increases in Ca2+ in 16HBE14o- cells (n = 4–5). b Similar results were obtained in primary HBE cells (n = 3–7). c G15 (1 μM) reversed the inhibitory effect of E2 (100 nM) on UTP (10 μM)- and UDP (100 μM)-induced increases in Ca2+ in 16HBE14o- cells (n = 4–6). d real-time PCR analyses of GPER expression in 16HBE14o- cells transfected with siRNA targeting GPER (siGPER) or scramble siRNA (SCR) (n = 5). e Protein band of GPER in 16HBE14o- cells transfected with siGPER or SCR. GAPDH was used as internal control (n = 4). f GFP and Fura-2 fluorescence in 16HBE14o- cells transfected with GFP-tagged siRNA. Fura-2 fluorescence intensity indicates Ca2+ density. g–i 16HBE14o- cells transfected with SCR or siGPER were stimulated with 10-μM UTP in the presence or absence of G1 (10 nM) for 10 min, and the maximal increase in the Fura-2 fluorescence ratio was quantified (n = 14–23 experiments in individual cells). Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to nucleotide alone
In addition to using a specific antagonist, we also used lentiviral vectors expressing siRNA to downregulate the GPER gene in 16HBE14o- cells. The knockdown efficiency of GPER was examined on both the mRNA and protein levels (Fig. 4d, e). The expression level of GPER in 16HBE14o- cells transfected with siRNA targeting GPER (siGPER) was 43.9 % lower than those transfected with a negative control siRNA encoding a scrambled sequence (SCR; Fig. 4f). The presence of GFP did not disturb Fura-2 fluorescence during the measurement of [Ca2+]i [5]. The successfully transfected cells were selected for calcium measurements. The data show that 10-nM G1 inhibited UTP-induced increases in Ca2+ in the SCR control group (Fig. 4g), whereas no significant inhibitory effect was observed in the siGPER group treated with G1 (Fig. 4h, i). These results indicate that the inhibitory effect of G1 was mediated by GPER.
Effects of E2 and G1 on nucleotide-induced Ca2+ release and influx
Intracellular increases in calcium could result from the release of ER stores or influx through store-operated calcium (SOC) channels regulated by stromal interaction molecule 1 (STIM1) [26]. To examine the effects of E2 and G1 on P2Y receptor-activated intracellular Ca2+ release (first phase) and influx (second phase), epithelia were exposed to Ca2+-free solution in the presence or absence of G1 or E2 for 10 min and then stimulated with UDP or UTP together with G1 or E2. Subsequently, Ca2+ (2.5 mM) was added back to the perfusate to induce Ca2+ influx. Typical recordings of fluorescence ratios in control (Fig. 5a) and E2-treated (Fig. 5b) cells show that both 100-nM E2 and 10-nM G1 inhibited UDP (100 μM)-induced Ca2+ release in 16HBE14o- cells (Fig. 5c). However, only E2 inhibited UDP (100 μM)-induced Ca2+ influx (Fig. 5d). Moreover, the inhibitory effect of 10-nM G1 on UDP (100 μM)- or UTP (10 μM)-induced Ca2+ release (first phase) was reversed by 1-μM G15 (Fig. 5e). Taken together, these data demonstrate that E2 inhibited nucleotide-induced Ca2+ release and Ca2+ influx, whereas the GPER agonist, G1, only inhibited P2Y receptor-mediated Ca2+ release.

Inhibitory effects of E2 and G1 on the two phases of nucleotide-induced Ca2+ increase. a, b Representative recordings of Fura-2 fluorescence ratios in control (a) and E2-treated (b) cells. 16HBE14o- cells were superfused with nominally Ca2+-free solution for 10 min in the absence (control) or presence of E2 (100 nM) and then exposed to UDP (100 μM). Once [Ca2+]i had returned to basal levels, external Ca2+ was subsequently restored (2.5 mM). c, d The inhibitory effect of E2 (c; 100 nM) and G1 (d; 10 nM) on UDP (100 μM)-induced Ca2+ release (first phase) and Ca2+ influx (second phase) in 16HBE14o- cells, respectively (n = 6–9). e Cells were stimulated with nucleotide (UTP or UDP) alone or in the presence of G1 (10 nM) with or without G15 (1 μM), and Ca2+ release (first phase) was quantified by Fura-2 fluorescence microscopy (n = 4–8). f–h Representative recordings and quantification of Mn2+ quenching of Fura-2 fluorescence. 16HBE14o- cells preincubated with E2 (100 nM) or G1 (10 nM) were stimulated with 10-μM UTP for 5 min before adding Mn2+ (1 mM). In f, maximal fluorescence (360 nm wavelength) was set to 100 % and fluorescence quench was measured 120 s after Mn2+ application. g Compiled data showing the percentage of fluorescence decrease 120 s after adding Mn2+ in cells treated with UTP alone or in the presence of E2 or G1. h Summarized data showing the change in slope before and after Mn2+ application. Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to nucleotide alone
The manganese quench technique was also applied to report calcium influx through plasma membrane channels. The quenching of Fura-2 fluorescence by Mn2+ was measured at the Ca2+-independent excitation wavelength of Fura-2 (360 nm). When preincubating the cells with 100-nM E2 for 10 min, the rate of change on Mn2+ quenching was significantly decreased compared to untreated or G1 pretreated 16HBE14o- cells (Fig. 5f, g). Similarly, the percentage decrease of Fura-2 fluorescence 120 s after Mn2+ application also dropped significantly in E2 pretreated cells but not in G1 pretreated cells (Fig. 5h). These results indicate that E2, but not G1, blocked calcium influx through the plasma membrane.
Calcium release from the ER mainly occurs via inositol 1,4,5-trisphosphate receptors (IP3R) in human airway epithelial cells, including 16HBE14o- cells [40]. The activation of various subtypes of P2Y receptors causes an increase in Ca2+ via the phospholipase C (PLC)-IP3 signaling cascade [25]. Because the lifetime of IP3 within the cell before it is transformed into IP2 and IP1 is very short, IP1 accumulation levels can be used to represent IP3 levels in cells. To induce IP1 accumulation by activating the P2Y receptor-mediated signaling pathway in 16HBE14o- cells, 10-μM UTP (Fig. S1a) or 100-μM UDP (Fig. S1b) was used. Dimethyl sulfoxide (DMSO), the solvent used for E2 and G1, did not affect IP1 accumulation. UTP- or UDP-induced IP1 could be blocked by U73122 (10 μM), an inhibitor of PLC, whereas E2 (10 and 100 nM) or G1 (10 nM) had no effect on UTP- or UDP-induced IP1 accumulation.
The effects of E2 and G1 on SOC influx was determined by fluorescence resonance energy transfer (FRET) using 16HBE14o- cells co-expressing CFP- and YFP-STIM1. STIM1 proteins are known to undergo oligomerization in response to Ca2+ depletion in the ER [21]. The addition of ATP triggered Ca2+ release from the ER in 16HBE14o- cells, leading to the oligomerization of CFP- and YFP-STIM1, thus generating FRET signals (Fig. 6a). In controls and in cells pretreated with 10-nM G1, the addition of ATP elicited approximately a 20 % increase in N FRET , whereas in cells pretreated with 100-nM E2, the ATP-induced N FRET signal was significantly attenuated (Fig. 6a, b). Prominent increase in FRET signal was observed due to the oligomerization and translocation of STIM1 near the plasma membrane in control and G1 pretreated 16HBE14o- cells (Fig. 6c). However, the FRET signal was greatly reduced in cells pretreated with E2 compared to controls and G1 pretreated cells (Fig. 6c). To further investigate that the increase in FRET signal was due to oligomerization of STIM1, we measured STIM1 puncta formation in response to ATP stimulation in the absence of extracellular Ca2+. Addition of ATP to control 16HBE14o- cells transfected with YFP-STIM1 generated significant amount of STIM1 puncta dots (Fig. 6d, e). ATP induced a comparable amount of STIM1 puncta formation in cells pretreated with 10-nM G1 (Fig. 6d, e). However, the STIM1 puncta formation was significantly reduced in cells pretreated with 100-nM E2 (Fig. 6d, e). Taken together, these results suggest that E2 attenuates SOC by inhibiting STIM1 oligomerization.

E2 impairs STIM1 oligomerization during Ca2+ depletion from the ER. a Time course of changes in N FRET in 16HBE14o- cells co-transfected with CFP- and YFP-STIM1. Cells were pretreated with 10-nM G1 or 100-nM E2 in Ca2+-free HEPES-HBSS for 15 min. Cells pretreated with DMSO vehicle were used as controls (CTRL). Ca2+ from the ER was depleted by the addition of 10-μM ATP at t = 15 min. The experiment was repeated five times, and N FRET of the cells was normalized to its initial value. b Statistical analysis of the changes in N FRET at t = 20 min using one-way ANOVA with Tukey’s multiple comparison test, **P < 0.01. c Representative micrographs highlighting STIM1 localization before (t = 15 min) and after (t = 0 min) Ca2+ depletion from the ER. Images were captured in the CFP (upper panel) and FRET (lower panel) channels. Scale bar, 10 μm. d Representative confocal images depict the formation of YFP-STIM1 puncta in 16HBE14o- cells after ER Ca2+ was depleted by ATP. Cell were pretreated with DMSO (CTRL), 10-nM G1, or 100-nM E2 in Ca2+-free HEPES-HBSS for 15 min followed by ER Ca2+ depletion induced with 10-μM ATP. e STIM1 puncta were determined by ImageJ, and the amount of STIM1 puncta was summarized as mean ± SEM from six isolated cells obtained from three individual experiments of each treatment. Images were taken in ×60 oil immersion objective, and the scale bar represents 10 μm
E2- and G1-induced cAMP production
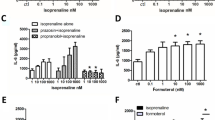
GPER activation may initiate the cAMP and protein kinase A (PKA) signaling pathways [50]. Thus, real-time cAMP levels were monitored in 16HBE14o- cells stimulated by G1. After the addition of 10-nM G1 to 16HBE14o- cells, the cAMP levels increased (Fig. 7a). This stimulatory effect of G1 on cAMP levels was blocked by the GPER antagonist, G15 (Fig. 7b), indicating that the effect was specific. Similarly, E2 (100 nM) also induced an increase in cAMP levels (change in emission ratio = 0.12 ± 0.02, n = 5). When the cells were treated with 1-μM G15 for 10 min before the addition of various concentrations of G1, cAMP production was significantly inhibited compared to G1 alone.

G1-stimulated cAMP mobilization and inhibition of increases in Ca2+ via cAMP-dependent PKA signaling in 16HBE14o- cells. a Real-time cAMP changes (represented by normalized CFP/FRET emission ratios) recorded in 16HBE14o- cells stimulated with G1 (10 nM). b 16HBE14o- cells were stimulated with different concentrations of G1 (1, 10, and 100 nM) alone or in the presence of G15 (1 μM). G15 was added 10 min before the addition of G1. Forskolin (25 μM) was used as a positive control to stimulate a maximal increase in cAMP levels (n = 4–8). c, d 16HBE14o- cells were treated with the adenylyl cyclase inhibitors, MDL 12330A or SQ 22536 (0.1, 1, and 10 μM), for 10 min followed by stimulation of the cells with G1 (10 nM) (n = 4–6). e, f Cells were treated with E2 (100 nM) or G1 (10 nM) in the presence or absence of the cAMP-dependent PKA inhibitor, H89 (10 μM), for 10 min, followed by stimulation of the cells with UTP (10 μM) or UDP (100 μM) (n = 3–5). Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to G1 or nucleotide alone
To demonstrate the involvement of adenylyl cyclase (AC) in cAMP production, two AC inhibitors, MDL 12330A and SQ 22536, were used. G1-induced cAMP production was significantly inhibited by 10-min treatments with MDL 12330A (0.1, 1, and 10 μM) in a concentration-dependent manner (Fig. 7c). Similar results were obtained with SQ 22536 (Fig. 7d). These results suggest that GPER is coupled to the activation of AC, likely via the Gs alpha subunit, to stimulate an increase in cAMP levels in 16HBE14o- cells.
The role of PKA in E2- and G1-mediated inhibition of calcium increases
We next determined whether the cAMP-dependent pathway was involved in the inhibitory effects of E2 and G1 on the P2Y-induced increase in Ca2+ in 16HBE14o- cells. H89, a PKA inhibitor, was used to inhibit downstream signaling targets of cAMP. The inhibitory effects of G1 (10 nM) on 10-μM UTP- or 100-μM UDP-induced increases in Ca2+ were reversed by co-incubation of the cells with H89 (10 μM) for 10 min (Fig. 7e). Similar results were obtained with 100-nM E2 (Fig. 7f). These results demonstrate that the inhibitory effects of E2 and G1 on P2Y receptor-induced Ca2+ signaling are mediated via the activation of a cAMP-dependent PKA pathway.
Effects of the GPER agonist, G1, on nucleotide- or poly-l-arginine-induced cytokine production in HBE cells
Our previous study indicates that the levels of two proinflammatory cytokines, interleukin 8 (IL-8) and IL-6, increase significantly after airway bronchial epithelial cells are stimulated by extracellular nucleotides when the cells are damaged by poly-l-arginine [20]. Therefore, the effects of G1 on nucleotide- or poly-l-arginine-induced IL-8 and IL-6 production were determined in HBE cells. ATPγS (10 μM) or poly-l-arginine (3 μM) was incubated with the cells for 6 h in the presence or absence of G1 (10 nM). Significant inhibition of ATPγS- or poly-l-arginine-stimulated IL-8 release was observed under these conditions (Fig. 8a, b). The addition of 10-nM G1 also showed a significant inhibitory effect on ATPγS-stimulated IL-6 release in 16HBE14o- cells (Fig. 8c). These results indicate that GPER may play an important role in inhibiting proinflammatory cytokine secretion stimulated by P2Y receptor activation in HBE cells.

G1 inhibited nucleotide-stimulated IL-8 and IL-6 release in 16HBE14o- cells. a–c 16HBE14o- cells were treated with ATPγS (a, c; 10 μM) or poly-l-arginine (b; 1 μM) for 6 h in the presence or absence of G1 (10 nM). The supernatant was then collected, and IL-8 or IL-6 was quantified using an ELISA kit. The levels of IL-6 and IL-8 were corrected against vehicle control alone and expressed as the net release. Data are expressed as the mean ± SEM. *P < 0.05 compared to ATPγS or poly-l-arginine alone (n = 3–8). d Schematic diagram showing the GPER function in regulating P2Y receptor-mediated Ca2+ signaling and cytokine secretion in human bronchial epithelia. In HBE cells, nucleotides, such as UTP, UDP, and ATP, activate P2Y receptors to modulate intracellular increases in Ca2+, which lead to the release of proinflammatory cytokines, such as IL-8 and IL-6. Activation of GPER by E2 or its specific agonist, G1, attenuates both the nucleotide-evoked increases in Ca2+ and the cytokine secretion. This inhibitory effect is likely due to the activation of a GPER-mediated cAMP/PKA pathway. Note that whether G1 and E2 can translocate through the plasma membrane to act on the receptor that needs to be verified
Discussion
GPER is known to play important roles in multiple tissues, including the heart, brain, lung, liver, skeletal muscle, and kidney [33]. However, few reports have described the expression and function of GPER in airway epithelial cells. Only one study described the relatively high expression of GPER in human nonsmall cell, lung cancer cell lines compared to immortalized normal lung bronchial epithelial cells [23]. This study therefore reports for the first time the expression and function of GPER as an anti-inflammatory component in human bronchial epithelia and highlights that GPER likely serves this role through its opposing effects on the proinflammatory pathway activated by the P2Y receptors in inflamed airway epithelia.
The mRNA and protein expression levels of GPER in primary HBE and 16HBE14o- cells were confirmed by RT-PCR and Western blotting, respectively. Although GPER localizes to the ER [35] and plasma membrane [17], its subcellular localization is still controversial. Overall, the localization of GPER appears to vary depending on the cell type. Our immunofluorescence studies showed partial colocalization between the GPER and the plasma membrane with no overlap between GPER and Golgi, or nucleus. Besides, a very small amount of GPER was localized in ER. These results were similar to those observed in osteocyte-like MLO-Y4 cells and transfected HEK-293 cells [17, 34]. Different tissues or cell types may have different subcellular GPER localizations, which may be due to the different roles GPER plays in various cell types, such as cell proliferation, apoptosis, and immune responses [27]. However, it should be noted that, even in the same cells, GPER could change its location via endocytotic processes. Two reports have demonstrated that GPER can be trafficked intracellularly from the plasma membrane [8, 38]. This unique mechanism could decrease the amount of GPER at the plasma membrane and protect cells from chronic signaling. Thus, we could not exclude this possibility, but the exact details of this intriguing membrane receptor trafficking pathway in human bronchial epithelia require further study.
In this study, activation of GPER by G1 did not alter basal [Ca2+]i levels, but it did significantly inhibit P2Y receptor-mediated increases in Ca2+. This inhibitory effect was not P2Y receptor subtype-specific, because G1 inhibited the Ca2+ responses elicited by different P2Y receptor subtype ligands (e.g., UTP, UDP, and ATPγS). The inhibitory effect could be reversed when pretreating with GPER-specific antagonist, G15. Besides, siRNA knockdown of GPER was also applied to further confirm the role of GPER. Various transfect reagents (e.g., lipofectamine 2000, lipofectamine RNAiMax, siPORT NeoFX, DharmaFECT 1 and 4) have been applied in our experiment for GPER silencing; however, the knockdown efficiency was low and inconsistent (data not shown). Therefore, lentiviral-mediated silencing approach was adopted and a stable silenced cell line was generated with better knockdown efficiency on GPER. To reveal whether G1 inhibited intracellular increases in Ca2+ by blocking Ca2+ release and/or Ca2+ influx, we characterized the two phases of Ca2+ increase by perfusing cells with a Ca2+-free Krebs-Henseleit Buffer, followed by a Ca2+-containing solution. The results showed that E2 inhibited both P2Y receptor-mediated Ca2+ release and Ca2+ influx, whereas G1 only inhibited Ca2+ release. However, the observed difference in Ca2+ signals could be due to regulation of Ca2+ pumps. The inhibitory effect of E2 on Ca2+ influx was further confirmed by the data obtained from Mn2+ quench experiments. Taken together, these data suggest that the activation of GPER only inhibits P2Y-activated IP3-mediated Ca2+ release, whereas classical E2 receptors activated by E2 played a role in regulating Ca2+ influx. FRET microscopy further confirmed that the inhibitory effect of E2, but not G1, on SOC influx was due to the inhibition of STIM1 oligomerization. Our findings were similar to those reported recently [39], which showed that E2 can signal nongenomically by inhibiting basal phosphorylation of STIM1, leading to a reduction of SOC entry in human airway cells.
The inhibition of Ca2+ release by G1 could occur via different pathways. For example, GPER activation might block the activity of PLCβ to diminish the synthesis of IP3, inhibit the activity of IP3R to release Ca2+ from ER, or stimulate Ca2+ uptake into stores by activating the endoplasmic Ca2+-ATPase pump. To investigate the detailed mechanism underlying the inhibitory effects of E2 and G1 on P2Y receptor-mediated Ca2+ mobilization, we conducted another series of studies to examine whether E2 or G1 could inhibit IP3 production. We measured IP1 accumulation to determine IP3 levels. IP1 accumulation induced by both UDP and UTP was significantly inhibited by the PLC inhibitor, U73122, whereas the addition of E2 or G1 showed no inhibitory effects. This result indicates that the E2- or G1-mediated inhibition of P2Y receptor-mediated Ca2+ signaling was not related to a change in IP3 levels. Thus, the observed inhibition may be due to the inhibition of IP3 independent of Ca2+ release. Alternatively, it may be due to an unidentified signaling pathway or molecule that interferes with the interaction between IP3 and IP3R [2].
GPER couples to different signaling pathway(s), including the cAMP/PKA pathway [50]. Our data suggest that GPER in human bronchial epithelia was coupled to AC, resulting in an increase in cAMP levels. Notably, blocking the downstream target of cAMP with H89 reversed the inhibitory effect of G1 on P2Y receptor-mediated Ca2+ signaling, and cAMP-dependent protein kinase reportedly inhibits IP3-induced Ca2+ release in human bone marrow cells [42]. The cAMP/PKA pathway may inhibit receptor-operated calcium entry (ROCE) via transient receptor potential canonical channel 6 (TRPC6). TRPC6 is expressed in both undifferentiated and differentiated primary HBE cells [12]. Calcium influx mediated by TRPC6 is functionally coupled to calcium-activated chloride channel activity in human airway epithelial cells [4] and can be regulated by P2Y receptor activation in mouse podocytes [37]. A recent study suggests that the cAMP/PKA signaling pathway can inhibit endothelin type A receptor-mediated ROCE via TRPC6 by phosphorylation of Ser28 site in human embryonic kidney 293 cells [22]. Although G1 did not have any significant effect on nucleotide-mediated Ca2+ influx, we did not explicitly examine P2Y receptor-mediated ROCE in this study. It would be interesting for future research to examine if GPR30 can inhibit P2Y receptor-mediated ROCE via TRPC6 in human airway epithelia. In addition to PKA, Epac is another downstream target of cAMP that transduces diverse cellular actions [7, 9]. The cAMP increases evoked by G1 are sufficient to activate Epac. Interestingly, our previous study demonstrates that both Epac 1 and Epac 2 are expressed in 16HBE14o- cells [24]. Therefore, we could not exclude the possibility that some of the observed inhibitory effects were mediated through activation of Epac. Moreover, our recent study demonstrates that the proinflammatory effect of nucleotides is mediated via an increase in [Ca2+]i after P2Y receptor activation. Treating 16HBE14o- cells with the intracellular Ca2+ chelator, BAPTA-AM, but not H89, inhibited P2Y receptor-mediated IL-6 and IL-8 secretion [20]. Taken together, GPER likely inhibits the P2Y receptor-mediated inflammatory response by downregulating [Ca2+]i in human airway epithelia. A recent study reported that Ca2+-dependent calmodulin can regulate GPER-dependent signaling at the receptor level [44]. Therefore, a P2Y receptor-mediated increase in Ca2+ could, in turn, regulate GPER function, but the details of the possible cross talk between the two receptors require further investigation. 16HBE14o- cells were cultured in MEM without phenol red in some experiments since phenol red may serve as a weak estrogen mimic. However, no significant differences have been observed in terms of GPER expression and the inhibitory effect of G1 on P2Y receptor-mediated cytokine secretion and Ca2+ increase (data not shown) in cells cultured in MEM with or without phenol red.
In summary, this study characterizes the expression, localization, and role of GPER, as well as its interaction with P2Y receptors, that were co-expressed in human bronchial epithelia. Activation of GPER by E2 or its specific agonist, G1, rapidly attenuated a nucleotide-evoked increase in Ca2+, whereas the specific GPER antagonist, G15, reversed this GPER-mediated inhibition. Furthermore, E2 and G1 also inhibited nucleotide-induced cytokine release. The inhibitory effects on P2Y receptor-mediated Ca2+ mobilization and cytokine secretion are likely due to GPER-mediated activation of a cAMP-dependent PKA pathway in human bronchial epithelia (Fig. 8d).
References
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Ahuja M, Jha A, Maleth J, Park S, Muallem S (2014) cAMP and Ca2+ signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55:385–393
Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, Prossnitz ER, Dun NJ, Jordan VC (2010) The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res 70:1184–1194
Bertrand J, Dannhoffer L, Antigny F, Vachel L, Jayle C, Vandebrouck C, Becq F, Norez C (2015) A functional tandem between transient receptor potential canonical channels 6 and calcium-dependent chloride channels in human epithelial cells. Eur J Pharmacol 765:337–345
Billing-Marczak K, Przybyszewska M, Kuznicki J (1999) Measurements of [Ca2+] using fura-2 in glioma C6 cells expressing calretinin with GFP as a marker of transfection: no Ca2+-buffering provided by calretinin. Biochim Biophys Acta 1449:169–177
Boittin FX, Petermann O, Hirn C, Mittaud P, Dorchies OM, Roulet E, Ruegg UT (2006) Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J Cell Sci 119:3733–3742
Borland G, Smith BO, Yarwood SJ (2009) EPAC proteins transduce diverse cellular actions of cAMP. Br J Pharmacol 158:70–86
Cheng SB, Graeber CT, Quinn JA, Filardo EJ (2011) Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids 76:892–896
Cheng X, Ji Z, Tsalkova T, Mei F (2008) Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin Shanghai 40:651–662
Chow AW, Liang JF, Wong JS, Fu Y, Tang NL, Ko WH (2010) Polarized secretion of interleukin (IL)-6 and IL-8 by human airway epithelia 16HBE14o- cells in response to cationic polypeptide challenge. PLoS One 5, e12091
Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, Fricks I, Young SL, Tarran R (2008) 17beta-Estradiol inhibits Ca2+-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia. J Clin Invest 118:4025–4035
Corteling RL, Li S, Giddings J, Westwick J, Poll C, Hall IP (2004) Expression of transient receptor potential C6 and related transient receptor potential family members in human airway smooth muscle and lung tissue. Am J Respir Cell Mol Biol 30:145–154
De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, Pezzi V, Belfiore A, Maggiolini M (2013) Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene 32:678–688
Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER (2009) In vivo effects of a GPR30 antagonist. Nat Chem Biol 5:421–427
Fasolato C, Hoth M, Matthews G, Penner R (1993) Ca2+ and Mn2+ influx through receptor-mediated activation of nonspecific cation channels in mast cells. Proc Natl Acad Sci U S A 90:3068–3072
Feige JN, Sage D, Wahli W, Desvergne B, Gelman L (2005) PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc Res Tech 68:51–58
Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P (2007) Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148:3236–3245
Gruszczynska-Biegala J, Pomorski P, Wisniewska MB, Kuznicki J (2011) Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS One 6, e19285
Haggerty CL, Ness RB, Kelsey S, Waterer GW (2003) The impact of estrogen and progesterone on asthma. Ann Allergy Asthma Immunol 90:284–291
Hao Y, Liang JF, Chow AW, Cheung WT, Ko WH (2014) P2Y6 receptor-mediated proinflammatory signaling in human bronchial epithelia. PLoS One 9, e106235
Hewavitharana T, Deng X, Wang Y, Ritchie MF, Girish GV, Soboloff J, Gill DL (2008) Location and function of STIM1 in the activation of Ca2+ entry signals. J Biol Chem 283:26252–26262
Horinouchi T, Higa T, Aoyagi H, Nishiya T, Terada K, Miwa S (2012) Adenylate cyclase/cAMP/protein kinase A signaling pathway inhibits endothelin type A receptor-operated Ca2+ entry mediated via transient receptor potential canonical 6 channels. J Pharmacol Exp Ther 340:143–151
Jala VR, Radde BN, Haribabu B, Klinge CM (2012) Enhanced expression of G-protein coupled estrogen receptor (GPER/GPR30) in lung cancer. BMC Cancer 12:624
Lau WK, Chow AW, Au SC, Ko WH (2011) Differential inhibitory effects of CysLT1 receptor antagonists on P2Y6 receptor-mediated signaling and ion transport in human bronchial epithelia. PLoS One 6, e22363
Lazarowski ER, Boucher RC (2009) Purinergic receptors in airway epithelia. Curr Opin Pharmacol 9:262–267
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15:1235–1241
Maggiolini M, Picard D (2010) The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol 204:105–114
Mortaz E, Folkerts G, Nijkamp FP, Henricks PA (2010) ATP and the pathogenesis of COPD. Eur J Pharmacol 638:1–4
Pan Z, Zhao X, Brotto M (2012) Fluorescence-based measurement of store-operated calcium entry in live cells: from cultured cancer cell to skeletal muscle fiber. J Vis Exp 60, e3415
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45
Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ (2008) Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol 70:165–190
Prossnitz ER, Barton M (2011) The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 7:715–726
Prossnitz ER, Sklar LA, Oprea TI, Arterburn JB (2008) GPR30: a novel therapeutic target in estrogen-related disease. Trends Pharmacol Sci 29:116–123
Ren J, Wu JH (2012) 17beta-estradiol rapidly activates calcium release from intracellular stores via the GPR30 pathway and MAPK phosphorylation in osteocyte-like MLO-Y4 cells. Calcif Tisse Int 90:411–419
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630
Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (1997) Gender gap in cystic fibrosis mortality. Am J Epidemiol 145:794–803
Roshanravan H, Dryer SE (2014) ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol 306:F1088–F1097
Sanden C, Broselid S, Cornmark L, Andersson K, Daszkiewicz-Nilsson J, Martensson UE, Olde B, Leeb-Lundberg LM (2011) G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol 79:400–410
Sheridan JT, Gilmore RC, Watson MJ, Archer CB, Tarran R (2013) 17beta-Estradiol inhibits phosphorylation of stromal interaction molecule 1 (STIM1) protein: implication for store-operated calcium entry and chronic lung diseases. J Biol Chem 288:33509–33518
Sienaert I, Huyghe S, Parys JB, Malfait M, Kunzelmann K, De Smedt H, Verleden GM, Missiaen L (1998) ATP-induced Ca2+ signals in bronchial epithelial cells. Pflugers Arch 436:40–48
Tas PW, Stossel C, Roewer N (2008) Inhibition of the histamine-induced Ca2+ influx in primary human endothelial cells (HUVEC) by volatile anaesthetics. Eur J Anaesthesiol 25:976–985
Tertyshnikova S, Fein A (1998) Inhibition of inositol 1,4,5-trisphosphate-induced Ca2+ release by cAMP-dependent protein kinase in a living cell. Proc Natl Acad Sci U S A 95:1613–1617
Thomas P, Pang Y, Filardo EJ, Dong J (2005) Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632
Tran QK, Vermeer M (2014) Biosensor-based approach identifies four distinct calmodulin-binding domains in the G protein-coupled estrogen receptor 1. PLoS One 9, e89669
Tutdibi O, Brinkmeier H, Rudel R, Fohr KJ (1999) Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J Physiol 515:859–868
van der Krogt GN, Ogink J, Ponsioen B, Jalink K (2008) A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS One 3, e1916
Wong AM, Chow AW, Au SC, Wong CC, Ko WH (2009) Apical versus basolateral P2Y6 receptor-mediated Cl− secretion in immortalized bronchial epithelia. Am J Respir Cell Mol Biol 40:733–745
Wong CH, Ko WH (2002) Stimulation of Cl− secretion via membrane-restricted Ca2+ signaling mediated by P2Y receptors in polarized epithelia. J Biol Chem 277:9016–9021
Xia Z, Liu Y (2001) Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J 81:2395–2402
Yu X, Li F, Klussmann E, Stallone JN, Han G (2014) G protein-coupled estrogen receptor 1 mediates relaxation of coronary arteries via cAMP/PKA-dependent activation of MLCP. Am J Physiol Endocrinol Metab 307:E398–E407
Zhang JY, Kowal DM, Nawoschik SP, Dunlop J, Pausch MH, Peri R (2010) Development of an improved IP1 assay for the characterization of 5-HT2C receptor ligands. Assay Drug Dev Technol 8:106–113
Acknowledgments
We thank Dr. D. C. Gruenert (University of Vermont, Burlington, USA) for the generous gift of the 16HBE14o- cells and Dr. K. Jarlink (The Netherlands Cancer Institute, Amsterdam, the Netherlands) for supplying the Epac sensor. We thank Dr. Bernard Lam, Department of Physiology, National University of Singapore, and Prof. Brian B. J. Harvey, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Ireland, for their expert advice.
Funding
This work was supported by a grant from the Research Grants Council General Research Fund (Ref. No. 466611) awarded to W.H. Ko.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1437 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hao, Y., Chow, A.W., Yip, W.C. et al. G protein-coupled estrogen receptor inhibits the P2Y receptor-mediated Ca2+ signaling pathway in human airway epithelia. Pflugers Arch - Eur J Physiol 468, 1489–1503 (2016). https://doi.org/10.1007/s00424-016-1840-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1840-7